

Abstract
PURPOSE
Rhodopsin mutations account for approximately 25% of human autosomal dominant retinal degenerations. However, the molecular mechanisms by which rhodopsin mutations cause photoreceptor cell death are unclear. Mutations in genes involved in the termination of rhodopsin signaling activity have been shown to cause degeneration by persistent activation of the phototransduction cascade. This study examined whether three disease-associated rhodopsin substitutions Pro347Ser, Lys296Glu, and the triple mutant Val20Gly, Pro23His, Pro27Leu (VPP) caused degeneration by persistent transducin-mediated signaling activity.
METHODS
Transgenic mice expressing each of the rhodopsin mutants were crossed onto a transducin α-subunit null (Trα−/−) background, and the rates of photoreceptor degeneration were compared with those of transgenic mice on a wild-type background.
RESULTS
Mice expressing VPP-substituted rhodopsin had the same severity of degeneration in the presence or absence of Trα. Unexpectedly, mice expressing Pro347Ser- or Lys296Glu-substituted rhodopsins exhibited faster degeneration on a Trα−/− background. To test whether the absence of α-transducin contributed to degeneration by favoring the formation of stable rhodopsin/arrestin complexes, mutant Pro347Ser+, Trα−/− mice lacking arrestin (Arr−/−) were analyzed. Rhodopsin/arrestin complexes were found not to contribute to degeneration.
CONCLUSIONS
The authors hypothesized that the decay of metarhodopsin to apo-opsin and free all-trans-retinaldehyde is faster with Pro347Ser-substituted rhodopsin than it is with wild-type rhodopsin. Consistent with this, the lipofuscin fluorophores A2PE, A2E, and A2PE-H2, which form from retinaldehyde, were elevated in Pro347Ser transgenic mice.
Autosomal dominant retinitis pigmentosa (ADRP) is a genetically heterogeneous group of inherited retinal degenerations that cause blindness in humans. Mutations in several genes encoding proteins of the phototransduction cascade have been causatively associated with ADRP.1 Rhodopsin mutations, more than 100 of which have been identified, collectively account for the most common known cause of ADRP.1 Thus, it is important to elucidate the molecular mechanisms underlying cell death in this class of mutations.
Previous research from our laboratory has described transgenic mouse mutants that cause degeneration by prolonged activation of the phototransduction cascade.2 Null mutations in the rhodopsin kinase3 and arrestin4 genes, each of which plays a role in terminating rhodopsin activity, caused light-dependent retinal degeneration. Complete protection from retinal degeneration was observed when either mutation was crossed onto a Trα−/− background.5 Retinal degeneration in Rpe65 null (Rpe65−/−) mutant mice6 was also blocked completely when placed on a Trα−/− background.7 Degeneration in Rpe65−/− mice results from persistent signaling by apo-opsin caused by impaired synthesis of the 11-cis-retinaldehyde (11-cis-RAL) chromophore,2 which functions as an inverse agonist.
Constitutively active rhodopsin mutants that activate transducin in a light-independent manner have previously been described under in vitro conditions.8–21 Constitutive signaling activity in Drosophila is also associated with retinal degeneration.22,23 Three activated rhodopsin mutants associated with congenital night blindness have been reported in humans.13,14,24,25 In this study, we investigated whether persistent photosignaling activity by rhodopsin mutants was also a cause of retinal degeneration.
The severity of retinal degeneration was compared in transgenic mouse lines carrying one of three mutant rhodopsin transgenes placed on either a wild-type (WT; Trα+/+) or a Trα−/− genetic background. If abnormal rhodopsin signaling caused retinal degeneration, we predicted that rhodopsin mutants on the Trα−/− background would be protected from degeneration. Importantly, retinas of Trα−/− mice did not degenerate except at advanced ages (6+ months), when less than 10% of photoreceptors were lost.26
We studied transgenic mice expressing three forms of substituted rhodopsin that cause autosomal dominant retinitis pigmentosa in humans. VPP transgenic mice express the disease-associated Pro23His plus two substitutions, Val20Gly and Pro27Leu. VPP-rhodopsin mRNA is expressed at levels equivalent to those of WT,27 although relative levels of substituted protein compared to WT protein are lower.28 Histologic analysis of VPP transgenic mice showed abnormal disc morphogenesis at the base of rod outer segments.29 Immunohisto-chemical methods localized most VPP-substituted rhodopsin to rod outer segment disks.28
The second transgenic mouse line we studied expressed Lys296Glu-substituted opsin (K296E),30 also associated with retinitis pigmentosa in humans.31 The Lys296 residue is the Schiff base attachment site for the 11-cis-RAL chromophore. Substitutions at this residue prevent association of apo-opsin with 11-cis-RAL to form rhodopsin. In vitro, Lys296Glu-substituted opsin constitutively activated α-transducin independently of light. It remains a point of controversy whether Lys296Glu-substituted opsin is phosphorylated by rhodopsin kinase and does8,30 or does not9 bind arrestin. In contrast to results from in vitro studies, the mutant opsin in Lys296Glu transgenic mice localized to rod outer segments was constitutively phosphorylated and bound to arrestin.30
The third transgenic mouse line we studied expressed disease-associated Pro347Ser-substituted rhodopsin.32 In vitro studies showed this mutant rhodopsin regenerated normally with 11-cis-RAL and, on light exposure, exhibited a spectral absorbance shift comparable to WT rhodopsin.33 Pro347Ser-substituted rhodopsin also activated α-transducin, was phosphorylated by rhodopsin kinase, and subsequently bound arrestin.34 Although Pro347Ser-substituted rhodopsin was present in outer segments of these transgenic mice, mutant rhodopsin was also present in inner segments and accumulated in submicrometer-sized extracellular vesicles near the junction between outer and inner segments.32
In the present study, we examined whether VPP-, Lys296Glu-, or Pro347Ser- substituted rhodopsins triggered α-transducin-mediated cell death by producing transgenic mice expressing these mutant rhodopsins on a Trα−/− genetic background. We showed that the absence of α-transducin had no effect on the rate of photoreceptor degeneration in VPP transgenic mice. Unexpectedly, we observed accelerated retinal degeneration in mice expressing Lys296Glu- and Pro347Ser-substituted rhodopsin mutants on a Trα−/− background. This contrasts with the protection conferred by the absence of α-transducin in rhodopsin kinase,5 arrestin,5 and rpe657 null mutant mice. Our results showed that persistent photosignaling was not a mechanism of retinal degeneration for these three rhodopsin mutations. Further, the accelerated retinal degeneration in Lys296Glu and Pro347Ser transgenic mice suggested a mechanism whereby α-transducin conferred a protective effect. Possible explanations for the accelerated degeneration are preferential formation of rhodopsin/arrestin complexes35–40 and the destabilization of light-activated mutant rhodopsin in the absence of α-transducin.
MATERIALS AND METHODS
Animals
All procedures were carried out in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and were within the guidelines of the Tufts-New England Medical Center Institutional Animal Care and Use Committee. Rhodopsin mutant mice on a Trα+/+ and Trα−/− genetic background were maintained as independent lines under normal cyclic light (5–100 lux, in-cage readings). The mutant rhodopsin transgenes were maintained and studied in the heterozygous state. Lys296Glu line A30 and Pro347Ser line C132 rhodopsin transgenic mice were used in these studies.
Genotyping
The α-transducin genotype was determined by Southern blot or PCR analysis. For Southern blot analysis, purified genomic DNA was digested with XbaI and was probed with a radiolabeled 1.6-kb XhoI/EcoRI fragment encompassing exons 3 to 6 of Trα. For PCR analysis, DNA primers used to amplify the Trα gene were 5′-TAT CCA CCA GGA CGG GTA TTC-3′ (forward primer) for Trα1, 5′-GGG AAC TTC CTG ACT AGG GGA GG-3′ (reverse primer) for Trα2, and 5′-GCG GAG TCA TTG AGC TGG TAT-3′ (reverse primer) for Trα3. The amplification yielded a 387- and a 273-bp gene product for the WT and the null mutant allele, respectively. Primers used to amplify the Arr gene were 5′CCATCTTGTTCAATGGCCGATCCC3′ for Neo8AR, 5′GACAATGGGACTGAGATGGTGGG3′ for Tail2, and 5′GGACAGACAGCATGGCAGCCTG3′ for E2B. Amplification yielded a 280-bp WT gene product and a 349-bp null mutant gene product.
Rhodopsin transgene-positive animals were identified by PCR analysis. Both the Pro347Ser and the Lys296Glu rhodopsin mutants expressed subcloned human rhodopsin genes and were identified using the same PCR amplification strategy. Primer pairs specific to human rhodopsin were 5′-CGT TCC AAG TCT CCT GGT GT-3′ for R6 and 5′-GAC CTA GGC TCT TGT TGC TG-3′ for R7, which produced an approximately 200-bp PCR product. Annealing for 1 minute was ramped from 65°C to 58°C for 7 cycles, dropping at a rate of 1°C per cycle, and then was denatured at 94°C for 35 seconds, annealed at 56°C for 1 minute, and extended at 72°C for 40 seconds for an additional 23 cycles. VPP mutant rhodopsin was amplified using primer pairs 5′-AA CCA TGG CAG TTC TCC ATG CT-3′ for Rh3 and 5′-GTC CTT GGC CTC TCT GAA C-3′ for OP2B specific to mouse rhodopsin. After denaturation, the annealing temperature was ramped from 70°C to 60°C for 1 minute, minus 1°C per cycle for 10 cycles. This was followed by 22 cycles of denaturation at 94°C for 30 seconds, annealing at 59°C for 1 minute, and extension at 72°C for 1 minute. To distinguish the mutant from the endogenous WT rhodopsin gene, the PCR product was digested with NcoI. WT rhodopsin was cleaved to produce a 200- and a 300-bp fragment, whereas the VPP mutant opsin, which lacked the NcoI restriction site, yielded a 500-bp fragment. All mice studied were homozygous for the Leu450 allele of the Rpe65 gene.
Histology
Mice were anesthetized with 0.017 mL avertin/g body weight before cardiac perfusion of 100 mL fixative (1% paraformaldehyde, 2% glutaraldehyde in 0.1 M phosphate buffer) at a rate of 200 mL fixative/h. Eyes were oriented at the superior-most point of the eye with a cauterizing pen at the ora serrata. Eyes were excised and rotated in fixative for 2 hours at room temperature, the anterior segment was removed, and eyes were fixed overnight at 4°C. Eyecups were cut into four quadrants marked as superior nasal, superior temporal, inferior nasal, and inferior temporal. Tissue quadrants were rinsed several times in 0.1 M phosphate buffer and were fixed in 1% osmium tetroxide in 0.1 M phosphate buffer for 1 hour. After fixation, tissue quadrants were rinsed several times in PBS and passed through a dehydrating series of ethanol, with alcohol concentrations increasing from 30% to 90% by increments of 10% for 5-minute intervals. Tissue quadrants were immersed in 100% EtOH for 10 minutes three times and then propylene oxide for 10 minutes three times. Retina quadrants were soaked for 30 minutes with 33% phenol,4,4′-(l-methylethylidene) bis-polymer with (chloromethyl) oxirane (Araldite 502 resin; Ted Pella Inc., Redding, CA) in propylene oxide, then for 90 minutes in 66% phenol,4,4′-(l-methylethylidene) bis-polymer with (chloromethyl) oxirane (Araldite 502 resin; Ted Pella Inc.) in propylene oxide. Finally, eyes were infiltrated in a solution of 100% phenol,4,4′-(l-methylethylidene) bis-polymer with (chloromethyl) oxirane (Araldite 502 resin; Ted Pella Inc.) containing dodecenyl succinic anhydride and 2,4,6-Tris [dimethyl-aminomethyl] phenol (DMP-30), and the resin was hardened for 48 hours in a 60°C oven. Half-micron sections were cut on an ultramicrotome (MT6000 Sorvall Ultramicrotome; DuPont, Wilmington, DE). Sections were stained with 1% toluidine blue, 1% sodium borate, in 0.1 M phosphate buffer.
Quantification of Retinal Degeneration
Retinal sections cut along the vertical meridian of the eye at the optic nerve head were analyzed. For each animal, several sections were examined. Rows of nuclei in the outer nuclear layer (ONL) were counted at the central, superior, and inferior midperipheral retina. Values were averaged for each animal, and the average value from several animals was averaged to obtain the mean and SEM.
Analysis of A2E and A2E Precursors
All manipulations were performed on ice under dim red light (Wratten 1A; Eastman Kodak, Rochester, NY). One mouse eyecup containing retina plus RPE was homogenized in 1 mL PBS, pH 7.2. Samples were homogenized further by adding 4 mL chloroform/methanol (2:1, vol/vol), extracted by the addition of 4 mL chloroform and 3 mL dH2O, and centrifuged at 1000g for 10 minutes. Chloroform extracts were dried under a stream of argon, and the residues were dissolved in 100 µL of 2-propanol for analysis by HPLC. Phospholipid extracts were analyzed by normal-phase HPLC on a silica column (Zorbax Rx-Sil 5 µm, 250 × 4.6 mm; Agilent, Palo Alto, CA) using a liquid chromatograph equipped with photodiode-array detector (model 1100; Agilent Technologies, Wilmington, DE). The mobile phase (hexane/2-propanol/ethanol/25 mM potassium phosphate/glacial acetic acid, 485:376:100:45:0.275, vol/vol) was filtered and pumped through the system at 0.5 to 1.4 mL/min. Column and solvent temperatures were maintained at 35°C.
Lipofuscin Granule Quantitation
Mice were fixed by vascular perfusion with 2% formaldehyde and 2.5% glutaraldehyde in 100 mM sodium phosphate buffer, pH 7.2. Secondary fixation was in 1% osmium tetroxide. Eyes were dissected into quadrants, dehydrated in ethanol, and embedded in phenol,4,4′-(l-methylethylidene) bis-polymer with (chloromethyl) oxirane (Araldite 502 resin; Ted Pella Inc.). Ultrathin sections for electron microscope viewing were cut on an ultramicrotome (Ultracut UCT; Leica Microsystems, Wetzlar, Germany) and were picked up on 200 mesh uncoated copper grids. Sections were stained with uranium and lead salts and were viewed with an electron microscope (Zeiss 910; Carl Zeiss, Thornwood, NY).
Pigment epithelial fields were imaged with a digital camera (Keen-View, Lakewood, CO). Eleven fields were collected from a control mouse 2 months old, and 10 fields were collected from a control mouse 3 months old. Thirty-four fields were collected from three experimental mice 2 months old, and 23 fields were collected from two experimental mice 3 months old. Measurements were made at a constant magnification of 16,000X. Using analySIS software, the pigment epithelial cytoplasm area and each lipofuscin body were outlined. For each field, total lipofuscin area was compared with the total pigment epithelial area. Each field was considered as n = 1. Results were presented as mean ± SD. Statistical analysis was performed using the Student’s t-test.
RESULTS
No Degeneration in α-Transducin Null Mice with Normal Rhodopsin
To determine whether rhodopsin mutants caused degeneration by aberrant photosignaling, we compared the degeneration rates of rhodopsin mutant mice reared in cyclic light on a Trα+/+ or Trα−/− background. For each of the three rhodopsin mutant lines studied, degeneration was examined at several time points using an end point of greater than 50% photoreceptor cell loss. Degeneration severity was assessed by counting rows of photoreceptor cell nuclei in the ONL. The contribution of the Trα−/− phenotype to degeneration was minimal. ONL thicknesses of Trα−/− mice at 1, 2, 3, 4, and 6 months of age were comparable to age-matched WT retinas (Fig. 1A), demonstrating that the Trα−/− phenotype did not contribute to degeneration. This is consistent with our initial published characterization of Trα−/− mice.26
FIGURE 1.
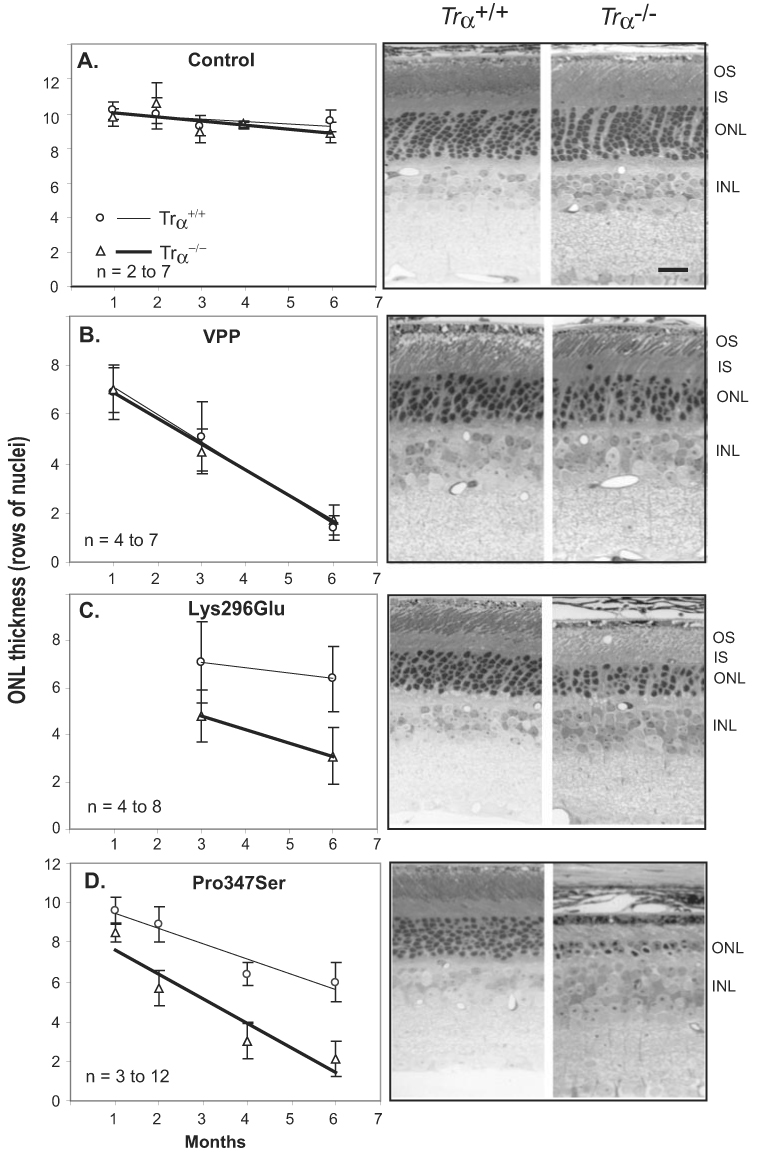
Comparison of rhodopsin mutant mice on Trα+/+ and Trα−/− genetic backgrounds. (A) Time course of degeneration in Trα+/+ (○) and Trα−/− (△) mice (left); retinal morphology at 3 months (right). (B) VPP, Trα+/+ (○) and VPP, Trα−/− (△) degeneration kinetics (left); retinal morphology at 3-months (right). (C) Lys296Glu, Trα+/+ (○) and Lys296Glu, Trα−/− (△) degeneration kinetics (left); retinal morphology at 3 months (right). (D) Pro347Ser, Trα+/+ (○) and Pro347Ser, Trα−/− (△) degeneration kinetics (left); retinal morphology at 6 months (right). OS, outer segment; IS, inner segment; ONL, outer nuclear layer; INL, inner nuclear layer. n = number of animals sampled at each time point. Scale bar, 10 µm.
Degeneration of VPP-Substituted Rhodopsin Mouse Retinas
Retinal morphologies of VPP mutant mice on Trα+/+ or Trα−/− backgrounds were compared at 1, 3, and 6 months of age (Fig. 1B). Degeneration severity increased with age. However, there was no difference in the rate of degeneration on the two different genetic backgrounds. Because the time course of retinal degeneration was similar on both the Trα+/+ and the Trα−/− backgrounds, these results indicated that VPP-substituted rhodopsin does not cause photoreceptor cell death by activating the visual transduction cascade.
Accelerated Retinal Degeneration of Lys296Glu-Substituted Rhodopsin Mouse Retinas in the Absence of α-Transducin
To determine whether the Lys296Glu rhodopsin mutation caused retinal degeneration by inappropriate photosignaling, we examined the retinal morphologies of mice expressing Lys296Glu-substituted opsin at 3 and 6 months of age on a Trα+/+ or a Trα−/− background (Fig. 1C). At 3 and 6 months of age, more rapid degeneration was observed in Lys296Glu-substituted rhodopsin mutants on the Trα−/− genetic background than on the Trα+/+ genetic background. These results demonstrated that transducin-mediated signaling does not cause degeneration for this rhodopsin mutation.
Accelerated Degeneration in Pro347Ser-Substituted Rhodopsin Mice in the Absence of α-Transducin
Retinal morphologies of Pro347Ser mutant mice32 on a Trα+/+ or a Trα−/− background were compared at 1, 2, 4, and 6 months of age (Fig. 1D). As with the Lys296Glu-substituted rhodopsin, the severity of degeneration was significantly accelerated with the Pro347Ser substitution on the Trα−/− genetic background compared with the Trα+/+ genetic background. These results demonstrated that for the Pro347Ser-substituted rhodopsin, aberrant transducin-mediated signaling was not a cause of degeneration.
Our observations refute transducin-mediated signaling as a mechanism of degeneration in the three rhodopsin mutations studied. However, the accelerated degeneration observed for the Lys296Glu- and Pro347Ser-substituted rhodopsins was not expected. There are several possible explanations for the observed results. We tested these possibilities in the Pro347Ser-substituted rhodopsin mutant mouse line because it degenerates faster than the Lys296Glu mutant mouse.
One possibility is that elevated levels of total rhodopsin may cause retinal degeneration, as previously observed with transgenic overexpression of WT rhodopsin.41 Combined levels of endogenous and transgene-encoded rhodopsin may contribute to degeneration. This seems unlikely, however, because we previously showed that the absence of Trα−/− does not alter rhodopsin levels.26 We also expect that the gene expression level of the Pro347Ser-substituted rhodopsin transgene would be the same whether expressed on a Trα−/− or a Trα+/+ genetic background because the transgene integration site is the same. Furthermore, direct measurement of rhodopsin levels by difference spectroscopy showed 1-month-old Pro347Ser, Trα−/− mutant mice retained 190 ± 20 pmol rhodopsin per retina (n = 3) compared with 320 ± 30 pmol rhodopsin per retina in Pro347Ser mutants on a WT Trα+/+ genetic background (n = 3; P < 0.002). The decrease in rhodopsin concentration in Pro347Ser, Trα−/− mice is likely attributable to the more rapid degeneration.
Effect of Rhodopsin/Arrestin Complex Formation on Retinal Degeneration in Pro347Ser Mutant Mice
The formation of stable rhodopsin/arrestin complexes causes retinal degeneration in Drosophila38,40 and mice.42 It is possible that Pro347Ser-substituted rhodopsin preferentially binds arrestin in the absence of α-transducin because arrestin and α-transducin bind competitively to phosphorylated rhodopsin.43 Furthermore, Pro347Ser-substituted rhodopsin peptide shows increased phosphorylation kinetics44 and can bind arrestin.34 This may favor the formation of stable rhodopsin/arrestin complexes. To test whether the rhodopsin/arrestin complex formation contributes to degeneration, we produced Pro347Ser rhodopsin mutant mice on a double null transducin/arrestin (Pro347Ser, Trα−/−, Arr−/−) genetic background.
We compared the severity of degeneration in 4-month-old Pro347Ser, Trα−/−, Arr+/+ and Pro347Ser, Trα−/−, Arr−/− mice reared in cyclic light (Fig. 2). We showed previously that Arr−/− mice placed on a Trα−/− genetic background were protected from light-induced degeneration.5 Evaluation of an independent set of animals again revealed accelerated degeneration in Pro347Ser rhodopsin mutant mice in the absence of α-transducin. Pro347Ser rhodopsin mutants on a Trα−/−, Arr+/+ background retained 2.7 ± 0.7 (n = 6) rows of nuclei, and P347S mutants on a Trα−/−, Arr−/− background retained 1.9 ± 0.8 (n = 9) rows of nuclei. We failed to observe protection from degeneration in the absence of arrestin protein, indicating that rhodopsin/arrestin complex formation did not contribute to retinal degeneration in the Pro347Ser-substituted rhodopsin mouse model.
FIGURE 2.
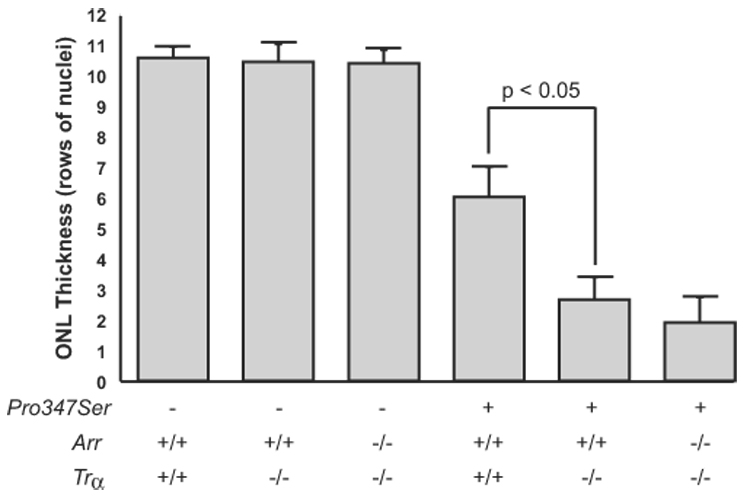
Role of rhodopsin/arrestin complexes in Pro347Ser rhodopsin mutant degeneration. The ONL thickness of 4-month-old Pro347Ser rhodopsin mutant mice in the presence or absence of arrestin and transducin was plotted. The absence of α-transducin or arrestin alone did not contribute to degeneration (compare 3 left columns). Loss of α-transducin in combination with the Pro347Ser rhodopsin mutation produced a statistically significant loss of ONL thickness (columns 4 and 5). The combined loss of α-transducin and arrestin did not protect from degeneration (last column). Error bars show SEM.
Slowed Degeneration in Pro347Ser, Trα−/− Mice by Dark Rearing
Another possible explanation for the accelerated degeneration in mice on the Trα−/− genetic background is stabilization of light-activated Pro347Ser-substituted metarhodopsin by binding of α-transducin. Given that transducin binding is initiated by light activation of rhodopsin, we assessed whether dark rearing Pro347Ser, Trα−/− mice provided protection from degeneration.
We compared the severity of degeneration in 4-month-old Pro347Ser, Trα−/− and Pro347Ser, Trα+/+ mice reared in cyclic light or complete darkness (Fig. 3). Dark-reared Pro347Ser, Trα−/− mice were protected from retinal degeneration (4.8 ± 0.8, n = 4) compared with light-reared Pro347Ser, Trα−/− mice (3.0 ± 0.9, n = 12, P < 0.01), supporting our hypothesis that light activation of the mutant Pro347Ser-substituted rhodopsin destabilized the mutant rhodopsin. In contrast, dark-reared Pro347Ser, Trα+/+ control mice conferred no additional protection (5.7 ± 1.0 rows of nuclei; n = 6) compared with cyclic light-reared mice (6.4 ± 0.6 rows of nuclei, n = 8). This observation is consistent with our hypothesis that α-transducin binding stabilizes the mutant metarhodopsin, providing a protective effect.
FIGURE 3.
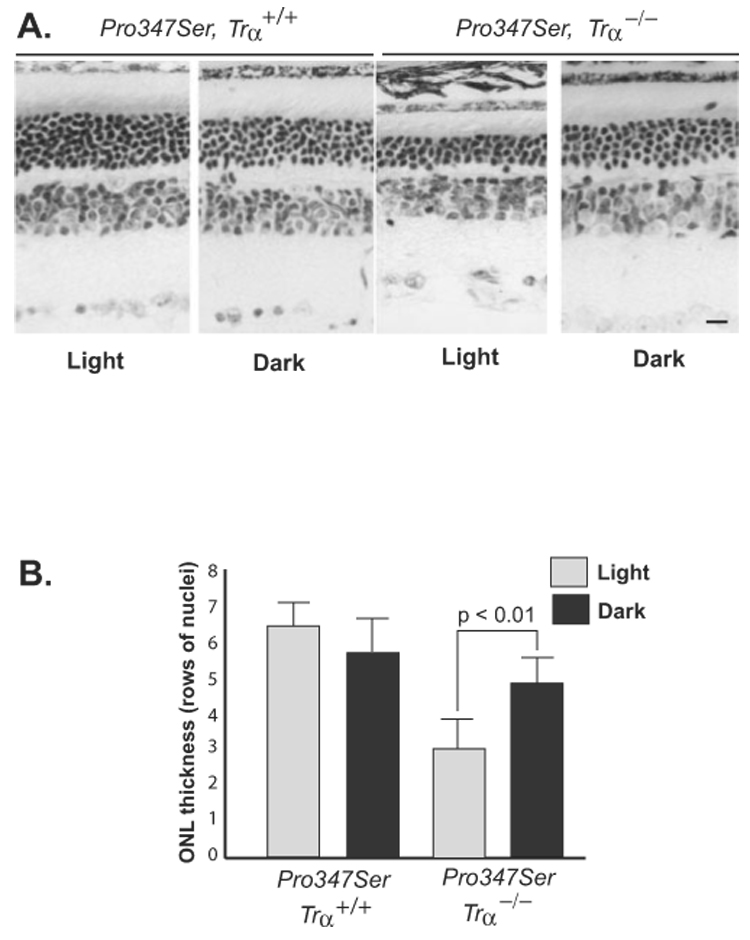
α-transducin stabilizes Pro347Ser metarhodopsin. (A) Retinal morphology of Pro347Ser rhodopsin mutant mice on a Trα+/+ or Trα−/− genetic background reared in cyclic light or dark reared. Pro347Ser, Trα+/+ mice showed similar degrees of degeneration whether reared in cyclic light or darkness. Dark rearing provided protection from degeneration but only in P347S, Trα−/− retinas. (B) Histogram comparing ONL thickness in cyclic light-reared (gray) and dark-reared (black) animals on the Trα+/+ or Trα−/− genetic background. Error bars show SEM. Scale bar, 10 µm.
Elevation of Lipofuscin Fluorophores in Pro347Ser Rhodopsin Mutant Mice
The accelerated degeneration observed in Pro347Ser transgenic mice may result from destabilization of light-activated Pro347Ser metarhodopsin. WT metarhodopsin decays slowly to yield apo-opsin and free all-trans-RAL. Free all-trans-RAL is highly cytotoxic. It is eliminated by reduction to all-trans-retinol. The reduction of all-trans-RAL in rods is a rate-limiting step in the visual cycle.45 Possibly, Pro347Ser-substituted metarhodopsin decays faster than all-trans-RAL clearance from the cell.
In photoreceptors, free all-trans-RAL condenses spontaneously with phosphatidylethanolamine in outer segment disks to form N-retinylidene-phosphatidylethanolamine (N-ret-PE). N-ret-PE can react with another all-trans-RAL to form a family of toxic bis-retinoid fluorophores that include A2PE-H2, A2PE, A2E, and iso-A2E.46–48 A2PE and A2E are photosensitizers subject to photooxidation, producing reactive moieties that can modify DNA and proteins and result in cell death. These fluorophores are formed in photoreceptor outer segments and accumulate in the RPE.
To test whether bis-retinoid fluorophores accumulated in the retinal pigment epithelium of Pro347Ser-substituted transgenic mice, we measured levels of A2E and its precursors in dark-adapted 2-month-old WT and Pro347Ser-transgenic mice on Trα+/+ and Trα−/− backgrounds (Fig. 4A). No significant difference was seen between N-ret-PE in Pro347Ser+, Trα+/+ and WT eyecups. This was expected because all-trans-retinal is not produced in dark-adapted mice and N-ret-PE forms in rapid equilibrium with all-trans-RAL. However, A2PE and A2E were elevated twofold to threefold in Pro347Ser+, Trα+/+ mutant eyecups compared with WT controls. Similarly, iso-A2E was elevated 40-fold and A2PE-H2 was elevated 27-fold relative to WT littermate control eyecups. Eight-week-old Pro347Ser+, Trα−/− transgenic and nontransgenic littermate Trα−/− mice were also assessed for accumulation of phospholipids. Similar to Pro347Ser, Trα+/+ mice, Pro347Ser, Trα−/− mice showed significantly elevated levels of A2PE, iso-A2E, and A2PE-H2 compared with Trα−/− controls (data not shown). These results reveal an accumulation of bis-retinoid fluorophores in the eyecups of Pro347Ser-substituted rhodopsin transgenic mice, supporting our hypothesis that light-activated Pro347Ser-substituted rhodopsin is destabilized.
FIGURE 4.
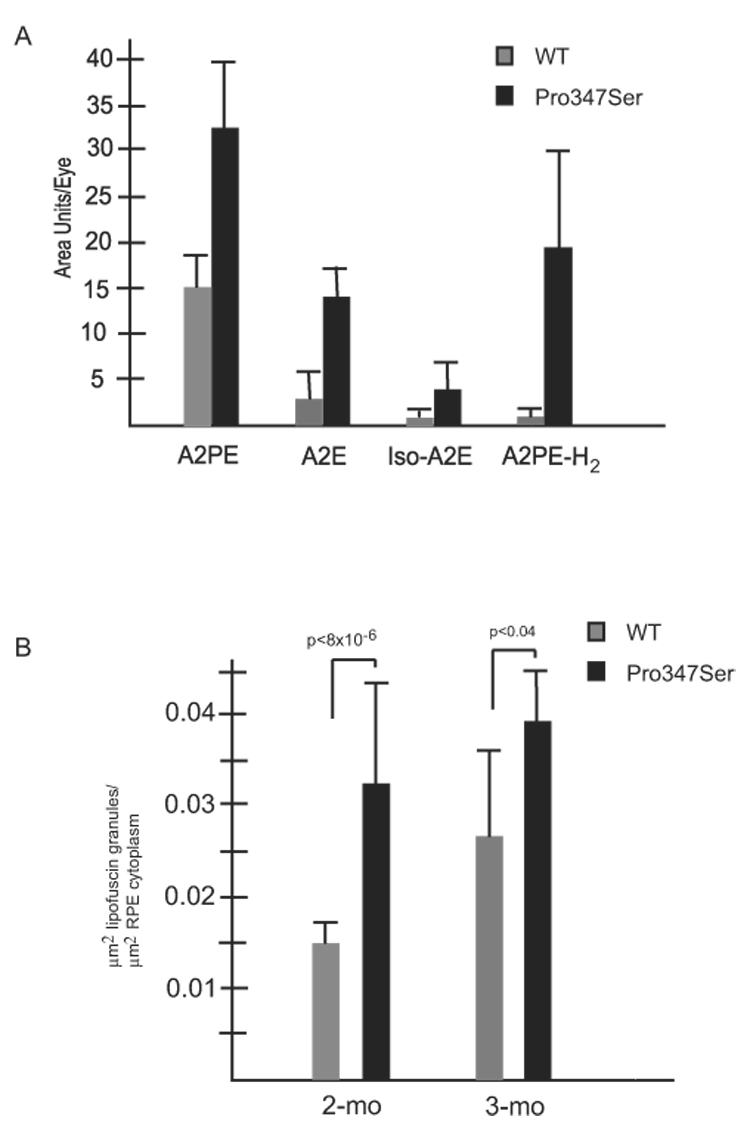
Comparison of A2E and A2E-precursor levels in (A) 61-day-old WT mice (gray) and littermate Pro347Ser, Trα+/+ transgenic mice (black) show statistically significant differences for four bis-retinoids. A2E was present at 4.8 versus 1 pmol/eye in Pro347Ser, Trα+/+ and WT mice, respectively. (B) Lipofuscin granule density. Four mice of each genotype were surveyed by measuring square microns of lipofuscin granule per square micron of RPE cytoplasm. P347S mutant mice (black) had a higher density of granules than WT control mice (gray). Error bars represent SD.
Higher Lipofuscin Granule Density in Pro347Ser Rhodopsin Mutant Mice
Excess A2E fluorophore accumulation in the RPE is associated with a corresponding increase in lipofuscin granule density. To determine whether the increase in A2PE, A2E, iso-A2E, and A2PE-H2 correlated with an increase in lipofuscin granules, lipofuscin granule density was compared in 2- and 3-month-old Pro347Ser-transgenic and littermate control mice by measuring square microns of lipofuscin granule per square micron of RPE cytoplasm (Fig. 4B). Ten to 12 fields were counted per mouse. Two-month-old Pro347Ser-transgenic mice (34 fields, n = 3) showed greater than twofold increased lipofuscin granule density over littermate WT controls (P < 8 × 10−6). Three-month-old animals also showed increased lipofuscin granule density (P < 0.04). Both data sets showed a corresponding increase in lipofuscin granule density associated with elevated levels of A2E and its precursors.
DISCUSSION
We have evaluated the contribution of α-transducin signaling to retinal degenerative disease in three mouse models of rhodopsin-mediated ADRP. Previous studies showed that null mutations in the rhodopsin kinase and arrestin genes, required for termination of the photoresponse, caused degeneration because of persistent photosignaling.5,7 We tested whether persistent signaling by mutant rhodopsins also caused degeneration. Of three rhodopsin mutants examined, none were protected from retinal degeneration when placed on an α-transducin null background. These results showed that persistent photosignaling is not a mechanism of retinal degeneration in transgenic mice expressing VPP-, Lys296Glu-, or Pro347Ser-substituted rhodopsins. VPP rhodopsin mutant mice, a model for the human Pro23His mutation, showed no change in the rate of degeneration on Trα+/+ compared with Trα−/− backgrounds. The absence of α-transducin, however, accelerated degeneration in Lys296Glu and Pro347Ser transgenic mice.
The similar rates of degeneration in VPP transgenic mice in the absence or presence of α-transducin indicated that photoreceptor cell apoptosis is unrelated to activation of the visual transduction cascade. Our results contrast with those of Samardzija et al.,49 who report that VPP, Trα−/− double-mutant mice experience protection from photoreceptor degeneration. The reasons for the discrepancy are unclear but likely stem from the effect of genetic modifiers. The Rpe65 genotype is a known genetic modifier of sensitivity to light damage. Mapping studies indicate the existence of additional modifiers of light damage sensitivity.50 The Rpe65 genotypes were assessed in both studies and did not account for the different results. Published data on the VPP mutant mouse are also consistent with the presence of genetic modifiers. The VPP mouse line shows increased susceptibility to light damage51 and partial, but incomplete, protection from retinal degeneration when dark reared,52 suggesting the role of light-dependent and signal-independent degenerative mechanisms. The light-independent component may relate to the faster degeneration observed in VPP mutant albino mice that is unrelated to increased retinal illumination because dark-reared albinos showed faster degeneration.53 Furthermore, the VPP mouse line used by Samardzija et al.27 degenerated nearly twice as quickly as the VPP subline used in our studies and the initially characterized mouse line. Identifying genetic modifiers that regulate disease susceptibility will be important for understanding disease mechanisms. Other reports describe concurrently operating degenerative mechanisms in other retinal degeneration models.5,22,23
Lys296Glu-transgenic mice on a Trα−/− genetic background did not show protection from degeneration. Our results agree with the conclusions of Li et al.30 that constitutive activation of the visual transduction cascade does not cause retinal degeneration in this animal model. However, we observed accelerated degeneration in 6-month-old Lys296Glu, Trα−/− mice that was less pronounced at 3 months of age. Chen et al.42 do not report accelerated degeneration in Lys296Glu, Trα−/− mice. They examined animals only up to 2.5 months of age, which may explain the apparent discrepancy, but also report that Lys296Glu transgenic mice undergo degeneration by the formation of stable rhodopsin/arrestin complexes.42 It is possible that the accelerated degeneration of Lys296Glu transgenic mice we observed in the absence of α-transducin was caused by the favored formation of rhodopsin/arrestin complexes because α-transducin and arrestin competitively bind phosphorylated rhodopsin.43
The Pro347Ser transgenic mouse line also showed accelerated degeneration in the absence of α-transducin. Photoreceptor degeneration was significantly slower in dark- than in cyclic light-reared Pro347Ser, Trα−/− mice, indicating that light activation of the mutant rhodopsin is an initiating event. However dark-reared Pro347Ser, Trα+/+ mice were not protected from degeneration compared with cyclic light-reared mice of the same genotype, suggesting that the presence of α-transducin provided protection. Rhodopsin/arrestin complex formation was not a major contributor to degeneration because placing the Pro347Ser mutation on a double α-transducin and arrestin null mutant background did not provide protection from retinal degeneration.
The Pro347Ser residue is part of the highly conserved VAPA C-terminal end of rhodopsin known to play an important role in trafficking rhodopsin to rod outer segments (for a review, see Deretic54). Mutations in this region result in mislocalization of rhodopsin to inner segments. The mislocalized opsin is thought to cause degeneration by aberrant activation of signaling pathways in the inner segment.55 Our results do not support transducin activation by mislocalized opsin, though they do not exclude signaling mediated by another G-protein. Tam et al.56 also report that mislocalized C-terminal rhodopsin mutants do not signal by transducin activation.
Our results are consistent with the stabilization of light-activated Pro347Ser metarhodopsin by α-transducin binding. In its absence, the mutant Pro347Ser metarhodopsin may decay more rapidly to its component parts, apo-opsin and all-trans-RAL. All-trans-RAL condenses with phosphatidylethanolamine to form N-ret-PE, which can react with a second molecule of all-trans-RAL to form toxic bis-retinoid fluorophores.46–48 Elevated levels of the fluorophores A2E, iso-A2E, and A2E-precursors in Pro347Ser transgenic mouse eyecups and the correlative increase in lipofuscin granules support our hypothesis. A2PE and A2E are subject to photo-oxidation and can cause cell death.57–59 Bis-retinoids are associated with normal aging of the human eye but accumulate at higher levels in association with some forms of human retinal degenerations, including Stargardt macular dystrophy, retinitis pigmentosa, and cone-rod dystrophy.60 Elevation of A2E and other lipofuscin fluorophores in RPE cells have also been reported in animal models of retinal and macular degeneration.47,61–68 Vitamin A supplementation can ameliorate disease severity for some retinal degenerations,69,70 but vitamin A supplementation may be detrimental to patients with destabilized rhodopsin mutations that decay rapidly and release all-trans-RAL.
Only one disease-associated mutation in the transducin α-subunit has been described that is associated with Nougaret night blindness.71,72 Therefore, it is unlikely that loss of transducin function constitutes a prevalent mechanism of retinal degeneration. However, it is very likely that a significant subset of rhodopsin mutations impairs transducin binding, and this may represent a highly disease-relevant mechanism of degeneration that is worthy of further exploration.
Acknowledgments
The authors thank Tiansen Li for providing the Pro347Ser and Lys296Glu rhodopsin mutant mouse lines and Wolfgang Baehr for providing the VPP mutant mice. The authors thank Flore Celestin of the Specialized Center of Research in Ischemic Heart Disease Histology Core for assistance with histologic sections and Kibibi Rwayitare for assistance with genotyping.
Supported by a Fight for Sight Student Fellowship (ERB); Foundation Fighting Blindness Grant (JL); National Institutes of Health Grants EY12008 (JL), F2EY12912A (ZW), EY00444 (DB), EY00331 (DB), EY11713 (GHT), EY015844 (GHT) and Core Grant P30 EY13078 (Tufts New England Medical Center); and a Research to Prevent Blindness Challenge Grant. DB is a recipient of the Dolly Green Chair endowment. GHT is the Charles Kenneth Feldman and Jules & Doris Stein Research to Prevent Blindness Professor.
Footnotes
Disclosure: E. Brill, None; K.M. Malanson, None; R.A. Radu, None; N.V. Boukharov, None; Z. Wang, None; H.-Y. Chung, None; M.B. Lloyd, None; D. Bok, None; G.H. Travis, None; M. Obin, None; J. Lem, None
References
- 1.Daiger SP. RetNet: Retinal Information Network. Houston: The University of Texas-Health Science Center; 1996–2007. [Google Scholar]
- 2.Lem J, Fain GL. Constitutive opsin signaling: night blindness or retinal degeneration? Trends Mol Med. 2004;10:150–157. doi: 10.1016/j.molmed.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 3.Chen CK, Burns ME, Spencer M, et al. Abnormal photoresponses and light-induced apoptosis in rods lacking rhodopsin kinase. Proc Natl Acad Sci USA. 1999;96:3718–3722. doi: 10.1073/pnas.96.7.3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu J, Dodd RL, Makino CL, Simon MI, Baylor DA, Chen J. Prolonged photoresponses in transgenic mouse rods lacking arrestin. Nature. 1997;389:505–509. doi: 10.1038/39068. [DOI] [PubMed] [Google Scholar]
- 5.Hao W, Wenzel A, Obin MS, et al. Evidence for two apoptotic pathways in light-induced retinal degeneration. Nat Genet. 2002;32:254–260. doi: 10.1038/ng984. [DOI] [PubMed] [Google Scholar]
- 6.Redmond TM, Yu S, Lee E, et al. Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat Genet. 1998;20:344–351. doi: 10.1038/3813. [DOI] [PubMed] [Google Scholar]
- 7.Woodruff ML, Wang Z, Chung HY, Redmond TM, Fain GL, Lem J. Spontaneous activity of opsin apoprotein is a cause of Leber congenital amaurosis. Nat Genet. 2003;35:158–164. doi: 10.1038/ng1246. [DOI] [PubMed] [Google Scholar]
- 8.Rim J, Oprian DD. Constitutive activation of opsin: interaction of mutants with rhodopsin kinase and arrestin. Biochemistry. 1995;34:11938–11945. doi: 10.1021/bi00037a035. [DOI] [PubMed] [Google Scholar]
- 9.Robinson PR, Buczylko J, Ohguro H, Palczewski K. Opsins with mutations at the site of chromophore attachment constitutively activate transducin but are not phosphorylated by rhodopsin kinase. Proc Natl Acad Sci USA. 1994;91:5411–5415. doi: 10.1073/pnas.91.12.5411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robinson PR, Cohen GB, Zhukovsky EA, Oprian DD. Constitutively active mutants of rhodopsin. Neuron. 1992;9:719–725. doi: 10.1016/0896-6273(92)90034-b. [DOI] [PubMed] [Google Scholar]
- 11.Zhukovsky EA, Robinson PR, Oprian DD. Transducin activation by rhodopsin without a covalent bond to the 11-cis-retinal chromophore. Science. 1991;251:558–560. doi: 10.1126/science.1990431. [DOI] [PubMed] [Google Scholar]
- 12.Cohen GB, Oprian DD, Robinson PR. Mechanism of activation and inactivation of opsin: role of Glu113 and Lys296. Biochemistry. 1992;31:12592–12601. doi: 10.1021/bi00165a008. [DOI] [PubMed] [Google Scholar]
- 13.Rao VR, Cohen GB, Oprian DD. Rhodopsin mutation G90D and a molecular mechanism for congenital night blindness. Nature. 1994;367:639–642. doi: 10.1038/367639a0. [DOI] [PubMed] [Google Scholar]
- 14.Rao VR, Oprian DD. Activating mutations of rhodopsin and other G protein-coupled receptors. Annu Rev Biophys Biomol Struct. 1996;25:287–314. doi: 10.1146/annurev.bb.25.060196.001443. [DOI] [PubMed] [Google Scholar]
- 15.Han M, Lou J, Nakanishi K, Sakmar TP, Smith SO. Partial agonist activity of 11-cis-retinal in rhodopsin mutants. J Biol Chem. 1997;272:23081–23085. doi: 10.1074/jbc.272.37.23081. [DOI] [PubMed] [Google Scholar]
- 16.Han M, Smith SO, Sakmar TP. Constitutive activation of opsin by mutation of methionine 257 on transmembrane helix 6. Biochemistry. 1998;37:8253–8261. doi: 10.1021/bi980147r. [DOI] [PubMed] [Google Scholar]
- 17.Kim JM, Altenbach C, Thurmond RL, Khorana HG, Hubbell WL. Structure and function in rhodopsin: rhodopsin mutants with a neutral amino acid at E134 have a partially activated conformation in the dark state. Proc Natl Acad Sci USA. 1997;94:14273–14278. doi: 10.1073/pnas.94.26.14273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramon E, del Valle LJ, Garriga P. Unusual thermal and conformational properties of the rhodopsin congenital night blindness mutant Thr-94>Ile. J Biol Chem. 2003;278:6427–6432. doi: 10.1074/jbc.M210929200. [DOI] [PubMed] [Google Scholar]
- 19.Sieving PA, Richards JE, Naarendorp F, Bingham EL, Scott K, Alpern M. Dark-light: model for nightblindness from the human rhodopsin Gly-90>Asp mutation. Proc Natl Acad Sci USA. 1995;92:880–884. doi: 10.1073/pnas.92.3.880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sieving PA, Fowler ML, Bush RA, et al. Constitutive “light” adaptation in rods from G90D rhodopsin: a mechanism for human congenital nightblindness without rod cell loss. J Neurosci. 2001;21:5449–5460. doi: 10.1523/JNEUROSCI.21-15-05449.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zvyaga TA, Fahmy K, Siebert F, Sakmar TP. Characterization of the mutant visual pigment responsible for congenital night blindness: a biochemical and Fourier-transform infrared spectroscopy study. Biochemistry. 1996;35:7536–7545. doi: 10.1021/bi960391n. [DOI] [PubMed] [Google Scholar]
- 22.Iakhine R, Chorna-Ornan I, Zars T, et al. Novel dominant rhodopsin mutation triggers two mechanisms of retinal degeneration and photoreceptor desensitization. J Neurosci. 2004;24:2516–2526. doi: 10.1523/JNEUROSCI.5426-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang T, Xu H, Oberwinkler J, Gu Y, Hardie RC, Montell C. Light activation, adaptation, and cell survival functions of the Na+/Ca2+ exchanger CalX. Neuron. 2005;45:367–378. doi: 10.1016/j.neuron.2004.12.046. [DOI] [PubMed] [Google Scholar]
- 24.Gross AK, Rao VR, Oprian DD. Characterization of rhodopsin congenital night blindness mutant T94I. Biochemistry. 2003;42:2009–2015. doi: 10.1021/bi020613j. [DOI] [PubMed] [Google Scholar]
- 25.Jin S, Cornwall MC, Oprian DD. Opsin activation as a cause of congenital night blindness. Nat Neurosci. 2003;6:731–735. doi: 10.1038/nn1070. [DOI] [PubMed] [Google Scholar]
- 26.Calvert PD, Krasnoperova NV, Lyubarsky AL, et al. Phototransduction in transgenic mice after targeted deletion of the rod transducin α-subunit. Proc Natl Acad Sci USA. 2000;97:13913–13918. doi: 10.1073/pnas.250478897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Naash MI, Hollyfield JG, Al-Ubaidi M, Baehr W. Simulation of human autosomal dominant retinitis pigmentosa in transgenic mice expressing a mutated murine opsin gene. Proc Natl Acad Sci USA. 1993;90:5499–5503. doi: 10.1073/pnas.90.12.5499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu TH, Ting TD, Okajima TI, et al. Opsin localization and rhodopsin photochemistry in a transgenic mouse model of retinitis pigmentosa. Neuroscience. 1998;87:709–717. doi: 10.1016/s0306-4522(98)00173-0. [DOI] [PubMed] [Google Scholar]
- 29.Liu X, Wu T-H, Stowe S, Matsushita A, Arikawa K, Naash MI. Defective phototransductive disk membrane morphogenesis in transgenic mice expressing opsin with a mutated N-terminal domain. J Cell Sci. 1997;110:2589–2597. doi: 10.1242/jcs.110.20.2589. [DOI] [PubMed] [Google Scholar]
- 30.Li T, Franson WK, Gordon JW, Berson EL, Dryja TP. Constitutive activation of phototransduction by K296E opsin is not a cause of photoreceptor degeneration. Proc Natl Acad Sci USA. 1995;92:3551–3555. doi: 10.1073/pnas.92.8.3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Owens SL, Fitzke FW, Inglehearn CF, et al. Ocular manifestations in autosomal dominant retinitis pigmentosa with a Lys-296-Glu rhodopsin mutation at the retinal binding site. Br J Ophthalmol. 1994;78:353–358. doi: 10.1136/bjo.78.5.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li T, Snyder WK, Olsson JE, Dryja TP. Transgenic mice carrying the dominant rhodopsin mutation P347S: evidence for defective vectorial transport of rhodopsin to the outer segments. Proc Natl Acad Sci USA. 1996;93:14176–14181. doi: 10.1073/pnas.93.24.14176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sung CH, Davenport CM, Nathans J. Rhodopsin mutations responsible for autosomal dominant retinitis pigmentosa: clustering of functional classes along the polypeptide chain. J Biol Chem. 1993;268:26645–26649. [PubMed] [Google Scholar]
- 34.Weiss ER, Hao Y, Dickerson CD, et al. Altered cAMP levels in retinas from transgenic mice expressing a rhodopsin mutant. Biochem Biophys Res Comm. 1995;216:755–761. doi: 10.1006/bbrc.1995.2686. [DOI] [PubMed] [Google Scholar]
- 35.Wang T, Montell C. Phototransduction and retinal degeneration in Drosophila. Pflugers Arch. 2007;454:821–847. doi: 10.1007/s00424-007-0251-1. [DOI] [PubMed] [Google Scholar]
- 36.Lee SJ, Xu H, Kang LW, Amzel LM, Montell C. Light adaptation through phosphoinositide-regulated translocation of Drosophila visual arrestin. Neuron. 2003;39:121–132. doi: 10.1016/s0896-6273(03)00390-8. [DOI] [PubMed] [Google Scholar]
- 37.Orem NR, Xia L, Dolph PJ. An essential role for endocytosis of rhodopsin through interaction of visual arrestin with the AP-2 adaptor. J Cell Sci. 2006;119:3141–3148. doi: 10.1242/jcs.03052. [DOI] [PubMed] [Google Scholar]
- 38.Alloway PG, Howard L, Dolph PJ. The formation of stable rhodopsin-arrestin complexes induces apoptosis and photoreceptor cell degeneration. Neuron. 2000;28:129–138. doi: 10.1016/s0896-6273(00)00091-x. [DOI] [PubMed] [Google Scholar]
- 39.Alloway PG, Dolph PJ. A role for the light-dependent phosphorylation of visual arrestin. Proc Natl Acad Sci USA. 1999;96:6072–6077. doi: 10.1073/pnas.96.11.6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kiselev A, Socolich M, Vinos J, Hardy RW, Zuker CS, Ranganathan R. A molecular pathway for light-dependent photoreceptor apoptosis in Drosophila. Neuron. 2000;28:139–152. doi: 10.1016/s0896-6273(00)00092-1. [DOI] [PubMed] [Google Scholar]
- 41.Olsson JE, Gordon JW, Pawlyk BS, et al. Transgenic mice with a rhodopsin mutation (Pro23His): a mouse model of autosomal dominant retinitis pigmentosa. Neuron. 1992;9:815–830. doi: 10.1016/0896-6273(92)90236-7. [DOI] [PubMed] [Google Scholar]
- 42.Chen J, Shi G, Concepcion FA, Xie G, Oprian D. Stable rhodopsin/arrestin complex leads to retinal degeneration in a transgenic mouse model of autosomal dominant retinitis pigmentosa. J Neurosci. 2006;26:11929–11937. doi: 10.1523/JNEUROSCI.3212-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krupnick JG, Gurevich VV, Benovic JL. Mechanism of quenching of phototransduction: binding competition between arrestin and transducin for phosphorhodopsin. J Biol Chem. 1997;272:18125–18131. doi: 10.1074/jbc.272.29.18125. [DOI] [PubMed] [Google Scholar]
- 44.Ohguro H. High levels of rhodopsin phosphorylation in missense mutations of C-terminal region of rhodopsin. FEBS Lett. 1997;413:433–435. doi: 10.1016/s0014-5793(97)00957-5. [DOI] [PubMed] [Google Scholar]
- 45.Saari JC, Garwin GG, Van Hooser JP, Palczewski K. Reduction of all-trans-retinal limits regeneration of visual pigment in mice. Vision Res. 1998;38:1325–1333. doi: 10.1016/s0042-6989(97)00198-3. [DOI] [PubMed] [Google Scholar]
- 46.Parish CA, Hashimoto M, Nakanishi K, Dillon J, Sparrow J. Isolation and one-step preparation of A2E and iso-A2E, fluorophores from human retinal pigment epithelium. Proc Natl Acad Sci USA. 1998;95:14609–14613. doi: 10.1073/pnas.95.25.14609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mata NL, Weng J, Travis GH. Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc Natl Acad Sci USA. 2000;97:7154–7159. doi: 10.1073/pnas.130110497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bui TV, Han Y, Radu RA, Travis GH, Mata NL. Characterization of native retinal fluorophores involved in biosynthesis of A2E and lipofuscin-associated retinopathies. J Biol Chem. 2006;281:18112–18119. doi: 10.1074/jbc.M601380200. [DOI] [PubMed] [Google Scholar]
- 49.Samardzija M, Wenzel A, Naash M, Reme CE, Grimm C. Rpe65 as a modifier gene for inherited retinal degeneration. Eur J Neurosci. 2006;23:1028–1034. doi: 10.1111/j.1460-9568.2006.04639.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Danciger M, Lyon J, Worrill D, et al. New retinal light damage QTL in mice with the light-sensitive RPE65 LEU variant. Mamm Genome. 2004;15:277–283. doi: 10.1007/s00335-003-2336-2. [DOI] [PubMed] [Google Scholar]
- 51.Wang M, Lam TT, Tso MOM, Naash MI. Expression of a mutant opsin gene increases the susceptibility of the retina to light damage. Vis Neurosci. 1997;14:55–62. doi: 10.1017/s0952523800008750. [DOI] [PubMed] [Google Scholar]
- 52.Naash MI, Peachey NS, Li Z-Y, et al. Light-induced acceleration of photoreceptor degeneration in transgenic mice expressing mutant rhodopsin. Invest Ophthalmol Vis Sci. 1996;37:775–782. [PubMed] [Google Scholar]
- 53.Naash MI, Ripps H, Li S, Goto Y, Peachey NS. Polygenic disease and retinitis pigmentosa: albinism exacerbates photoreceptor degeneration induced by the expression of a mutant opsin in transgenic mice. J Neurosci. 1996;16:7853–7858. doi: 10.1523/JNEUROSCI.16-24-07853.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deretic D. A role for rhodopsin in a signal transduction cascade that regulates membrane trafficking and photoreceptor polarity. Vision Res. 2006;46:4427–4433. doi: 10.1016/j.visres.2006.07.028. [DOI] [PubMed] [Google Scholar]
- 55.Alfinito PD, Townes-Anderson E. Activation of mislocalized opsin kills rod cells: a novel mechanism for rod cell death in retinal disease. Proc Natl Acad Sci USA. 2002;99:5655–5660. doi: 10.1073/pnas.072557799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tam BM, Xie G, Oprian DD, Moritz OL. Mislocalized rhodopsin does not require activation to cause retinal degeneration and neurite outgrowth in Xenopus laevis. J Neurosci. 2006;26:203–209. doi: 10.1523/JNEUROSCI.3849-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fishkin NE, Sparrow JR, Allikmets R, Nakanishi K. Isolation and characterization of a retinal pigment epithelial cell fluorophore: an all-trans-retinal dimer conjugate. Proc Natl Acad Sci USA. 2005;102:7091–7096. doi: 10.1073/pnas.0501266102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim SR, Nakanishi K, Itagaki Y, Sparrow JR. Photooxidation of A2-PE, a photoreceptor outer segment fluorophore, and protection by lutein and zeaxanthin. Exp Eye Res. 2006;82:828–839. doi: 10.1016/j.exer.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 59.Jang YP, Matsuda H, Itagaki Y, Nakanishi K, Sparrow JR. Characterization of peroxy-A2E and furan-A2E photooxidation products and detection in human and mouse retinal pigment epithelial cell lipofuscin. J Biol Chem. 2005;280:39732–39739. doi: 10.1074/jbc.M504933200. [DOI] [PubMed] [Google Scholar]
- 60.Sparrow J, Boulton M. RPE lipofuscin and its role in retinal pathobiology. Exp Eye Res. 2005;80:595–606. doi: 10.1016/j.exer.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 61.Weng J, Mata NL, Azarian SM, Tzekov RT, Birch DG, Travis GH. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt’s disease from the phenotype in abcr knockout mice. Cell. 1999;98:13–23. doi: 10.1016/S0092-8674(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 62.Maeda A, Maeda T, Imanishi Y, et al. Role of photoreceptor-specific retinol dehydrogenase in the retinoid cycle in vivo. J Biol Chem. 2005;280:18822–18832. doi: 10.1074/jbc.M501757200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Karan G, Lillo C, Yang Z, et al. Lipofuscin accumulation, abnormal electrophysiology, and photoreceptor degeneration in mutant ELOVL4 transgenic mice: a model for macular degeneration. Proc Natl Acad Sci USA. 2005;102:4164–4169. doi: 10.1073/pnas.0407698102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Katz ML, Eldred GE, Robison WG., Jr. Lipofuscin autofluorescence: evidence for vitamin A involvement in the retina. Mech Ageing Dev. 1987;39:81–90. doi: 10.1016/0047-6374(87)90088-1. [DOI] [PubMed] [Google Scholar]
- 65.Liu J, Itagaki Y, Ben-Shabat S, Nakanishi K, Sparrow JR. The biosynthesis of A2E, a fluorophore of aging retina, involves the formation of the precursor, A2-PE, in the photoreceptor outer segment membrane. J Biol Chem. 2000;275:29354–29360. doi: 10.1074/jbc.M910191199. [DOI] [PubMed] [Google Scholar]
- 66.Rakoczy PE, Zhang D, Robertson T, et al. Progressive age-related changes similar to age-related macular degeneration in a transgenic mouse model. Am J Pathol. 2002;161:1515–1524. doi: 10.1016/S0002-9440(10)64427-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang D, Brankov M, Makhija MT, et al. Correlation between inactive cathepsin D expression and retinal changes in mcd2/mcd2 transgenic mice. Invest Ophthalmol Vis Sci. 2005;46:3031–3038. doi: 10.1167/iovs.04-1510. [DOI] [PubMed] [Google Scholar]
- 68.Ambati J, Anand A, Fernandez S, et al. An animal model of age-related macular degeneration in senescent Ccl-2- or Ccr-2-deficient mice. Nat Med. 2003;9:1390–1397. doi: 10.1038/nm950. [DOI] [PubMed] [Google Scholar]
- 69.Berson EL, Rosner B, Sandberg MA, et al. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993;111:761–772. doi: 10.1001/archopht.1993.01090060049022. [DOI] [PubMed] [Google Scholar]
- 70.Li T, Sandberg MA, Pawlyk BS, et al. Effect of vitamin A supplementation on rhodopsin mutants threonine-17> methionine and proline-347> serine in transgenic mice and in cell cultures. Proc Natl Acad Sci USA. 1998;95:11933–11938. doi: 10.1073/pnas.95.20.11933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dryja TP, Hahn LB, Reboul T, Arnaud B. Missense mutation in the gene encoding the alpha subunit of rod transducin in the Nougaret form of congenital stationary night blindness. Nat Genet. 1996;13:358–360. doi: 10.1038/ng0796-358. [DOI] [PubMed] [Google Scholar]
- 72.Muradov KG, Artemyev NO. Loss of the effector function in a transducin-alpha mutant associated with Nougaret night blindness. J Biol Chem. 2000;275:6969–6974. doi: 10.1074/jbc.275.10.6969. [DOI] [PubMed] [Google Scholar]