

Abstract
The endocannabinoid, N-arachidonylethanolamine (AEA) is accumulated by neurons via a process that has been characterized biochemically but not molecularly. Inhibitors of AEA accumulation have been characterized individually but have not been compared in a single study. Our purpose was to compare the potency of five previously described compounds (AM404, AM1172, VDM11, OMDM-2 and UCM707) both as inhibitors of AEA and N-palmitoylethanolamine (PEA) accumulation by cerebellar granule neurons and as inhibitors of AEA hydrolysis. The compounds all inhibited AEA accumulation; AM404, VDM11 and OMDM-2 with IC50 values of approximately 5 μM, while AM1172 and UCM707 exhibited IC50 values of 24 and 30 μM, respectively. The compounds also inhibited PEA accumulation; AM404 being the most potent with an IC50 of 6 μM, while the other compounds had IC50 values in the range of 30-70 μM. All of the compounds potently inhibited AEA hydrolysis by brain membranes; the KI values for AM404, VDM11 and UCM707 were less than 1 μM; AM1172 and OMDM-2 exhibited KI values of 3 and 10 μM, respectively. The IC50 values for inhibition of AEA accumulation were compared to the IC50 values for PEA accumulation and AEA hydrolysis using linear regression. None of the regressions were significant. These data indicate that inhibition of AEA accumulation by neurons is not a result of inhibition of endocannabinoid hydrolysis and is a process different from the accumulation of PEA. These studies support the hypothesis that the cellular AEA accumulation beyond simple equilibrium between intracellular and extracellular concentrations occurs because AEA binds to an intracellular protein that is not FAAH but that also recognizes the AEA uptake inhibitors.
INTRODUCTION
While the endocannabinoid, N-arachidonylethanolamine (AEA), is accumulated by cells, the mechanism for this accumulation has been elusive (Hillard and Jarrahian, 2003). Most studies support the concept that the accumulation is driven by the concentration gradient for AEA between intracellular and extracellular compartments; however, it is not clear whether the translocation of AEA across the membrane bilayer involves a protein carrier or occurs via diffusion. The process of AEA accumulation is saturable and can be inhibited by structurally similar molecules. These data support a specific interaction between AEA and the competitors of its uptake at some critical stage in the accumulation process. However, the site of interaction is not known. It has been suggested that fatty acid amide hydrolase (FAAH) activity is responsible for maintenance of the concentration gradient and that accumulation inhibitors are functionally relevant because they inhibit FAAH (Glaser et al., 2003). Whereas FAAH is likely important in some cell types (such as C6 glioma (Hillard and Jarrahian, 2005)), it is not required for AEA accumulation by neurons (Fegley et al., 2004; Ortega-Gutierrez et al., 2004).
In recent years, five structurally different inhibitors of AEA accumulation have been developed and characterized. These compounds include 4 modified arachidonates: N-arachidonyl-4-hydroxyphenylamine (AM404) (Beltramo et al., 1997b); N-(5Z, 8Z, 11Z, 14Z eicosatetraenyl)-4-hydroxybenzamide (AM1172), (Fegley et al., 2004); N-arachidonyl-2-methyl, 4-hydroxyphenylamine (VDM11); N-arachidonyl-3-furylmethylamine (UCM707) (Lopez-Rodriguez et al., 2001); and (S)-1′-(4-hydroxybenzyl) oleoylethanolamide (OMDM-2) (Ortar et al., 2003). These compounds provide an opportunity to further our understanding of the mechanism for AEA accumulation since they have structural diversity and differential affinities for inhibition of accumulation. However, the data regarding the potency of the inhibitors have not always been consistent and, perhaps more importantly, the inhibitory profiles of the compounds for other metabolic pathways have been unclear. The purposes of the studies reported herein were: (1) to compare the potencies of the five analogs to inhibit AEA accumulation in cerebellar granule neurons using an assay for which a relatively large structure activity profile has been published (Jarrahian et al., 2000; Hillard and Jarrahian, 2005); (2) to compare the potencies for inhibition of AEA accumulation to inhibition of PEA accumulation in the same cell preparations; and (3) to determine IC50 values for the inhibition of FAAH. Finally, we compared, using regression analyses, the IC50 values for each compound to inhibit AEA accumulation and to inhibit FAAH and PEA accumulation. In no case was a significant correlation found. We conclude that the accumulation of AEA by cerebellar granule cells is not dependent upon hydrolysis of AEA and occurs via a different from that mediating PEA accumulation.
MATERIALS AND METHODS
Materials
Male, ICR mice (21-24 g), used as a source of hydrolytic enzymes, and Sprague-Dawley timed-pregnant rats were obtained from Harlan Company (Madison, WI, U.S.A.). Cerebellar granule neurons (CGN) were isolated from Sprague-Dawley rat pups of either gender (7-10 days of age) and were maintained in culture as described previously (Hillard et al., 1997). Neurons were seeded at 106 cells/incubation and were used for accumulation studies at 6-8 days in vitro. AM404, VDM11, OMDM-2, PEA and AEA were purchased from Tocris Cookson (Ellisville, MO, U.S.A). UCM707 was purchased from Cayman Chemical Company (Ann Arbor, MI, U.S.A). Radiolabeled AEA used in the accumulation studies ([3H] labeled in the arachidonyl moiety) was obtained from the Research Resources Drug Supply System of the National Institute on Drug Abuse; PEA (palmitoyl-9,10-[3H]) and AEA used in the FAAH assays ([3H] labeled in the ethanolamine moiety) were purchased from American Radiolabeled Chemicals (Missouri, MO, U.S.A). All other salts and buffers were purchased from Sigma Chemical Company (St. Louis, MO, U.S.A).
Synthesis of AM1172
Mesyl chloride (35 μL, 0.448 mmol) followed by triethylamine (62 μL, 0.448 mmol) were added to a 0°C solution of arachidonyl alcohol (0.065 g, 0.224 mmol) in dichloromethane (2 mL) under an argon atmosphere. After 1 h, the reaction mixture was diluted with dichloromethane (10 mL) and then washed with water (2 × 10 mL), brine (1 × 10 mL), dried over Na2SO4 and concentrated in vacuo to give arachidonyl mesylate as a colorless oil (0.080 g, quantitative), which was utilized for the next reaction without purification. TLC: 30% EtOAc/hexanes, Rf ∼ 0.50.
Sodium azide (0.028 g, 0.435 mmol) was added to the above mesylate (0.080 g, 0.217 mmol) in dry dimethylformamide (2 mL) under an argon atmosphere. After stirring overnight at 40°C, the reaction mixture was diluted with dichloromethane (20 mL), washed with water (3 × 10 mL), brine (1 × 10 mL), dried over Na2SO4 and concentrated in vacuo. The residue was purified by SiO2 column chromatography to give arachidonyl azide (0.061 g, 87%) as a colorless oil. TLC: 10% EtOAc/hexanes, Rf ∼ 0.83; 1H NMR (CDCl3, 400 MHz) δ 5.44-5.30 (m, 8H), 3.27 (t, 2H, J = 7.0, 6.7 Hz), 2.87-2.79 (m, 6H), 2.14-2.02 (m, 4H), 1.66-1.58 (m, 2H), 1.50-1.24 (m, 8H), 0.89 (t, 3H, J = 7.0, 6.7 Hz); IR (neat) 2096 cm-1.
Triphenylphosphine (0.037 g, 0.143 mmol) was added to the above azide (0.045 g, 0.143 mmol) in a mixture of THF (1 mL) and water (2 drops) under an argon atmosphere.
After stirring overnight, the reaction mixture was diluted with dichloromethane (2 mL), dried over Na2SO4, and evaporated in vacuo to give arachidonyl amine (0.041 g, quantitative) as a colorless oil that was utilized for the next step without further purification. TLC: 30% EtOAc/hexanes, Rf ∼ 0.20.
4-(Tetrahydro-2H-pyran-2-yloxy)benzoic acid (0.034 g, 0.156 mmol), N,N’-dicyclohexylcarbodiimide (0.016 g, 0.077 mmol), and 4-dimethylaminopyridine (4 mg) were added sequentially to a solution of the above arachidonyl amine (0.041 g, 0.142 mmol) in anhydrous dichloromethane (4 mL) under an argon atmosphere. After stirring overnight, the reaction mixture was diluted with dichloromethane (20 mL), washed with water (2 × 10 mL), brine (1 × 10 mL), dried over Na2SO4 and concentrated in vacuo. Purification of the residue via SiO2 column chromatography (4% EtOAc/hexanes) furnished N-arachidonyl 4-(tetrahydro-2H-pyran-2-yloxy)benzamide (0.058 g, 82%). TLC: 30% EtOAc/hexanes, Rf ∼ 0.47.
p-Toluenesulfonic acid (4 mg) was added to the above amide (0.058 g, 0.117 mmol) in anhydrous dichloromethane (3 mL) under an argon atmosphere. After 1 h, the reaction mixture was diluted with dichloromethane (20 mL), washed with water (2 × 10 mL), brine (1 × 10 mL), dried over Na2SO4 and concentrated in vacuo. Purification of the residue via SiO2 column chromatography furnished N-arachidonyl 4-hydroxybenzamide (AM1172; 0.040 g, 81%). TLC: 50% EtOAc/hexanes, Rf ∼ 0.44; 1H NMR (CDCl3, 300 MHz) δ 8.79 (bs, 1H), 7.60 (d, 2H, J = 8.5 Hz), 6.87(d, 2H, J = 8.5 Hz), 6.26(t, 1H, J = 5.8 Hz), 5.44-5.28 (m, 8H), 3.44 (q, 2H, J = 7.0, 5.8 Hz), 2.88-2.75 (m, 6H), 2.14-2.00 (m, 4H), 1.68-1.57 (m, 2H), 1.50-1.23 (m, 8H), 0.88 (t, 3H, J = 6.7 Hz); 13C NMR (CDCl3, 75 MHz) δ 168.6, 160.5, 130.7, 129.8, 128.9, 128.8, 128.5, 128.4, 128.3, 128.1, 127.7, 125.5, 115.8, 40.4, 31.7, 29.5, 29.4, 27.4, 27.1, 27.0, 25.8, 22.7, 14.3.
Preparation of 4-(tetrahydro-2H-pyran-2-yloxy)benzoic acid: A solution of p-hydroxybenzoic acid (1.0 g, 7.25 mmol), 2,3-dihydropyran (3.3 mL, 36.2 mmol), and p-toluenesulfonic acid (4 mg) in anhydrous THF (10 mL) was stirred under an argon atmosphere. After 5 hours, triethylamine (1 mL) was added and all volatiles were evaporated in vacuo. The resultant oily residue was dissolved in acetone (10 mL) and NaOH (1 M aq. solution, 10 mL) was added. After stirring overnight, the acetone was evaporated, water (5 mL) was added, and the alkaline solution was washed with dichloromethane (3 × 10 mL). The remaining aqueous solution was acidified by NaHSO4 (1 M aq. solution, 5 mL) and extracted with dichloromethane (4 × 10 mL). The combined organic extracts were dried over Na2SO4 and concentrated in vacuo to yield 4-(tetrahydro-2H-pyran-2-yloxy)benzoic acid (1.363 g, 85%), which was used in the subsequent reactions without further purification. TLC: 60% EtOAc/hexanes, Rf ∼ 0.3; 1H NMR (CDCl3, 400 MHz) δ 8.05 (d, 2H, J = 8.8 Hz), 7.10 (d, 2H, J = 8.8 Hz), 5.53 (t, 1H, J = 3.0 Hz), 3.90-3.82 (m, 1H), 3.66-3.60 (m, 1H), 2.08-1.96 (m, 1H), 1.92-1.86 (m, 2H), 1.78-1.58 (m, 3H).
Assays
The accumulation of AEA and PEA by CGN were determined as reported previously (Hillard and Jarrahian, 2005). The concentration of AEA or PEA was 0.65 nM; incubation time was 2 min. Each individual experiment was carried out in triplicate using 6 concentrations of competitor made in dimethylsulfoxide (DMSO; final concentration, 0.1%). The neurons were preincubated with the competitors for 10 min prior to the addition of AEA or PEA. Nonspecific association of AEA or PEA were determined in the presence of 100 μM of the corresponding unlabeled solute. Individual IC50 values were determined from each experiment using non-linear regression to fit the data to a single site competition equation. The IC50 values reported are the mean of 3 or 4 separate experiments.
Studies of the inhibition of FAAH by the compounds were carried out using an in vitro assay as reported previously (Jarrahian et al., 2000). The source of FAAH were P2 membranes prepared from whole mouse brain (Hillard et al., 1995). Membranes (10 μg protein) were incubated in a final volume of 0.5 ml of TME buffer (50 mM Tris-HCl, 3.0 mM MgCl2, and 1.0 mM EDTA, pH 7.4) containing 1.0 mg/ml fatty acid-free bovine serum albumin and 2 nM [3H]AEA. Incubations were carried out at 30°C for 10 min and were stopped with the addition of 2 ml of chloroform/methanol (1:2). After standing at ambient temperature for 30 min, 0.67 ml of chloroform and 0.6 ml of water were added. Aqueous and organic phases were separated by centrifugation at 1,000 rpm for 10 min. The amount of [3H] in both the aqueous and organic phases was determined by liquid scintillation counting and hydrolysis was calculated as the ratio aqueous dpm/(aqueous + organic dpm).
RESULTS
All five of the compounds studied (structures shown in Figure 1) completely inhibited the accumulation of AEA by CGN (Figure 2 and Table 1). The potencies of AM404, OMDM-2 and VDM11 were all very similar (approximately 5 μM) and did not differ significantly from one another. However, AM1172 and UCM707 had significantly lower potency, closer to the inhibitory potency of AEA itself in these neurons (Hillard et al., 1997).
Figure 1.
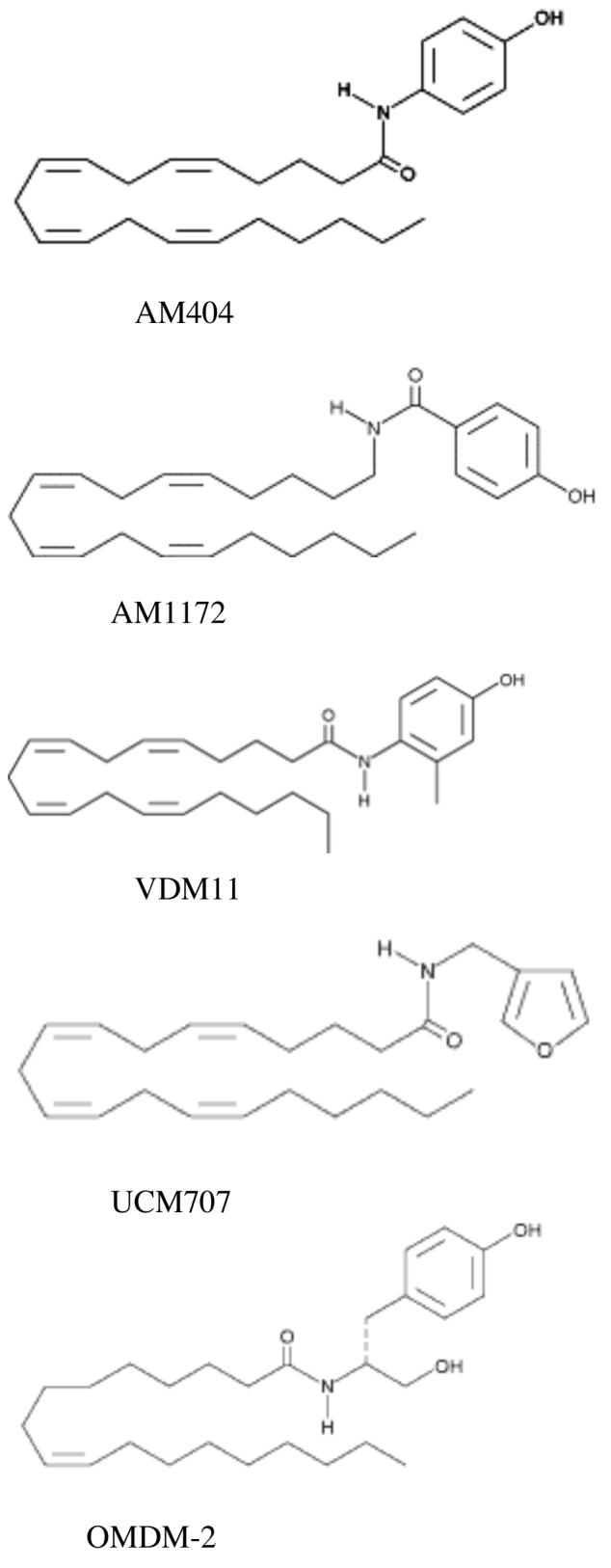
Structures of the compounds used in this study.
Figure 2.

Concentration response curves for the inhibition of AEA accumulation by CGN. Cells were washed, equilibrated in buffer for 30 min, then preincubated for 5 min with the inhibitor. [3H]AEA was added (final concentration, 0.65 nM) and the incubation was continued for exactly 2 min. Buffer was removed and cells were scraped in 0.5 ml of water. Buffer and cells were counted and the fraction accumulated by the cells was calculated as: (dpm in cells)/(dpm in cells + dpm in media). Nonspecific association was defined as the fraction accumulated in the presence of 100 μM AEA. Each experiment was carried out in triplicates\ and the entire concentration responses curves were repeated in at least 3 different preparations of CGN. Points represent the mean, vertical lines represent the S.E.M. Lines drawn are the best fit of the data points to a single site, competition equation using non-linear regression.
Table 1.
Summary of the effects of AEA accumulation inhibitors on AEA accumulation by CGN, metabolism of AEA by FAAH and PEA accumulation by CGN. Each value is the mean of at least 3 separate determinations, shown ± S.E.M.
| Compound | Inhibition of AEA accumulation (IC50, μM) | Inhibition of FAAH (KI, μM) | Inhibition of PEA accumulation (IC50, μM) |
|---|---|---|---|
| AM404 | 4.87 ± 1.73 | 0.60 ± 0.08 | 6.70 ± 1.39 |
| AM1172 | 24.0 ± 1.9 | 3.18 ± 0.10 | 36.0 ± 5.9 |
| UCM 707 | 30.3 ± 9.3 | 0.37 ± 0.10 | 44.1 ± 13.0 |
| VDM11 | 5.5 ± 1.2 | 0.44 ± 0.06 | 70.3 ± 30.0 |
| OMDM-2 | 4.9 ± 0.5 | 9.7 ± 2.0 | 32.8 ± 15.6 |
PEA is a highly abundant fatty acyl ethanolamide in brain that is also accumulated by CGN, although to a lesser degree than AEA (Hillard and Jarrahian, 2005). The accumulation of PEA is inhibited by AEA (Jacobsson and Fowler, 2001; Hillard and Jarrahian, 2005) with an IC50 value of about 1 μM (Jacobsson and Fowler, 2001). PEA accumulation is also inhibited by the AEA accumulation inhibitors investigated here (Table 1). PEA accumulation was less sensitive to the inhibitors than AEA accumulation, with the exception of AM404. There was no correlation between the potencies of the compounds to inhibit AEA accumulation and the potencies to inhibit PEA accumulation (r2 = 0.015; p = 0.84).
It has been suggested that hydrolysis of AEA by FAAH can enhance the accumulation of AEA by maintaining its concentration gradient (Day et al., 2001). It has been further suggested that inhibitors of AEA accumulation are inhibitors of FAAH-mediated hydrolysis and reduce accumulation because they collapse the concentration gradient (Glaser et al., 2003). Although this hypothesis has been largely discounted through the use of cells that are devoid of FAAH (Fegley et al., 2004; Ortega-Gutierrez et al., 2004), we examined the effects of the inhibitors on FAAH activity in mouse brain membranes. All five of the inhibitors tested function as inhibitors of FAAH (Figure 3 and Table 1). In fact, under our assay conditions, which are designed to be very sensitive to inhibitors, AM404, VDM11 and UCM707 all inhibit AEA hydrolysis with KI values less than 1 μM. While AM1172 and OMDM-2 are 5-10 fold less potent, they are still effective inhibitors in the micromolar concentration range. Although all of the accumulation inhibitors are also FAAH inhibitors, there is not a significant correlation between inhibitory potency for AEA accumulation and inhibition of AEA hydrolysis (r2 = 0.088; p = 0.63).
Figure 3.

Concentration response curves for the inhibition of AEA hydrolysis by mouse brain membrane-derived FAAH. Membranes were preincubated for 5 min with the inhibitor. [3H]AEA was added (final concentration, 2 nM) and the incubation was continued for exactly 10 min. The reaction was stopped by the addition of chloroform:methanol (1:2) followed by extraction and separation of aqueous and organic phases. Radioactivity was determined in both phases and the percent hydrolysis was calculated. Each experiment was carried out in triplicate and the entire concentration responses curves were repeated in at least 3 different membrane preparations. Points represent the mean, vertical lines represent the S.E.M. Lines drawn are the best fit of the data points to a single site, competition equation using non-linear regression.
DISCUSSION
We have confirmed that the five compounds examined are inhibitors of the accumulation of AEA by CGN. However, the potencies obtained for several of the compounds differ from what has been observed in other cell types. We found that AM404 inhibited AEA accumulation with an IC50 of 4.87 μM, which is very similar to the values that we obtained using CGN and C6 glioma in earlier studies (Jarrahian et al., 2000; Hillard and Jarrahian, 2005) and the values that have been reported by others (Beltramo et al., 1997a; De Petrocellis et al., 2000; Fowler et al., 2004a). As we have reported previously (Jarrahian et al., 2000), AM404 is a potent inhibitor of FAAH-mediated hydrolysis of AEA with an IC50 value of 600 nM. However, AM404 is itself hydrolyzed by FAAH at a rate only about 16% of that of AEA (Lang et al., 1999) suggesting that while AM404 has relatively high affinity for the substrate binding pocket of FAAH, it is not readily hydrolyzed and therefore functions as a competitive inhibitor of AEA hydrolysis by the enzyme. Therefore, AM404 could theoretically increase synaptic AEA content by two mechanisms: inhibition of cellular accumulation (which would sequester AEA from the CB1 receptor) and inhibition of FAAH-mediated catabolism.
AM1172 is structurally similar to AM404 except that the carboxamide position is reversed. In agreement with the report of Fegley and colleagues (Fegley et al., 2004) in cortical neurons, AM1172 inhibits AEA accumulation by CGN; but is about 6 fold less potent than AM404. It has an IC50 value of 24 μM, which is very similar to the IC50 value for AEA in CGN (Hillard and Jarrahian, 2005). While the IC50 value for AM1172 determined in cortical neurons is lower than seen here (between 1 and 10 μM (Fegley et al., 2004)), the IC50 for AEA is also lower in these cells (Beltramo et al., 1997b), suggesting that there is heterogeneity in the AEA accumulation process among neuronal populations.
In contrast to the findings of Fegley and colleagues (Fegley et al., 2004), we find that AM1172 is an inhibitor of FAAH-mediated metabolism of AEA. While the IC50 value obtained (3 μM) is 5-fold higher than the IC50 value obtained for AM404, AM1172 is also about 5-6 fold less potent as an inhibitor of AEA accumulation. Interestingly, AM1172 is also about 5-6 fold less potent than AM404 as an inhibitor of PEA accumulation by CGN. Therefore, in CGN, AM1172 does not provide any improvement over AM404 as a selective inhibitor of AEA accumulation versus inhibition of FAAH or PEA accumulation.
In our experimental models, VDM11 is very similar to AM404; i.e. it is a good inhibitor of both AEA accumulation and of FAAH. The IC50 values for inhibition of AEA accumulation by CGN is 2-fold less than reported for the same compound in C6 glioma and RBL-2H3 (De Petrocellis et al., 2000), suggesting that VDM11 is more potent in neurons than other cell types. The inhibitory effect of VDM11 on FAAH is in agreement with recent reports of Fowler and colleagues (Fowler et al., 2004a; Vandevoorde and Fowler, 2005) that VDM11 is both an inhibitor and substrate for FAAH, much like AM404. These results differ from those originally presented by Di Marzo and colleagues (De Petrocellis et al., 2000), likely because of methodological differences. For example, we have employed a very low concentration of radiolabeled AEA in our studies, in part to mimic the low concentrations of AEA in the brain and to enhance our ability to detect inhibition. In addition, our FAAH assay involves preincubation of the tissue with inhibitors for 10 min prior to the addition of radiolabeled AEA while the Di Marzo method adds both inhibitor and substrate simultaneously. Interestingly, it is likely that this issue is responsible for the difference in result since it is also the procedure used by Fowler and colleagues when inhibition of FAAH by VDM11 was seen (Fowler et al., 2004a). Perhaps VDM11 binds preferentially to FAAH that has not seen substrate.
The fourth compound examined, UCM707 is a substituted arachidonate like AM404, AM1172, and VDM11. However, a furan is used as the head group rather than a phenyl ring. While our previous structure-activity profile did not include any furan derivatives, we found that benzene rings in the head group region, with or without hydroxyl substitutions, were the most potent inhibitors of AEA accumulation in CGN (Jarrahian et al., 2000). In agreement with that original conclusion, UCM707 is not a potent inhibitor of AEA accumulation in CGN, with an IC50 value of 30 μM. Early reports from other laboratories regarding UCM707 have been contradictory. UCM707 reportedly inhibits AEA accumulation in U937 cells with an IC50 value of 800 nM (Lopez-Rodriguez et al., 2001) and in CGN with an IC50 value of 4 μM (Ortega-Gutierrez et al., 2004). On the other hand, the IC50 value for UCM707 was reported as 25-42 μM in RBL-2H3 cells (Fowler et al., 2004a) and inhibited accumulation in PC-3 cells by only 30% at a concentration of 100 μM (Ruiz-Llorente et al., 2004). Our current results indicate that UCM707 is not a potent inhibitor of AEA accumulation by CGN.
UCM707 is a very potent inhibitor of FAAH-mediated hydrolysis of AEA, with a KI value of 380 nM, the lowest of the five analogs investigated. This finding is also consistent with our earlier SAR study which demonstrated that amides of arachidonic acid are generally good inhibitors of FAAH, particularly when the head group substitution is not large (Jarrahian et al., 2000). To our knowledge, this is the first report that UCM707 is a potent inhibitor of FAAH; two earlier studies report IC50 values of 30 to greater than 50 μM (Lopez-Rodriguez et al., 2003; Fowler et al., 2004b). It is possible that the methodological differences described above also account for these discrepancies.
The final compound examined, OMDM-2, is a oleic acid derivative with a modified phenyl head group substitution. In agreement with an IC50 of 3 μM in RBL-2H3 cells (Ortar et al., 2003), OMDM-2 is a potent inhibitor of AEA accumulation by CGN, with an IC50 value of approximately 5 μM. This result is consistent with the demonstration by Piomelli and colleagues that the length of the acyl chain is not a critical determinant of inhibitory potency provided that at least one double bond is present (Piomelli et al., 1999). We find that OMDM-2 is also an inhibitor of FAAH, with a KI value of 9.7 μM. These data are in contradiction to other studies in which IC50 values greater than 50 μM were reported (Ortar et al., 2003; Fowler et al., 2004a).
We have investigated the effects of the five inhibitors on the accumulation of PEA by CGN. Like AEA, the accumulation of PEA is saturable, temperature-dependent and occurs in multiple cell types (Jacobsson and Fowler, 2001; Hillard and Jarrahian, 2005). However, there are a few hints that PEA accumulation is mechanistically different from AEA accumulation. First, the amount of PEA accumulated is much less than the amount of AEA (Hillard and Jarrahian, 2005). Second and more significantly, while PEA accumulation is inhibited by AEA, AEA accumulation is not inhibited by PEA in any cell type investigated (Jacobsson and Fowler, 2001; Hillard and Jarrahian, 2005), which is strong evidence that the mechanisms for PEA and AEA accumulation are not overlapping.
In the present study, the structure-activity profiles provide further evidence that the processes are not overlapping. AM404 is a good inhibitor of the accumulation of PEA by CGN, in agreement with earlier studies using Neuro-2a and RBL-2H3 cells (Jacobsson and Fowler, 2001). However, none of the other compounds examined are as potent as AM404. In particular, OMDM-2 and VDM11, which inhibit AEA accumulation at concentrations nearly identical to AM404, are 5-10 fold less potent inhibitors of PEA accumulation. We find that there is no correlation between the IC50 values for PEA accumulation and IC50 values for FAAH inhibition by these compounds. This finding does not support a hypothesis that we put forth earlier that PEA accumulation by CGN is dependent upon FAAH (Hillard and Jarrahian, 2005). Unfortunately, our current lack of understanding of the molecular mechanisms of AEA and PEA accumulation make interpretation of these data difficult. However, the differences could become clear as more data regarding relative effects of inhibitors of the accumulation of these two very similar solutes are gathered. Furthermore, some insights could be gleaned from studies of structurally similar molecules. As discussed in the session by Hamilton, unesterified fatty acids diffuse rapidly across a lipid membrane and do not require a specialized transporter. The AEA molecule does not have a charged head group, and the energy barrier for translocation is expected to be low, leading to the prediction that this molecule also would not have a requirement for a protein-mediated transporter, or flippase. However, the structure activity data among inhibitors of AEA accumulation suggest that specific and saturable binding to a protein is involved in AEA accumulation. The challenge is to identify the protein or proteins involved.
In conclusion, we have examined the potencies of five available compounds of inhibitors of AEA and PEA accumulation by CGN and as inhibitors of FAAH-mediated hydrolysis of AEA. We find that all of the compounds are good inhibitors of FAAH, and certainly will inhibit FAAH to some extent at concentrations at which they effectively inhibit AEA accumulation. However, there is no correlation between the IC50 values for inhibition of AEA accumulation and FAAH inhibition, which argues against the inhibition of FAAH as the primary mechanism for inhibition of AEA accumulation. As was recently concluded by Vandervoode and Fowler (Vandevoorde and Fowler, 2005), biochemical dissection of the mechanism of AEA accumulation by cells is clouded by a lack of specific inhibitors.
ACKNOWLEDGMENTS
The authors gratefully acknowledge the technical assistance of LeAnna DeLoache and Craig Roelke. These studies were supported by NIH grant DA09155 (CJH and WBC) and NIH GM31278 and the Robert A. Welch Foundation (JRF).
REFERENCES
- Beltramo M, di Tomaso E, Piomelli D. Inhibition of anandamide hydrolysis in rat brain tissue by (E)-6- (bromomethylene) tetrahydro-3-(1-naphthalenyl)-2H-pyran-2-one. FEBS Lett. 1997a;403:263–267. doi: 10.1016/s0014-5793(97)00061-6. [DOI] [PubMed] [Google Scholar]
- Beltramo M, Stella N, Calignano A, Lin SY, Makriyannis A, Piomelli D. Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science. 1997b;277:1094–1097. doi: 10.1126/science.277.5329.1094. [DOI] [PubMed] [Google Scholar]
- Day TA, Rakhshan F, Deutsch DG, Barker EL. Role of fatty acid amide hydrolase in the transport of the endogenous cannabinoid anandamide. Mol Pharmacol. 2001;59:1369–1375. doi: 10.1124/mol.59.6.1369. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Bisogno T, Davis JB, Pertwee RG, Di Marzo V. Overlap between the ligand recognition properties of the anandamide transporter and the VR1 vanilloid receptor: inhibitors of anandamide uptake with negligible capsaicin-like activity. FEBS Lett. 2000;483:52–56. doi: 10.1016/s0014-5793(00)02082-2. [DOI] [PubMed] [Google Scholar]
- Fegley D, Kathuria S, Mercier R, Li C, Goutopoulos A, Makriyannis A, Piomelli D. Anandamide transport is independent of fatty-acid amide hydrolase activity and is blocked by the hydrolysis-resistant inhibitor AM1172. Proc Natl Acad Sci U S A. 2004;101:8756–8761. doi: 10.1073/pnas.0400997101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler CJ, Tiger G, Ligresti A, Lopez-Rodriguez ML, Di Marzo V. Selective inhibition of anandamide cellular uptake versus enzymatic hydrolysis-a difficult issue to handle. Eur J Pharmacol. 2004a;492:1–11. doi: 10.1016/j.ejphar.2004.03.048. [DOI] [PubMed] [Google Scholar]
- Fowler CJ, Tiger G, Ligresti A, Lopez-Rodriguez ML, Di Marzo V. Selective inhibition of anandamide cellular uptake versus enzymatic hydrolysis--a difficult issue to handle. Eur J Pharmacol. 2004b;492:1–11. doi: 10.1016/j.ejphar.2004.03.048. [DOI] [PubMed] [Google Scholar]
- Glaser ST, Abumrad NA, Fatade F, Kaczocha M, Studholme KM, Deutsch DG. Evidence against the presence of an anandamide transporter. Proc Natl Acad Sci U S A. 2003;100:4269–4274. doi: 10.1073/pnas.0730816100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillard CJ, Jarrahian A. Cellular accumulation of anandamide: consensus and controversy. Br J Pharmacol. 2003;140:802–808. doi: 10.1038/sj.bjp.0705468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillard CJ, Jarrahian A. Accumulation of anandamide: Evidence for cellular diversity. Neuropharmacology. 2005;48:1072–1078. doi: 10.1016/j.neuropharm.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Hillard CJ, Wilkison DM, Edgemond WS, Campbell WB. Characterization of the kinetics and distribution of N-arachidonylethanolamine (anandamide) hydrolysis by rat brain. Biochim Biophys Acta. 1995;1257:249–256. doi: 10.1016/0005-2760(95)00087-s. [DOI] [PubMed] [Google Scholar]
- Hillard CJ, Edgemond WS, Jarrahian A, Campbell WB. Accumulation of N-arachidonoylethanolamine (anandamide) into cerebellar granule cells occurs via facilitated diffusion. J. Neurochem. 1997;69:631–638. doi: 10.1046/j.1471-4159.1997.69020631.x. [DOI] [PubMed] [Google Scholar]
- Jacobsson SO, Fowler CJ. Characterization of palmitoylethanolamide transport in mouse Neuro-2a neuroblastoma and rat RBL-2H3 basophilic leukaemia cells: comparison with anandamide. Br J Pharmacol. 2001;132:1743–1754. doi: 10.1038/sj.bjp.0704029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrahian A, Manna S, Edgemond WS, Campbell WB, Hillard CJ. Structure-activity relationships among N-arachidonylethanolamine (Anandamide) head group analogues for the anandamide transporter. J Neurochem. 2000;74:2597–2606. doi: 10.1046/j.1471-4159.2000.0742597.x. [DOI] [PubMed] [Google Scholar]
- Lang W, Qin C, Lin S, Khanolkar AD, Goutopoulos A, Fan P, Abouzid K, Meng Z, Biegel D, Makriyannis A. Substrate specificity and stereoselectivity of rat brain microsomal anandamide amidohydrolase. J Med Chem. 1999;42:896–902. doi: 10.1021/jm980461j. [DOI] [PubMed] [Google Scholar]
- Lopez-Rodriguez ML, Viso A, Ortega-Gutierrez S, Lastres-Becker I, Gonzalez S, Fernandez-Ruiz J, Ramos JA. Design, synthesis and biological evaluation of novel arachidonic acid derivatives as highly potent and selective endocannabinoid transporter inhibitors. J Med Chem. 2001;44:4505–4508. doi: 10.1021/jm015545y. [DOI] [PubMed] [Google Scholar]
- Lopez-Rodriguez ML, Viso A, Ortega-Gutierrez S, Fowler CJ, Tiger G, de Lago E, Fernandez-Ruiz J, Ramos JA. Design, synthesis, and biological evaluation of new inhibitors of the endocannabinoid uptake: comparison with effects on fatty acid amidohydrolase. J Med Chem. 2003;46:1512–1522. doi: 10.1021/jm0210818. [DOI] [PubMed] [Google Scholar]
- Ortar G, Ligresti A, De Petrocellis L, Morera E, Di Marzo V. Novel selective and metabolically stable inhibitors of anandamide cellular uptake. Biochem Pharmacol. 2003;65:1473–1481. doi: 10.1016/s0006-2952(03)00109-6. [DOI] [PubMed] [Google Scholar]
- Ortega-Gutierrez S, Hawkins EG, Viso A, Lopez-Rodriguez ML, Cravatt BF. Comparison of anandamide transport in FAAH wild-type and knockout neurons: evidence for contributions by both FAAH and the CB1 receptor to anandamide uptake. Biochemistry. 2004;43:8184–8190. doi: 10.1021/bi049395f. [DOI] [PubMed] [Google Scholar]
- Piomelli D, Beltramo M, Glasnapp S, Lin SY, Goutopoulos A, Xie XQ, Makriyannis A. Structural determinants for recognition and translocation by the anandamide transporter. Proc Natl Acad Sci. 1999;96:5802–5807. doi: 10.1073/pnas.96.10.5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Llorente L, Ortega-Gutierrez S, Viso A, Sanchez MG, Sanchez AM, Fernandez C, Ramos JA, Hillard C, Lasuncion MA, Lopez-Rodriguez ML, Diaz-Laviada I. Characterization of an anandamide degradation system in prostate epithelial PC-3 cells: synthesis of new transporter inhibitors as tools for this study. Br J Pharmacol. 2004;141:457–467. doi: 10.1038/sj.bjp.0705628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandevoorde S, Fowler CJ. Inhibition of fatty acid amide hydrolase and monoacylglycerol lipase by the anandamide uptake inhibitor VDM11: evidence that VDM11 acts as an FAAH substrate. Br J Pharmacol. 2005;145:885–893. doi: 10.1038/sj.bjp.0706253. [DOI] [PMC free article] [PubMed] [Google Scholar]