

Abstract
Most replicases are multi-subunit complexes. DNA polymerase epsilon from Saccharomyces cerevisiae is composed of four subunits: Pol2p, Dpb2p, Dpb3p, and Dpb4p. Pol2p and Dpb2p are essential. To investigate a possible role for the Dpb2p subunit in maintaining the fidelity of DNA replication, we isolated temperature-sensitive mutants in the DPB2 gene. Several of the newly isolated dpb2 alleles are strong mutators, exhibiting mutation rates equivalent to pol2 mutants defective in the 3′ → 5′ proofreading exonuclease (pol2-4) or to mutants defective in mismatch repair (msh6). The dpb2 pol2-4 and dpb2 msh6 double mutants show a synergistic increase in mutation rate, indicating that the mutations arising in the dpb2 mutants are due to DNA replication errors normally corrected by mismatch repair. The dpb2 mutations decrease the affinity of Dpb2p for the Pol2p subunit as measured by two-hybrid analysis, providing a possible mechanistic explanation for the loss of high-fidelity synthesis. Our results show that DNA polymerase subunits other than those housing the DNA polymerase and 3′ → 5′ exonuclease are essential in controlling the level of spontaneous mutagenesis and genetic stability in yeast cells.
UNDERSTANDING of the mechanisms that control the generation of mutations on normal and damaged DNA templates is crucial in studies of carcinogenesis and mutagenesis. The mutator hypothesis for the origins of cancer suggests that both early and late stages of tumor progression are connected to expression of a mutator phenotype (Loeb 2001; Loeb et al. 2003; Bielas et al. 2006). One likely source of spontaneous mutations are errors occurring during DNA replication. Thus, understanding of the mechanisms controlling DNA replication fidelity has become a major challenge of current molecular biology.
The accuracy of DNA synthesis is maintained by three highly conserved processes: correct base selection by the DNA polymerases, removal of base insertion errors by 3′ → 5′ exonucleolytic proofreading activity of DNA polymerases, and postreplication correction of polymerase errors by the DNA mismatch repair system (MMR). Thus, DNA polymerases are central to replication fidelity. Most of the major replicative DNA polymerases, often called replicases, are multi-subunit holoenzymes (HE). In eukaryotic cells, DNA replication is executed by at least three DNA polymerases: Pol α, Pol δ, and Pol ɛ. Pol ɛ HE is a four-subunit complex composed of a DNA polymerase/exonuclease subunit, Pol2p, and three auxiliary subunits: Dpb2p, Dpb3p, and Dpb4p (Hamatake et al. 1990; Dua et al. 2000; Chilkova et al. 2003; Asturias et al. 2006). This composition of subunits is conserved from yeast to humans (Kesti et al. 1993; Li et al. 1997; Jokela et al. 1998; Li et al. 2000; Feng et al. 2003; Spiga and D'Urso 2004). Two subunits, Pol2p and Dpb2p, are essential in yeast (Morrison et al. 1990; Araki et al. 1991a). Dpb3p and Dpb4p are not essential (Araki et al. 1991b; Ohya et al. 2000). Pol2p is essential for leading-strand DNA replication and plays a role in linking DNA replication and the S-phase checkpoint in Saccharomyces cerevisiae (Araki et al. 1992; Budd and Campbell 1993; Navas et al. 1995, 1996; Dua et al. 1998, 1999; Pursell et al. 2007). Strains carrying a temperature-sensitive mutation in the DPB2 gene (dpb2-1) suggest that Dpb2p is also essential for DNA replication (Araki et al. 1991a). At the restrictive temperature, dpb2-1 cells are dumbbell shaped, which is characteristic for a DNA replication defect. Dpb2p interacts with Pol2p (Sugino 1995; Dua et al. 2000) and is phosphorylated by Cdc28p, a cyclin-dependent protein kinase, in a cell-cycle-dependent manner (Kesti et al. 2004). Nevertheless, the precise cellular function of Dpb2p is unknown.
Strains carrying exonuclease-deficient Pol δ (pol3-01) or Pol ɛ (pol2-4) are mutators. Using URA3 forward mutation or his7-2 reversion assays, Morrison and Sugino (1994) demonstrated that the pol3-01 allele elevates spontaneous-mutation rates by ∼100-fold, as compared to an only 10-fold increase for the pol2-4 allele. However, the fidelity of purified exonuclease-deficient Pol δ HE is reduced by ∼10-fold compared to wild type, while the fidelity of exonuclease-deficient Pol ɛ HE is reduced by ∼100-fold (Shimizu et al. 2002; Hashimoto et al. 2003). These differences may suggest that the in vivo spontaneous-mutation rates of pol3-01 and pol2-4 strains are modulated by some factor(s). One important factor is the status of the replication checkpoint, since pol3-01 and pol2-4 have similar mutation rates in checkpoint-defective dun1Δ mutants (Datta et al. 2000). There are other examples suggesting that noncatalytic DNA polymerase subunits may contribute to polymerase fidelity (Araki et al. 1991b; Huang et al. 2002; Pham et al. 2006). The absence of Pol32p, a nonessential subunit of Pol δ HE of S. cerevisiae, causes increases in genomic deletions of sequences flanked by short direct repeats (Huang et al. 2002). In addition, deletion of the Dpb3p subunit of Pol ɛ HE (dpb3Δ) elevated the spontaneous-reversion rate by factors 2.2, 2.6, and 20 at his1-7, ade2-1, and lys1-1, respectively (Araki et al. 1991b). In Escherichia coli, mutations in the dnaX gene, encoding the τ-subunit of DNA Pol III HE, lead to a mutator phenotype (Pham et al. 2006).
In this work we have investigated whether Dpb2p contributes to the fidelity of Pol ɛ. Using random mutagenesis, we identified a set of temperature-sensitive DPB2 mutated strains (dpb2ts). Among them, we identified several strains that dramatically increase the level of spontaneous mutations. For several tested markers, this mutator effect is even greater than that observed previously for Pol ɛ mutants defective in the 3′ → 5′ exonuclease proofreading activity (pol2-4) and is comparable to that observed in certain strains defective in DNA mismatch repair (msh6). We also provide evidence that the observed mutator phenotype is a consequence of an increase in replication errors in the dpb2 mutants. Our results indicate that noncatalytic replicase subunits may play an important role in maintaining the fidelity of DNA replication in eukaryotes, as has been shown previously in prokaryotic systems (Pham et al. 2006).
MATERIALS AND METHODS
Media and growth conditions:
S. cerevisiae strains are listed in Table 1. Strains were grown in standard media (Adams et al. 1997). Yeast complete medium (YPD), containing 1% yeast extract, 1% peptone, and 2% glucose, was routinely used for culturing yeast strains when nutrition selection was not required. For yeast transformations and mutagenesis assays, yeast strains were grown in SD minimal medium (0.67% yeast nitrogen base without amino acids, 3% glucose) supplemented with appropriate l-amino acids and nucleotides. To measure frequency of forward mutations at the CAN1 locus, SD plates were additionally supplemented with l-canavanine (60 mg/liter), an analog of arginine. Whereas, SD medium containing, in addition, uracil and 0.1% of 5-fluoroorotic acid (5-FOA) was used for selection of ura3 mutants.
TABLE 1.
Yeast strains used in this study
| Strain | Genotype | Source |
|---|---|---|
| BY4743 dpb2∷kanMX4/DPB2 | MATa/α his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 ura3Δ0/ura3Δ lys2Δ0/LYS2 MET15/met15Δ dpb2∷kanMX4/DPB2 | EUROSCARF accession no. Y25590 |
| ΔI(-2)I-7B-YUNI300 | MATatrp1-289 his7-2 leu2-Δ∷kanMX4 ura3-Δ ade2-1 lys2-ΔGG2899-2900 CAN1 | Pavlov et al. (2002) |
| ΔI(-2)I-7B-YUNI300 msh6∷hisG | MATatrp1-289 his7-2 leu2-Δ∷kanMX4 ura3-Δ ade2-1 lys2-ΔGG2899-2900 CAN1 msh6∷hisG | Y. Pavlov |
| ΔI(-2)I-7B-YUNI300 pol2-4 | MATatrp1-289 his7-2 leu2-Δ∷kanMX4 ura3-Δ ade2-1 lys2-ΔGG2899-2900 CAN1 pol2-4 | Y. Pavlov |
| FF18733 | MATatrp1-289 his7-2 leu2-3,112 ura3-52 lys1-1 | F. Fabre |
| S288C | MATα gal2 mal mel flo1 flo8-1 hap1 SUC2 | Mortimer and Johnston (1986) |
| SC11 | MATα his3Δ1 leu2Δ0 ura3Δ0 lys2Δ0 dpb2∷kanMX4 [pMJDPB2 (DPB2 URA3)] | This study; derivative of Y25590 |
| Y190 | MATatrp1-901 his3-200 leu2-3,112 ura3-52 ade2-101 lys2-801 gal4Δ gal80Δ cyh2 LYS2∷GAL1UAS- HIS3TATA box-HIS3 URA3∷GAL1UAS-GAL1TATA box-lacZ | Harper et al. (1993) |
| W303 dpb2-1 | MATatrp1-1 his3-11,15 leu2-3,112 ura3-1 ade2-1 can1-100 dpb2-1 | H. Araki |
Construction of plasmids:
Generally, plasmids were constructed according to the standard protocols as described by Sambrook et al. (1989). Propagation of plasmids was performed in DH5α or XL1-Blue MRF′ E. coli strains. All PCR products and fusion junctions were confirmed by DNA sequencing.
Cloning of the DPB2 gene:
Genomic DNA from strain FF18733 was used as a template to clone the DPB2 gene by PCR amplification with primers 5′-CGTTTGAGGAAGCTTAGGATACTTGGCGTAG-3′ (the HindIII site is underlined) and 5′-GTCCCCATGGATCCCATATATTGTATGCCG-3′ (BamHI is underlined). The 3.1-kb PCR product was HindIII/BamHI-cloned into URA3-containing YCplac33 (Gietz and Sugino 1988), resulting in plasmid pMJDPB2. To create a HIS3-containing plasmid, isogenic with plasmids carrying mutated DPB2 alleles (described below), a PCR amplification was performed using pMJDPB2 as a template and primers 5′-CATCTGCGGTCGACCCCCATACAAAC-3′ (SalI underlined) and 5′-CAAAAAGTATGGATCCAAATAGATGGCAG-3′ (BamHI underlined). Finally, the 2606-bp SalI–BamHI fragment, containing the DPB2 gene, was cloned into SalI/BamHI-digested pRS313 (Sikorski and Hieter 1989), yielding pGJ2.
Reconstruction of the dpb2-1 allele:
We rescued the previously described temperature-sensitive dpb2-1 allele (Araki et al. 1991a) on a plasmid to test its influence on replication fidelity and to compare with dpb2ts alleles generated ourselves. The full-length dpb2-1 ORF was isolated from W303 dpb2-1 yeast cells (kindly provided by H. Araki) by the gap-repair method using EcoRI/StuI-linearized pKF106 (described below). The resulting plasmid was named pKF161 and the dpb2-1 allele obtained was confirmed by sequencing.
Construction of integrating plasmids:
Construction of the plasmid for integration into the DPB2 genomic locus was a multi-step procedure. The ClaI-STOP part of DPB2 and its 901-bp 3′ flanking region were PCR amplified using genomic DNA of strain S288C as a template and the primer pair 5′-CCGCAAGATCCAATTCCTAGTG-3′ and 5′-GTAGTCGACGAGACGCTTGTGTGTGCTTGATTCTCC-3′ (SalI underlined). The 1477-bp PCR product was ClaI/SalI-digested and the resulting 1233-bp fragment was ligated into pRS313 at ClaI and XhoI sites, yielding pKF100. This destroyed the XhoI site of pRS313, making the XhoI site of DPB2 unique in derivatives of the resulting vector. The pKF100 plasmid was then digested with SmaI and ClaI, and a full-length PDPB2-DPB2-TDPB2 cassette (549-2079-901 bp) was regenerated by inserting the 2303-bp FspI–ClaI fragment of pMJDPB2. The resulting plasmid, pKF107, was subsequently converted to pKF106 by replacement of the XhoI–ClaI DPB2 fragment with a synthetic double-stranded DNA linker named plomba: 5′-CTCGAGCAATTTGCTGCAGTGCCTACACAAGGCATAACTTCGTATAGCATACATTATACGAAGTTATCCGATATCGAT-3′. The 799-bp ClaI–HindIII fragment of pKF100, comprising the 3′ fragment of the DPB2 ORF and the 5′-end of the DPB2 terminator (the ‘DPB2-TDPB2’ sequence; apostrophes indicate the sides of truncation), was excised and filled in with Klenow fragment and ligated into XbaI-linearized and Klenow-treated pKF106. A clone, containing the insert in the same orientation as the full-length DPB2 gene (the XbaI site recreated), was isolated and named pKF108. The pKF115 vector was created by ligation of the 1470-bp SmaI–Ecl136II part of pAG60 (Goldstein et al. 1999; EUROSCARF accession no. P30111) into the NaeI-linearized pRS303 (Sikorski and Hieter 1989). This resulted in insertion of the heterologous CaURA3MX4 cassette, containing the URA3 ORF of Candida albicans under the control of both promoter and terminator sequences of the Ashbya gossypii TEF gene. A clone bearing CaURA3MX4 in inverted orientation with respect to the HIS3 gene was chosen as pKF115. Finally, the 2911-bp ‘DPB2-TDPB2’-PDPB2-dpb2∷plomba-TDPB2 sequence was ApaI/SacI subcloned from pKF108 into pKF115, yielding an integrative plasmid named pKF117, which served as an acceptor for mutated DPB2 variants. The presence of the short (73 bp) plomba sequence instead of a larger fragment (1558 bp) of the DPB2 ORF allowed for unambiguous identification of the desired recombinant plasmids by restriction digestion. The map of pKF117 and the use of the vector for integration into the DPB2 locus is shown in supplemental Figure S1 at http://www.genetics.org/supplemental/. A control integrative plasmid, pKF120, was constructed in the same way as dpb2-containing pKF117 derivatives, but the wild-type DPB2 sequence was inserted in place of the mutated dpb2 gene.
Construction of two-hybrid plasmids:
Plasmids for the two-hybrid system (Fields and Song 1989) are based on pGBT9 and pGAD424 multi-copy vectors (Clontech), which encode the Gal4p DNA-binding (BDGAL4) or transcription activation domain (ADGAL4), respectively. pGBT9 and pGAD424 were converted to pKF75 and pKF80, respectively, by EcoRI–PstI digestion and by replacing the 26-bp excised sequence with a new polylinker compatible with a common series of bacterial and E. coli/yeast cloning vectors, i.e., pBluescript (Stratagene, La Jolla, CA) and pRS (Sikorski and Hieter 1989). All restriction sites introduced were placed in frame with the corresponding GAL4 domain. pKF75 and pKF80 were tested for lack of self-activation of expression of the Gal4p-dependent genes and were used for construction of fusions of genes of interest with the GAL4 domains for the directed two-hybrid assay. The synthetic linker used and the plasmids obtained are listed in Figure 1. For cloning of the DPB2 alleles, a sequence encoding ORF of the dpb2∷plomba allele was PCR amplified from pKF106 with primers  and
and 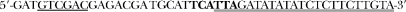 (ORF-coding sequence is double underlined, restriction sites are underlined, and nucleotides corresponding to the ATG and STOP codons are in boldface). The BamHI–SalI fragment of the PCR product was cloned into pKF75 and pKF80, giving pKF133 and pKF137 plasmids, respectively. The expected wild-type or mutated variants of DPB2 were subsequently created by replacing the plomba sequence with subcloned fragments of the respective alleles.
(ORF-coding sequence is double underlined, restriction sites are underlined, and nucleotides corresponding to the ATG and STOP codons are in boldface). The BamHI–SalI fragment of the PCR product was cloned into pKF75 and pKF80, giving pKF133 and pKF137 plasmids, respectively. The expected wild-type or mutated variants of DPB2 were subsequently created by replacing the plomba sequence with subcloned fragments of the respective alleles.
Figure 1.—

Genetic constructs for two-hybrid system. (A) Scheme of the polylinker that was built in pKF75 and pKF80 vectors. All restriction sites are in frame with the respective GAL4 domain. Unique restriction sites are indicated by checks (√), while both original EcoRI and PstI sites destroyed in the construction are crossed out. (B) Plasmids used in the two-hybrid assays.
The positive control for two-hybrid assays was the centromeric pCL1 plasmid (Fields and Song 1989), which contains the entire Gal4p-coding region (881 aa). As a negative control, we used pGBT9dnaE+ and pGAD424dnaE+ (Jonczyk et al. 1998).
Random mutagenesis of the DPB2 gene and selection of temperature-sensitive dpb2 alleles:
A library of mutated variants of DPB2 was created on centromeric plasmids using random mutagenesis procedures based on hydroxylamine treatment or mutagenic PCR. The hydroxylamine mutagenesis was performed using the pGJ2 plasmid and a standard protocol as described by Sambrook et al. (1989). The Diversify PCR random mutagenesis kit (Clontech) was used to introduce mutations by PCR. The mutagenic PCR amplifications were done under reaction conditions nos. 5–8 of the manufacturer's protocol and using the pMJDPB2 plasmid as a template and the same primers as used for construction of pGJ2. The PCR products were SalI/BamHI-digested, cloned into SalI/BamHI-linearized pRS313, and then used directly for yeast transformation.
To screen for plasmids carrying temperature-sensitive dpb2 alleles, strain SC11 was constructed. A diploid strain, Y25590, carrying wild-type and disrupted chromosomal alleles of DPB2 (BY4743 DPB2/dpb2∷kanMX4), was transformed with pMJDPB2 and Ura+ transformants were selected. After sporulation and tetrad dissection, a haploid disruptant strain (dpb2∷kanMX4 [pMJDPB2]) was obtained and named SC11. Strain SC11 was transformed with the library of mutated DPB2 variants on the pRS313 plasmid (HIS3), plated on SD medium supplemented with lysine and leucine, and incubated at 23° for 7–10 days. The His+ transformants were isolated and toothpicked twice at 23° onto plates additionally containing uracil and 5-FOA (a selective agent against Ura+ cells; Boeke et al. 1984) to remove the pMJDPB2 plasmid bearing the wild-type URA3 and DPB2 genes. About 15,000 Ura− colonies were selected and screened for temperature sensitivity at 37°. Several independent temperature-sensitive clones were identified. To confirm that temperature sensitivity resulted from the presence of the library clone, the plasmid DNA was isolated from each strain and retransformed into SC11 and the screening procedure was repeated as described above. Temperature-sensitive phenotype was confirmed for all isolated dpb2 strains. DNA sequence analysis of mutant alleles showed that all contained multiple missense mutations within the DPB2 gene causing amino acid alterations at the protein level. For further studies, two mutated alleles were chosen: one generated by PCR (pMJ25) and another generated by hydroxylamine treatment (pMJ36).
Next, fragments of dpb2-coding sequence from pMJ25, containing identified mutations, were excised and used to replace appropriate fragments of the wild-type DPB2 gene present in the plasmid pGJ2 (pRS313 HIS3 DPB2), generating three plasmids: pMJ100, pMJ101, and pMJ102 (Table 2). Since, in the case of pMJ36, the whole plasmid was treated with hydroxylamine, the DPB2 ORF was also subcloned into untreated plasmid, resulting in pMJ103 (Table 2), and the plasmid-dependent temperature-sensitive growth was confirmed again to exclude the possibility that the ts phenotype was caused by mutated vector, e.g., by mutations in the HIS3 marker gene.
TABLE 2.
Amino acid substitutions in the alleles of dpb2
| Original plasmid | Final plasmid | dpb2 allele | Amino acid changea | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pMJ25 | pMJ100 | dpb2-100 | L284P | T345A | ||||||||
| pMJ25 | pMJ101 | dpb2-101 | L284P | |||||||||
| pMJ25 | pMJ102 | dpb2-102 | T345A | |||||||||
| pMJ36 | pMJ103 | dpb2-103 | T342I | S343F | T345I | P347S | P348S | |||||
| pKF161 | dpb2-1 | D300N | K521R | V565F | G662R | |||||||
The amino acid numbering system is according to the Saccharomyces Genome Database (http://www.yeastgenome.org/); i.e., the second ATG is chosen as the translation initiation codon.
Integration into the DPB2 locus:
The DPB2 alleles were introduced into the chromosome by transplacement. The pKF117-derivative vectors were linearized at unique BamHI and XbaI sites positioned between PDPB2 and an additional fragment of the DPB2 terminator. The linearized plasmids were transformed (as described in Gietz and Woods 1998) into strain ΔI(-2)I-7B-YUNI300 (Pavlov et al. 2002) and its two derivatives deficient in mismatch repair (msh6∷hisG) or Pol ɛ proofreading exonuclease (pol2-4), respectively (Table 1). Transformants were selected for uracil prototrophy on plates, which were incubated at 23° for up to 10 days. Colonies were replica plated onto new SD −Ura plates to confirm the Ura+ phenotype. Additionally, these plates were duplicated and incubated at 37° for 3 days to check for temperature-sensitive growth. In the cases of alleles that caused a ts phenotype, only ts Ura+ transformants were further analyzed; otherwise all Ura+ colonies were checked for correct integration. The genomic DNA was isolated and used as a template in a PCR reaction with the primers 5′-TGTAAAACGACGGCCAGT-3′ (-21 M13 universal primer) and 5′-GAATACTGGCTTACCGAG-3′, which recognized the vector sequence and the chromosomal sequence downstream of DPB2, respectively, generating a 1361-bp band in case of integration into the aimed locus. The presence of the expected mutation(s) was verified by a second round of PCR with DPB2-specific primers and DNA sequencing of the PCR product. In view of the fact that most of the DPB2 alleles used, as well as both msh6∷hisG and pol2-4 strains, may contribute to a mutator phenotype, the number of passages was minimized and the confirmed integrants were immediately frozen in liquid nitrogen and stored at −80°. For that reason, the removing of the residual vector sequence from the chromosome was omitted. The DPB2 control strains were generated by integration of the pKF120 plasmid. The resultant control strains contained the wild-type DPB2 gene and the same residual vector sequence as in the strains constructed with the dpb2-containing derivatives of pKF117. In control studies, where we removed the vector sequences, we did not observe any difference in the rate of mutagenesis between DPB2 and DPB2∷vector alleles.
Measurement of spontaneous-mutation frequency and calculation of mutation rates:
The assays were performed in different genetic backgrounds. The DPB2 variants were introduced on plasmids (SC11 derivatives; BY genetic background) or integrated into the DPB2 chromosomal locus [ΔI(-2)I-7B-YUNI300 derivatives]. To determine spontaneous-mutation frequencies, 10–15 independent cultures of individual clones were used. Colonies were taken from two to three independent isolates of each strain and inoculated in 3–15 ml of liquid SD medium supplemented with required amino acids and nucleotides. The cultures were grown at 23° to stationary phase; yeast cells were collected by centrifugation, washed, and finally resuspended in water. Aliquots of undiluted cultures and appropriate dilutions were plated on selective and nonselective plates and incubated for 7–10 days at 23°, and the appeared colonies were counted. The frequency of forward mutations was measured at the CAN1 locus in both genetic backgrounds. Also, the reversion frequencies of his7-2, trp1-289, and lys2-ΔGG2899-2900 were measured in the ΔI(-2)I-7B-YUNI300 derivatives. Each experiment was repeated three times. Mutant frequency was determined by dividing the median mutant count by the median total cell count. The mutation rates were calculated using the equation μ = f/ln(Nμ) (where μ is the mutation rate per replication, f is the mutant frequency, and N is the total population size), which was solved by iteration (Drake 1991).
Immunoblot analysis of yeast extracts:
Yeast strains were grown at 23° in SD minimal medium, supplemented with required amino acids and nucleotides, until OD600nm reached 0.8 unit. Cells from 150-ml cultures were collected by centrifugation, and pellets were frozen in liquid nitrogen and stored at −80°. Cells were thawed and then lysed by addition of 100 μl of freshly prepared alkaline lysis buffer (50 mm NaOH, 2% SDS, 10% glycerol, 5% 2-mercapthoethanol, 2 mm EDTA, 0.04% bromophenol blue), and the samples were boiled for 5 min. Extracts were neutralized with 1 n HCl (4.5 μl/100 μl of cell extract), and cell extracts were centrifuged for 5 min at 4°. Proteins were separated by 7% SDS–PAGE and transferred to nitrocellulose membrane (Hybond-C Extra, Amersham Biosciences). BDGal4-Dpb2 fusion proteins (∼97 kDa) were detected with rabbit anti-BDGal4p antibodies (Sigma, St. Louis) followed by incubation with the ImmunoPure goat anti-rabbit IgG antibodies conjugated with horseradish peroxidase (HRP; Pierce, Rockford, IL). Bands were visualized using chemiluminescent substrates for HRP (SuperSignal WestPico, Pierce) and the Fluorchem SP Imager (Alpha Innotech).
Preparation of samples for flow cytometry analysis:
Selected strains were cultured in 50 ml of liquid YPDA medium (YPD supplemented with adenine) at permissive temperature (23°) until OD600 nm reached 0.4 unit. Samples (1 ml) were collected for further processing. Cells were treated with α-factor (10 μg/ml) and further incubated with shaking at 23° until 95% of the cells were synchronized (3 hr). Synchronized cultures were incubated with α-factor for an additional 1 hr at restrictive temperature (37°). Cells were harvested and released from α-factor by washing twice with prewarmed (37°) YPDA. After washing, cells were resuspended in 75 ml of fresh YPDA at 37°, and 1-ml samples were immediately collected for processing. The remaining cultures were further incubated with shaking at 37° and subsequent samples were collected every 15 min for 2.5 hr. All collected samples were fixed in 70% ethanol and prepared for flow cytometry as described previously (Boronat and Campbell 2007; Reis and Campbell 2007).
Two-hybrid assay:
To monitor protein–protein interactions, the yeast two-hybrid system was used (Fields and Song 1989). The directed assay was performed in Y190 strain (Harper et al. 1993) transformed with appropriate plasmids, and the lacZ genetic reporter was utilized to indicate the interactions. The interactions were assessed using both a filter assay with 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal) (Vojtek et al. 1993) and a quantitative in vitro β-galactosidase assay with O-nitrophenyl β-galactoside (ONPG) as a substrate (Rose et al. 1990). For the latter, the yeast strains were grown for 1 day at 23° in SD medium supplemented with required amino acids and nucleotides. The cultures were diluted 10 times with fresh SD medium and incubated for an additional 36 hr. The β-galactosidase activity was determined as described previously (Rose et al. 1990).
RESULTS
Generation of temperature-sensitive alleles of DPB2:
To test the role of the Dpb2p subunit of Pol ɛ HE in maintaining the fidelity of DNA replication, we isolated new dpb2ts mutants. If the Dpb2p subunit is involved in this process, we would expect to isolate variants of Dpb2p that exhibit a mutator phenotype. As the Dpb2p subunit is essential for yeast growth, the approach was to isolate a set of mutants with a temperature-sensitive phenotype and screen among them for mutators. Plasmids carrying the DPB2 gene were mutagenized by mutagenic PCR or by hydroxylamine treatment as described in materials and methods. Temperature-sensitive alleles were obtained by plasmid shuffling between wild-type and mutagenized DPB2 genes in the dpb2Δ strain (see materials and methods). After loss of the plasmid carrying wild-type DPB2, we screened 15,000 clones for temperature sensitivity (loss of growth at 37°). Thirty independent clones were identified and further assayed for growth at 23°, 30°, and 37°. Two clones that exhibited clear temperature sensitivity, that were strong mutators in our preliminary tests, and that had the smallest number of amino acid changes as determined by DNA sequencing, were chosen for further studies. These strains carry plasmids pMJ25 (dpb2-100), derived by PCR mutagenesis, and pMJ36 (dpb2-103), derived by hydroxylamine treatment. The nucleotide changes in the DPB2 gene, as identified by DNA sequencing, are shown in Table 2 and Figure 2. Two additional mutants, dpb2-101 and dpb2-102, each containing only one of the two amino acid substitutions carried by pMJ25 (dpb2-100), were created by subcloning. The dpb2-101 mutation, L284P, lies in a highly conserved motif within the gene (Figure 2) and confers moderate temperature sensitivity (Table 3). The dpb2-102 mutation, T345A, did not confer temperature sensitivity (nor a mutator phenotype, as shown below) and was therefore not characterized in detail. The temperature-sensitive dpb2-103 allele contains five clustered changes between amino acid residues 342 and 348 in the proline-rich region separating consensus regions III (OB-fold) and IV (calcineurin motifs) of this family of DNA polymerase-associated B subunits (Figure 2) (Makiniemi et al. 1999). We also determined the DNA sequence of the previously identified dpb2-1 allele (Araki et al. 1991a), which had not previously been sequenced, and found four amino acid changes, none of which occurred in any of the mutants in our collection (Figure 2, Table 2).
Figure 2.—

BLAST search for alignment of S. cerevisiae Dpb2p region of 241–689 aa to different species. Identical amino acids are shown against a solid background and similar amino acids (+) are shown against a shaded background. Amino acid substitutions in the mutant forms of the yeast Dpb2p are indicated above the sequence. Mutations identified in the dpb2-1 allele are underlined. Sc, S. cerevisiae; Sp, Schizosaccharomyces pombe; Ce, Caenorhabditis elegans; Xl, Xenopus laevis; Mm, Mus musculus; Hs, Homo sapiens.
TABLE 3.
Mutation rates and temperature sensitivity of the haploid strains carrying the chromosomal dpb2∷kanMX4 disruption and respective dpb2 allele on a centromeric plasmid
| DPB2 allele on centromeric plasmid | Mutation rates (CanR/107)a | Temperature sensitivityb
|
||
|---|---|---|---|---|
| 23° | 30° | 37° | ||
| DPB2 | 5 (±1) | + | + | + |
| dpb2-100 | 27 (±5) | + | ± | − |
| dpb2-101 | 13 (±3) | + | + | ± |
| dpb2-102 | 7 (±3) | + | + | + |
| dpb2-103 | 26 (±10) | + | ± | − |
| dpb2-1 | 12 (±2) | + | + | + |
The strain genotype of the haploid strains is MATα his3Δ1 leu2Δ0 ura3Δ0 lys2Δ0 dpb2∷kanMX4 (BY genetic background). Mutation rates were measured at 23°.
“±” indicates standard deviations from three experiments.
“+,” normal growth; “−,” no growth; “±,” slow growth.
The dpb2 mutants reveal phenotypes typical of DNA replication mutants:
The dpb2-100, dpb2-101, and dpb2-103 mutant alleles, as well as wild-type DPB2, were then integrated into the chromosome of strain ΔI(-2)I-7B-YUNI300, replacing the endogenous DPB2 locus, using pKF117 derivatives and the pKF120 plasmid, respectively. The mutants display varying degrees of temperature sensitivity (Figure 3A). As shown in Figure 3B, the dpb2-100 and dpb2-103 cells incubated at the nonpermissive temperature arrested with dumbbell morphology and with the nucleus between mother and daughter cells, the terminal phenotype of mutants with S-phase defects and of dpb2-1 (Pringle and Hartwell 1981; Araki et al. 1991a). The dpb2-101 mutant, which displays a weak temperature-sensitive phenotype (Figure 3A), exhibits almost wild-type nuclear and cell morphology (Figure 3B). We also analyzed S-phase progression by flow cytometry in dpb2-103 mutant cells, which had the most severe temperature-sensitive growth defect. Cells were arrested in G1 phase with α-factor and then released into a synchronous cell cycle at 37°. As shown in Figure 3C, the mutant cells appeared to enter S phase, but progressed much more slowly than the wild-type cells through S phase at 37° and arrested with a nearly 2C DNA content or greater. This pattern is typical of temperature-sensitive DNA replication elongation mutants and indicates that bulk DNA synthesis can occur slowly but that replication is defective or incomplete. We conclude that the dpb2-103 mutant may have a primary defect in chromosomal DNA replication at the restrictive temperature. The temperature-sensitive dpb2 mutants were examined for viability at 23°. The mutants exhibit decreased viability—between 50 and 80% viability compared to wild-type cells (Figure 4). These observations confirm that the DPB2 gene is important for chromosomal DNA replication.
Figure 3.—

Temperature sensitivity of dpb2 strains. DPB2 (control) and dpb2 alleles were integrated into the DPB2 chromosomal locus of ΔI(-2)I-7B-YUNI300. (A) Viability of the dpb2 mutants grown at 23°, 30°, and 37°. Cells were grown overnight at 23° on SD plates supplemented with required amino acids and nucleotides. Cultures were diluted to identical initial concentrations and 10-fold serial dilutions were spotted in 10-μl portions on SD plates. The ability to grow was subsequently tested at different temperatures, as indicated. (B) Cellular and nuclear morphology of dpb2 strains. Cells were grown at permissive temperature (23°) to log phase and subsequently incubated for 6 hr at 37°. Differential interference microscopy (DIC) images of representative cells of the DPB2 and dpb2 strains and DAPI fluorescence images of cells with DAPI-stained DNA. (C) Cell-cycle analysis of cells carrying DPB2 and dpb2-103 alleles. Yeast cells were grown in YPDA medium at 23° to 0.4 OD. Cultures were synchronized with α-factor (3 hr at 23° and 1 hr at 37°). Samples of cells released from α-factor were collected every 15 min and subjected to flow cytometry analysis. DNA was stained with propidium iodide.
Figure 4.—

Survival of S. cerevisiae strain ΔI(-2)I-7B-YUNI300 carrying integrated dpb2 alleles at 23°. The cultures were grown to stationary phase; yeast cells were collected by centrifugation, washed, and resuspended in water. Samples of appropriate dilutions were plated on SD plates and incubated for 7 days at 23°, and colonies were counted. Survival is calculated as a percentage of the titer of the strain carrying the wild-type allele of DPB2.
Mutator phenotypes of the dpb2 mutants are due to mutations in the DPB2 gene:
To quantify the mutation rates of the dpb2 alleles and to establish that any mutator effects found were due to dpb2 mutations, we assayed the forward mutation rate at the CAN1 locus in strain SC11 carrying HIS3 plasmids encoding DPB2, dpb2-100, dpb2-101, dpb2-102, and dpb2-103, as well as dpb2-1. Relevant plasmids were introduced into strain SC11 (which carries a deletion of the DPB2 chromosomal locus but contains DPB2 on plasmid pMJDPB2) by transformation, and pMJDPB2 was then eliminated from the transformants by growth on 5-FOA. The HIS3 plasmid-containing strains were used to determine the level of spontaneous mutagenesis at 23° by measuring canavanine resistance (CanR) as described in materials and methods. Wild-type cells are sensitive to canavanine, an analog of arginine. Any mutation that inactivates the arginine permease encoded by CAN1 results in CanR. The strain carrying the plasmid-encoded dpb2-102 allele (T345A), which was not temperature sensitive, did not cause any increase in mutability (Table 3). Strains bearing the remaining four mutated DPB2 alleles, however, elevate the rate of mutations at CAN1 two- to approximately fivefold compared to the strain bearing wild-type DPB2. Strains carrying the plasmid-borne mutants display different degrees of temperature sensitivity (Table 3), as do the integrated mutant alleles presented below. The degree of temperature sensitivity correlates well with the frequency of appearance of CanR mutants except the dpb2-1 mutation, which did not confer temperature sensitivity in the BY genetic background (Table 3). Mutagenesis was also determined at 30° for two mutants, dpb2-101 and dpb2-1, which are able to grow at this temperature. However, we did not observe any significant differences in mutation rate compared to results obtained at 23° (data not shown). We confirmed that the mutator phenotypes were Dpb2p dependent by replacing wild-type sequences in DPB2 with restriction fragments carrying the dpb2-100 and dpb2-103 mutations and showing that they conferred the mutator behavior.
Mutator effects of dpb2 mutations integrated into the chromosome:
To confirm that the effect in dpb2 mutants on mutation rates was not an artifact of expression from the plasmid or the genetic background, we tested the mutator effect in the strains containing chromosomal copies of the dpb2-100, dpb2-101, and dpb2-103 alleles. We have been unable to obtain viable integrants of the dpb2-1 allele on the chromosome of the strains used here. In the ΔI(-2)I-7B-YUNI300 strain, mutation frequencies can be measured at four different genetic loci, which allow parallel examination of the specificity of the introduced mutations with respect to base substitution and frameshift mutations (Pavlov et al. 2002). Forward mutation to canavanine resistance reflects a wide range of mutations, including base substitutions, frameshifts, and more complex mutations (Chen and Kolodner 1999). Also, ΔI(-2)I-7B-YUNI300 harbors the his7-2 mutation-reporter allele, consisting of a single base deletion in a run of eight A·T base pairs, which reverts to His+ mainly by addition of 1 bp or loss of 2 bp by insertion or deletion, respectively (Pavlov et al. 2001). The lys2-ΔGG2899-2900 allele reverts via −1 frameshifts at a run of seven A·T base pairs (Pavlov et al. 2002). Finally, ΔI(-2)I-7B-YUNI300 possesses the trp1-289 nonsense allele, which reverts mainly via a broad range of base-pair substitution mutations converting the premature STOP codon to an amino-acid-coding codon (Pavlov et al. 2001).
All three dpb2 mutants tested show an elevated rate of spontaneous mutagenesis for all markers tested (Table 4). The strongest effect, exerted by the dpb2-100 mutation, elevates the mutation rate 7-, 19-, 4-, and 6-fold at the CAN1, his7-2, trp1-289, and lys2-ΔGG2899-2900 markers, respectively. The weakest mutator, dpb2-101, elevates mutation rates ∼4-, 6-, 3-, and 3-fold at the same four markers, respectively. These data indicate that the respective dpb2 mutants are spontaneous mutators for both base substitution and frameshift mutations.
TABLE 4.
Mutation rates in strains with different chromosomal dpb2 alleles and in dpb2 msh6 double mutants
| Mutation ratesb,c
|
||||
|---|---|---|---|---|
| DPB2 allelesa | CanR/107 | His+/108 | Trp+/108 | Lys+/108 |
| DPB2 | 5 (±1) [1] | 5 (± 5) [1] | 5 (±5) [1] | 8 (±6) [1] |
| dpb2-100 | 36 (±12) [7] | 97 (±38) [19] | 18 (±12) [4] | 44 (±3) [6] |
| dpb2-101 | 18 (±4) [4] | 30 (±7) [6] | 13 (±8) [3] | 23 (±9) [3] |
| dpb2-103 | 25 (±6) [5] | 53 (±21) [11] | 24 (±13) [5] | 55 (±22) [7] |
| msh6 DPB2 | 47 (±12) [9] | 13 (±6) [3] | 18 (±4) [4] | 12 (±7) [1.5] |
| msh6 dpb2-100 | 222 (±53) [44] | 175 (±74) [35] | 150 (±60) [30] | 254 (±44) [32] |
| msh6 dpb2-101 | 191 (±22) [38] | 78 (±9) [16] | 145 (±52) [29] | 213 (±41) [27] |
| msh6 dpb2-103 | 235 (±74) [47] | 125 (±6) [25] | 139 (±69) [28] | 237 (±12) [30] |
dpb2 alleles integrated into the DPB2 chromosomal locus of ΔI(-2)I-7B-YUNI300 and ΔI(-2)I-7B-YUNI300 msh6∷hisG, respectively.
“±” indicates standard deviations from three experiments.
Within brackets are folds presenting the increase of mutability (the rate of mutagenesis in the respective dpb2 mutant is divided by the rate of mutagenesis in the strain carrying the wild-type DPB2 gene in the MSH6 background).
Mutator effects of dpb2 mutations in mismatch-repair-defective strains:
The mutation rate data presented above suggest that mutant forms of the Dpb2p subunit influence the fidelity of DNA replication. Since mismatches resulting from replication errors are corrected by the mismatch repair system (Schaaper 1993; for MMR review, see Jiricny 2006), strains deficient in mismatch repair allow a more direct assay of replication errors. In yeast cells, Msh2p-Msh3p and Msh2p-Msh6p heterodimers form the initiation complexes for two partially redundant repair pathways that act to repair replication errors. The Msh2p-Msh3p heterodimer primarily corrects small loop mismatches while the Msh2p-Msh6p heterodimer primarily corrects nucleotide substitutions and small loop mismatches (for review, see Jiricny 2006). The msh6 strain has a partial defect in mismatch repair that does not affect the survival of the cells and exhibits a moderate increase of spontaneous mutagenesis compared to msh2 strains that show a very strong mutator phenotype (Flores-Rozas and Kolodner 1998). In our studies, to avoid possible decrease of viability due to error catastrophy (Schaaper and Radman 1989), we used a strain lacking the Msh6p activity. To assess replication errors in the dpb2 mutants, the endogenous DPB2 gene in the mismatch-repair-defective strain ΔI(-2)I-7B-YUNI300 msh6∷hisG was replaced with the wild-type and mutant dpb2 alleles using pKF120 and derivatives of pKF117, respectively. We then measured mutation rates at the same four loci as above (Table 4). If mutations in the Dpb2p subunit affect fidelity of replication, we expect a significant increase in mutability in the dpb2 msh6 background. In the DPB2 msh6∷hisG control strain, the rate of spontaneous mutagenesis at the CAN1, his7-2, trp1-289, and lys2-ΔGG2899-2900 genes is, as expected, considerably higher than in the mismatch-repair-proficient strain, presumably due to lack of repair of errors generated by the wild-type replicative polymerases (Table 4). It is interesting that the dpb2 single mutants (Table 4) are as strong mutators for some alleles as the msh6∷hisG single mutant. In addition, the mutability of the dpb2 msh6∷hisG double mutants is significantly higher than the mutability of either dpb2 or msh6∷hisG single mutants (Table 4). The dpb2-100 msh6∷hisG double mutant was a 6-fold mutator for CanR and a 2-fold mutator for His+, 8-fold for Trp+, and 6-fold for Lys+ compared to dpb2-100. The dpb2-101 msh6∷hisG double mutant had an 11-fold and 3-fold higher rate of forward CanR mutations and frameshift reversion to His+ and an 11- and 9-fold higher rate of Trp+ and Lys+ mutations, respectively, than dpb2-101 by itself. The dpb2-103 msh6∷hisG had 9- and 2-fold higher rate of forward CanR mutations and frameshift reversion to His+ and a 6- and 4-fold higher rate of Trp+ and Lys+ mutations than dpb2-103 by itself. In addition, the values in brackets in Table 4 show that the dpb2 and msh6∷hisG mutator effects were synergistic for all markers tested. These effects in the msh6∷hisG strain suggest that many of the errors arising in the dpb2 mutator strains are corrected in mismatch-repair-proficient strains, as expected for DNA replication errors.
Mutator effects of dpb2 mutations in Pol ɛ proofreading-defective strains:
In addition to comparing mutagenicity of the dpb2 alleles in wild-type and MMR-defective strains, we also examined mutagenesis under conditions where Pol ɛ proofreading is not contributing to error avoidance. The pol2-4 mutation inactivates the intrinsic 3′ → 5′ exonuclease activity of Pol2p (Morrison and Sugino 1994; Shimizu et al. 2002). If Pol ɛ HE, containing mutant forms of Dpb2p, generates errors, which are subject to 3′ → 5′ proofreading, the dpb2 pol2-4 double mutants should exhibit a greatly elevated mutational rate relative to single dpb2 mutants. We introduced the mutated dpb2 alleles into strain ΔI(-2)I-7B-YUNI300 pol2-4. As expected, the haploid strain harboring the DPB2 and pol2-4 alleles exhibited an elevated level of mutability for three genetic markers tested (Table 5). CanR- and His+-causing mutations were increased by ∼8-fold and 3-fold, respectively, and Trp+ reversion was increased by 2-fold compared to the wild-type strain. The dpb2 single mutants each showed a level of forward CanR mutations comparable to the pol2-4 single mutant (Table 5). In the pol2-4 dpb2 double mutants, the rates of spontaneous mutagenesis at all three reporter genes were significantly greater than observed with either single mutant alone. The dpb2-100 pol2-4 double mutant increases the rate of mutagenesis to CanR, His+, and Trp+ 12-, 34-, and 5-fold, respectively, compared to the single pol2-4 mutant (Table 5). Therefore, the presence of pol2-4 showed significant synergy with dpb2 mutants in the his7-2 and trp1-289 reversion assays or in the CAN1 forward mutation assay. The synergistic effect strengthens the hypothesis that Dpb2p may control fidelity of replication by Pol ɛ and provides an additional argument that Pol ɛ participates in yeast chromosomal replication.
TABLE 5.
Synergistic mutator effect of pol2-4 and dpb2 alleles
| Relevant POL2 and DPB2 allelesa | Mutation ratesb
|
||
|---|---|---|---|
| CanR/107 | His+/108 | Trp+/108 | |
| POL2 DPB2 | 4 (±2) | 7 (±6) | 4 (±1) |
| pol2-4 DPB2 | 31 (±6) | 22 (±17) | 9 (±4) |
| POL2 dpb2-100 | 33 (±11) | 92 (±23) | 14 (±9) |
| pol2-4 dpb2-100 | 374 (±37) | 740 (±144) | 46 (±15) |
| POL2 dpb2-101 | 18 (±3) | 23 (±15) | 17 (±11) |
| pol2-4 dpb2-101 | 111 (±21) | 209 (±70) | 30 (± 14) |
| POL2 dpb2-103 | 31 (±9) | 50 (±28) | 14 (±6) |
| pol2-4 dpb2-103 | 109 (±17) | 329 (±96) | 38 (±10) |
dpb2 alleles integrated into the DPB2 chromosomal locus of ΔI(-2)I-7B-YUNI300 and ΔI(-2)I-7B-YUNI300 pol2-4, respectively.
“±” indicates standard deviations from three experiments.
Interaction of the C terminus of Pol2p with mutant Dpb2 proteins:
By interaction with other subunits of the holoenzyme, Dpb2p may direct assembly of the complex and stabilize the function of Pol ɛ. It was shown previously that Dpb2p binds to the Pol2p (Dua et al. 2000; Tsubota et al. 2006). If interaction of Pol2p with Dpb2p has significance in maintaining the high fidelity of DNA replication, one may expect to find a correlation between mutator phenotype and the ability to interact with Pol2p in certain dpb2 mutants. To test this hypothesis, we used the yeast two-hybrid system. We cloned wild-type and mutated variants of DPB2 into the pKF133 vector encoding the Gal4p DNA-binding domain as described in materials and methods. The resulting plasmids contain the complete DNA coding sequence of the DPB2 or dpb2 alleles, each fused to BDGAL4 at the first ATG codon. It was shown previously that the C-terminal half of Pol2p (aa 1265–2222) interacts strongly with Dpb2p but aa 2163–2222 fails to interact (Dua et al. 1998, 2000). On the basis of this observation we reasoned that the Pol2p C-terminal fragment (aa 2090–2222) would be sufficient for Pol2p-Dpb2p interaction. To test this, we cloned this fragment in the Gal4p activation domain-coding vector (pKF80) and carried out a directed two-hybrid assay at 23°, the same temperature that we used in mutagenesis experiments. Additionally, we performed a two-hybrid assay (filter assay) at 30° (the optimal temperature for yeast growth) and 33° (the highest permissive temperature for the Y190 strain). As expected, the Pol2p(K2090-I2222) interacted strongly with Dpb2p but not with the E. coli dnaE gene product (Figure 5A). Two mutants, dpb2-103 and dpb2-1, showed weaker Pol2p-Dpb2p interaction at higher temperature. Dpb2p-100, which causes the strongest mutator phenotype and is also temperature sensitive for growth, does not interact detectably with Pol2p(K2090-I2222); and Dpb2p-103, also a strong mutator and temperature sensitive for growth, shows a 50% reduction in β-galactosidase activity. Dpb2p-101, the weakest mutator, shows slightly (∼20%), but reproducibly, reduced interaction, similar to its partial temperature sensitivity; and Dpb2p-102, which does not show a mutator phenotype, interacts with Pol2p(K2090-I2222) normally. Control experiments demonstrate that the various BDGal4-Dpb2 fusion proteins are expressed at similar levels in S. cerevisiae cells (Figure 5B), excluding the possibility that the reduced interactions were due to dramatically reduced protein expression levels. Dpb2p-1 appears to be more severely defective than Dpb2p-101 in interaction with the Pol2p C terminus but has about the same level of mutagenesis (Figure 3). The weak interaction of Dpb2p-1 is in keeping with the reported inability to detect Dpb2p-1 in association with Pol2p purified from strain dpb2-1 (Araki et al. 1991a). As shown in Table 3, the dpb2-1 allele present on the plasmid does not show temperature-sensitive colony-forming ability, however. In summary, all of the strong dpb2 mutator alleles show reduced interaction with Pol2p.
Figure 5.—


Interaction of various Dpb2p mutants with Pol2p C terminus by two-hybrid assay. (A) Two-hybrid interactions. The Y190 strain was transformed with indicated plasmids and the ability of particular Dpb2p variants to interact with the C terminus of Pol2p was tested using the lacZ genetic reporter. Strains carrying empty or bacterial polymerase dnaE gene-coding plasmids were used as negative controls, whereas the strain bearing the wild-type GAL4 gene served as a strong positive control. The β-galactosidase expression levels were determined as described in materials and methods. Plate assays were carried out as described by Vojtek et al. (1993), while the in vitro assay was conducted according to Rose et al. (1990). (B) Expression of BDGal4p-Dpb2p fusion variants. The Y190 strain bearing the indicated plasmids for the two-hybrid assay was prepared and Western blotting was performed as described in materials and methods. The indicated Dpb2p variants are BDGal4p fusions and Pol2p(K2090-I2222) is an ADGal4p fusion. (Top) Blots probed with anti-BDGal4p antibodies. The 97-kDa band indicates the BDGal4p-Dpb2p fusions. (Bottom) The same blots probed with anti-Hts1p (histidyl-tRNA synthetase, 60 kDa; Chiu et al. 1992) antibodies as a gel loading control.
DISCUSSION
We have investigated the role of the DPB2 gene product in controlling the fidelity of chromosomal DNA replication in S. cerevisiae. We have isolated new temperature-sensitive dpb2 alleles. Our results show that the dpb2 alleles strongly increase spontaneous mutability (Tables 3–5). The dpb2 alleles carry mutations in different regions of the DPB2 gene, and they result in enhancing both base substitutions and frameshift mutations. Because the dpb2-dependent mutations are subject to correction by the mismatch repair system, we conclude that the dpb2-dependent mutations are replication errors. This conclusion is consistent with our further demonstration that a significant portion of the dpb2-dependent errors appear to be proofread by 3′ → 5′ exonuclease of Pol ɛ. Therefore, Dpb2p is essential not only for cell viability but also for fidelity of DNA replication.
All of the dpb2 mutator alleles have reduced viability compared to wild-type DPB2 (Figures 3 and 4 and Table 3) and show a similar nuclear and cellular morphology at the nonpermissive temperature (Figure 3). Interestingly, all of the proteins encoded by dpb2 mutator alleles interact less strongly with Pol2p than the wild-type Dpb2p. Finally, we have also shown that amino acids 2090–2222 of Pol2p are sufficient for interaction between Pol2p and Dpb2p, although our previous work suggests that additional Pol2p sequences modulate the Pol2p-Dpb2p interaction (Dua et al. 2000).
What is the role of Dpb2p in preventing replication errors? Dpb2p may affect fidelity of replication in several ways. First, by stabilizing the holoenzyme, it may indirectly control the insertion fidelity of Pol ɛ. We have shown by two-hybrid assays that there is a defect in interaction, even at 23°, between Pol2p and Dpb2p in the mutants studied here, including dpb2-1. Proline-rich regions, such as those in which the clustered dpb2 mutations fall (dpb2-103), often mediate transient and nonstoichiometric protein–protein interactions that are often important for function rather than for formation of a stable complex (Williamson 1994). This is interesting, given the flexibility of the Pol2p-Dpb2p structure indicated by the different conformations of the dimer observed in cryo-electron microscopy reconstructions. Such regions also play structural roles, however. The interaction with the OB-fold domain (Dpb2p-100, Dpb2p-101, Dpb2p-1) may be a stronger structural interaction but not be as relevant to fidelity without the second mutation.
Second, it is possible that Dpb2p influences the switch of the primer terminus between the polymerase and exonuclease site of Pol ɛ. However, this is rather unlikely because, on the basis of the results presented in Table 5, dpb2-100, dpb2-101, and dpb2-103 mutations show significant synergy with the pol2-4 proofreading mutation. The synergistic effects between pol2-4 and dpb2 mutator mutations for three genetic markers is more consistent with the model that mutations in DPB2 increase the number of errors that are subject to proofreading than with a model in which these mutations decrease the efficiency of proofreading. Such errors might arise due to increased incorporation of inappropriate nucleotides and/or increased extension of mispairs by the mutated Pol ɛ HE. Interestingly, not all previously described mutator alleles affecting Pol ɛ are synergistic with pol2-4 (Pavlov et al. 2004). For example, the pol2-Y831A mutation, falling in conserved polymerase region III of the Pol2p subunit, causes a mutator phenotype that is actually suppressed in the pol2-Y831A, 4 double-mutation-containing strain, suggesting that the double mutation in this case is copying a smaller fraction of the genome than either single mutant alone (Pavlov et al. 2004). While it is difficult to judge from our data alone, the synergy that we observe suggests that in the dpb2 pol2-4 double mutant Pol ɛ is still participating in replication of a significant fraction of the genome and that the errors are due to Pol ɛ and are not introduced by a compensating DNA polymerase, such as Pol δ.
A third possibility is that Dpb2p may influence processivity of the holoenzyme and may promote and/or stabilize optimum interaction of holoenzyme with DNA. The structure of Pol ɛ obtained by cryo-electron microscopy by Asturias et al. (2006) suggests that the holoenzyme is built of a globular catalytic Pol2p subunit and a more extended region that includes the Dpb2p, Dpb3p, and Dpb4p subunits (Dpb tail). Asturias et al. (2006) suggest that this Dpb tail domain contributes directly to Pol ɛ processivity by mediating engagement of Pol ɛ with the DNA template, either instead of or in addition to the polymerase clamp, PCNA. This model is consistent with the observation that the processivity of the intact, four-subunit Pol ɛ is higher than processivity of a 140-kDa N-terminal fragment of Pol2p, which lacks the Dpb subunits (Hamatake et al. 1990; Maki et al. 1998). The influence of proteins responsible for DNA polymerase processivity on fidelity of DNA synthesis has been tested (mainly in vitro, using primer extension assays) in several laboratories. Processivity factors such as gp45 of T4 (Kroutil et al. 1998) and RB69 (Bebenek et al. 2002, 2005), thioredoxin of T7 (Kunkel et al. 1994), the β-clamp of bacterial polymerases (Bloom et al. 1997; Kobayashi et al. 2002), the 55-kDa accessory subunit of mitochondrial Pol γ (Johnson and Johnson 2001; Longley et al. 2001; Fan et al. 2006), and eukaryotic PCNA (Mozzherin et al. 1996; Chen et al. 2000; Hashimoto et al. 2003) enhance binding of polymerase by decreasing the dissociation of the polymerase from DNA. The impact of processivity factors on fidelity, however, presents a complex picture, with processivity having both positive and negative effects on fidelity in in vitro studies.
Extension of mispairs is generally difficult for polymerases, particularly for more accurate, replicative enzymes (Pham et al. 1998, 1999; Beard and Wilson 2003). A possible source of errors is that, upon committing a misinsertion error, Pol ɛ may be temporarily stalled. While stalling might be expected to provide an increased chance for proofreading, it would also provide increased probability of enzyme dissociation. If mutant Pol ɛ dissociates more often from the mispair, it allows participation of low-fidelity polymerases in DNA replication. The mechanisms by which other DNA polymerases are recruited to the DNA growing point are under investigation (Wagner et al. 2002; Lopez de Saro et al. 2003) and it should be considered whether Dpb2p plays a role in the processivity of Pol ɛ and polymerase switch at the replication fork. The Dpb2p-Dpb3p-Dpb4p tail may prevent dissociation from the mismatched primer terminus, thus decreasing the probability that other, e.g., error-prone, polymerases may extend the mispaired terminus. Mutated Dpb2p may increase dissociation of the holoenzyme by destabilization of the interaction between the catalytic Pol2p domain and the Dpb2p-Dpb3p-Dpb4p tail. Indirect measurements of the binding between Dpb2p and Pol2p by the two-hybrid assay show that the two mutator mutants, Dpb2p-100 and Dpb2p-103, have a significantly impaired Dpb2p-Pol2p interaction while the interaction of the weak mutator Dpb2p-101 is less affected (Figure 5). It is reasonable to assume that the mutator phenotype of dpb2-100, dpb2-101, and dpb2-103 mutations directly reflects the decreased communication between the Dpb2p and Pol2p subunits. At the same time, reduced interaction may not necessarily lead to a high mutagenesis rate, as illustrated by the behavior of the dpb2-1 allele, so the alleles that we describe must affect a very specific type of interaction. The dpb2-1 allele may be so defective in interaction that another, error-free polymerase, such as Pol δ, substitutes for Pol ɛ, as is thought to occur in the pol2-16 mutant (Dua et al. 1999; Kesti et al. 1999). The Dpb2p subunit might modulate the activity of the Pol2p subunit and/or act as a protein that ensures proper communication between other (Dpb3p and Dpb4p) subunits of Pol ɛ HE. Additionally, we cannot exclude that Dpb2p facilitates interaction of the Pol ɛ complex with other DNA-replication-associated proteins and/or modulates the interaction of the holoenzyme with DNA.
Summarizing, we hypothesize that the Dpb2p subunit influences the fidelity of replication in S. cerevisiae cells by one or several of the following mechanisms: (i) stabilizing the structure of Pol ɛ HE; (ii) influencing the activity of other subunits like Pol2p; (iii) controlling the processivity of Pol ɛ HE; (iv) controlling/preventing the involvement of error-prone polymerases in replication; and (v) influencing interactions with other proteins participating in DNA replication.
The results presented in this study may have important implications for our understanding of the source of genetic instability in all eukaryotes. Interestingly, the human homolog of Dpb2p (POLE2) has 44% overall amino acid homology to the yeast Dpb2 protein (Li et al. 1997). It was previously shown that MMR defects destabilizing DNA occur in several tumor cell lines, particularly those from colon cancers (Kolodner 1996; Kolodner and Marsischky 1999; Jacob and Praz 2002). Several cancer susceptibility syndromes that are due to inherited mutations in genes whose products function in response to DNA damage and DNA recombination/repair have been described. Such a genetic defect could result in higher frequencies of spontaneous mutagenesis and/or DNA-damage-induced chromosome aberrations (Khanna and Jackson 2001). Our demonstration that dpb2 mutants exhibit a mutator phenotype comparable to that of mismatch-repair-defective strains (msh6) suggests that defects in a noncatalytic DNA polymerase subunit may increase genomic instability and the risk of diseases and carcinogenesis.
Acknowledgments
We thank Katarzyna Bebenek of the National Institute of Environmental Health Sciences (Research Triangle Park) and Zygmunt Ciesla of the Institute of Biochemistry and Biophysics Polish Academy of Sciences (Warsaw) for their critical reading of the manuscript; Anna Gajda for her excellent technical assistance; Youri Pavlov of the Eppley Institute for Research in Cancer, University of Nebraska Medical Center (Omaha); and Hiroyuki Araki, Division of Microbial Genetics, National Institute of Genetics, Research Organization of Information and Systems (Shizuoka, Japan) for providing S. cerevisiae strains. We thank also T. Mason (Department of Biochemistry, University of Massachusetts, Amherst, MA) for anti-Hts1p antibody. This work was supported by grant 2P04A05126 from the Polish Ministry of Science and Higher Education to M.J., K.F., P.J., and I.J.F; and by U. S. Public Health Service grants TW006463 [Fogarty International Collaboration Award (FIRCA)] to I.J.F., P.J., and J.L.C. and GM25508 to J.L.C.
References
- Adams, A., D. E. Gottschling, C. A. Kaiser and T. Stearns, 1997. Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Araki, H., R. K. Hamatake, L. H. Johnston and A. Sugino, 1991. a DPB2, the gene encoding DNA polymerase II subunit B, is required for chromosomal replication in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 88 4601–4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki, H., R. K. Hamatake, A. Morrison, A. L. Johnson, L. H. Johnston et al., 1991. b Cloning DPB3, the gene encoding the third subunit of DNA polymerase II of Saccharomyces cerevisiae. Nucleic Acids Res. 19 4867–4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki, H. P., P. A. Ropp, A. L. Johnson, L. H. Johnson, A. Morrison et al., 1992. DNA polymerase II, the probable homolog of mammalian DNA polymerase epsilon, replicates chromosomal DNA in the yeast Saccharomyces cerevisiae. EMBO J. 11 733–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asturias, F. J., I. K. Cheung, N. Sabouri, O. Chilkova, D. Wepplo et al., 2006. Structure of Saccharomyces cerevisiae DNA polymerase epsilon by cryo-electron microscopy. Nat. Struct. Mol. Biol. 13 35–43. [DOI] [PubMed] [Google Scholar]
- Beard, W. A., and S. H. Wilson, 2003. Structural insights into the origins of DNA polymerase fidelity. Structure 11 489–496. [DOI] [PubMed] [Google Scholar]
- Bebenek, A., G. T. Carver, H. K. Dressman, F. A. Kadyrov, J. K. Haseman et al., 2002. Dissecting the fidelity of bacteriophage RB69 DNA polymerase: site-specific modulation of fidelity by polymerase accessory proteins. Genetics 162 1003–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebenek, A., G. T. Carver, F. A. Kadyrov, G. E. Kissling and J. W. Drake, 2005. Processivity clamp gp45 and ssDNA-binding-protein gp32 modulate the fidelity of bacteriophage RB69 DNA polymerase in a sequence-specific manner, sometimes enhancing and sometimes compromising accuracy. Genetics 169 1815–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielas, J. H., K. R. Loeb, P. B. Rubin, L. D. True and L. A. Loeb, 2006. Human cancers express a mutator phenotype. Proc. Natl. Acad. Sci. USA 103 18238–18242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom, L. B., X. Chen, D. K. Fygenson, J. Turner, M. O'Donnell et al., 1997. Fidelity of Escherichia coli DNA polymerase III holoenzyme. The effects of beta, gamma complex processivity proteins and epsilon proofreading exonuclease on nucleotide misincorporation efficiencies. J. Biol. Chem. 272 27919–27930. [DOI] [PubMed] [Google Scholar]
- Boeke, J. D., F. Lacroute and G. R. Fink, 1984. A positive selection for mutants lacking orotidine-5-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol. Gen. Genet. 197 345–346. [DOI] [PubMed] [Google Scholar]
- Boronat, S., and J. L. Campbell, 2007. Mitotic Cdc6 stabilizes APC substrates by a partially Cdc28-independent mechanism and this stabilization is suppressed by deletion of Cdc55. Mol. Cell. Biol. 27 1158–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budd, M. E., and J. L. Campbell, 1993. DNA polymerases delta and epsilon are required for chromosomal replication in Saccharomyces cerevisiae. Mol. Cell. Biol. 13 496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C., and R. D. Kolodner, 1999. Gross chromosomal rearrangement in Saccharomyces cerevisiae replication and recombination deficient mutants. Nat. Genet. 23 81–85. [DOI] [PubMed] [Google Scholar]
- Chen, X, S. Zuo, Z. Kelman, M. O'Donnell, J. Hurwitz et al., 2000. Fidelity of eucaryotic DNA polymerase delta holoenzyme from Schizosaccharomyces pombe. J. Biol. Chem. 275 17677–17682. [DOI] [PubMed] [Google Scholar]
- Chilkova, O., B. H. Jonsson and E. Johansson, 2003. The quaternary structure of DNA polymerase epsilon from Saccharomyces cerevisiae. J. Biol. Chem. 278 14082–14086. [DOI] [PubMed] [Google Scholar]
- Chiu, M. J., T. L. Mason and G. H. Fink, 1992. HTS1 encodes both the cytoplasmic and mitochondrial histidyl-tRNA synthetase of Saccharomyces cerevisiae: mutations alter the specificity of compartmentation. Genetics 132 987–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta, A, J. L. Schmeits, N. S. Amin, P. J. Lau, K. Myung et al., 2000. Checkpoint-dependent activation of mutagenic repair in Saccharomyces cerevisiae pol3-01 mutants. Mol. Cell 6 593–603. [DOI] [PubMed] [Google Scholar]
- Drake, J. W., 1991. A constant rate of spontaneous mutation in DNA-based microbes. Proc. Natl. Acad. Sci. USA 88 7160–7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dua, R., D. L. Levy and J. L. Campbell, 1998. Role of the putative zinc finger domain of Saccharomyces cerevisiae DNA polymerase ɛ in DNA replication and S/M checkpoint pathway. J. Biol. Chem. 273 30046–30055. [DOI] [PubMed] [Google Scholar]
- Dua, R., D. L. Levy and J. L. Campbell, 1999. Analysis of the essential functions of the C-terminal protein/protein interaction domain of Saccharomyces cerevisiae pol ɛ and its unexpected ability to support growth in the absence of DNA polymerase domain. J. Biol. Chem. 274 22283–22288. [DOI] [PubMed] [Google Scholar]
- Dua, R., S. Edwards, D. L. Levy and J. L. Campbell, 2000. Subunit interactions within the Saccharomyces cerevisiae DNA polymerase ɛ (pol ɛ) complex. J. Biol. Chem. 275 28816–28825. [DOI] [PubMed] [Google Scholar]
- Fan, L., S. Kim, C. L. Farr, K. T. Schaefer, K. M. Randolph et al., 2006. A novel processive mechanism for DNA synthesis revealed by structure, modeling and mutagenesis of the accessory subunit of human mitochondrial DNA polymerase. J. Mol. Biol. 358 1229–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, W., L. Rodriguez-Menocal, G. Tolun and G. D'Urso, 2003. Schizosaccharomyces pombe Dpb2 binds to origin DNA early in S phase and is required for chromosomal DNA replication. Mol. Biol. Cell 14 3427–3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields, S., and O. Song, 1989. A novel genetic system to detect protein-protein interactions. Nature 340 245–246. [DOI] [PubMed] [Google Scholar]
- Flores-Rozas, H., and R. D. Kolodner, 1998. The Saccharomyces cerevisiae MLH3 gene functions in MSH3-dependent suppression of frameshift mutations. Proc. Natl. Acad. Sci. USA 95 12404–12409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz, R. D., and A. Sugino, 1988. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base-pair restriction sites. Gene 74 527–534. [DOI] [PubMed] [Google Scholar]
- Gietz, R. D., and R. A. Woods, 1998. Transformation of yeast by the lithium acetate/single-stranded carrier DNA/PEG method. Methods Microbiol. 26 53–66. [Google Scholar]
- Goldstein, A. L., X. Pan and J. H. McCusker, 1999. Heterologous URA3MX cassettes for gene replacement in Saccharomyces cerevisiae. Yeast 15 507–511. [DOI] [PubMed] [Google Scholar]
- Hamatake, R. K., H. Hasegawa, A. B. Clark, K. Bebenek, T. A. Kunkel et al., 1990. Purification and characterization of DNA polymerase II from the yeast Saccharomyces cerevisiae. J. Biol. Chem. 265 4072–4083. [PubMed] [Google Scholar]
- Harper, J. W., G. R. Adami, N. Wei, K. Keyomarsi and S. J. Elledge, 1993. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75 805–813. [DOI] [PubMed] [Google Scholar]
- Hashimoto, K, K. Shimizu, N. Nakashima and A. Sugino, 2003. Fidelity of DNA polymerase delta holoenzyme from Saccharomyces cerevisiae: the sliding clamp proliferating cell nuclear antigen decreases its fidelity. Biochemistry 42 14207–14213. [DOI] [PubMed] [Google Scholar]
- Huang, M. E., A. G. Rio, M. D. Galibert and F. Galibert, 2002. Pol32, a subunit of Saccharomyces cerevisiae DNA polymerase δ, suppresses genomic deletions and is involved in the mutagenic bypass pathway. Genetics 160 1409–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob, S., and F. Praz, 2002. DNA mismatch repair defects: role in colorectal carcinogenesis. Biochimie 84 27–47. [DOI] [PubMed] [Google Scholar]
- Jiricny, J., 2006. The multifaceted mismatch-repair system. Nat. Rev. 7 335–345. [DOI] [PubMed] [Google Scholar]
- Johnson, A., and K. A. Johnson, 2001. Exonuclease proofreading by human mitochondrial DNA polymerase. J. Biol. Chem. 276 38097–38107. [DOI] [PubMed] [Google Scholar]
- Jokela, M., M. Makiniemi, S. Lehtonen, C. Szpirer, U. Hellman et al., 1998. The small subunits of human and mouse DNA polymerase epsilon are homologous to the second largest subunit of the yeast Saccharomyces cerevisiae DNA polymerase epsilon. Nucleic Acids Res. 26 730–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonczyk, P., A. Nowicka, I. J. Fijalkowska, R. M. Schaaper and Z. Ciesla, 1998. In vivo protein interactions within the Escherichia coli DNA polymerase III core. J. Bacteriol. 180 1563–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesti, T., H. Frantti and J. E. Syvaoja, 1993. Molecular cloning of the cDNA for the catalytic subunit of human DNA polymerase epsilon. J. Biol. Chem. 268 10238–10245. [PubMed] [Google Scholar]
- Kesti, T., K. Flick, S. Keränen, J. E. Syväoja and C. Wittenberg, 1999. DNA polymerase epsilon catalytic domains are dispensable for DNA replication, DNA repair, and cell viability. Mol. Cell 3 679–685. [DOI] [PubMed] [Google Scholar]
- Kesti, T., W. H. McDonald, J. R. Yates, III and C. Wittenberg, 2004. Cell cycle-dependent phosphorylation of DNA polymerase epsilon subunit, Dpb2, by the cdc28 cyclin-dependent protein kinase. J. Biol. Chem. 279 14245–14255. [DOI] [PubMed] [Google Scholar]
- Khanna, K. K., and S. P. Jackson, 2001. DNA double-strand breaks: signaling, repair and the cancer connection. Nat. Genet. 27 247–254. [DOI] [PubMed] [Google Scholar]
- Kobayashi, S., M. R. Valentine, P. Pham, M. O'Donnell and M. F. Goodman, 2002. Fidelity of Escherichia coli DNA polymerase IV. Preferential generation of small deletion mutations by dNTP-stabilized misalignment. J. Biol. Chem. 277 34198–34207. [DOI] [PubMed] [Google Scholar]
- Kolodner, R., 1996. Biochemistry and genetics of eukaryotic mismatch repair. Genes Dev. 10 1433–1442. [DOI] [PubMed] [Google Scholar]
- Kolodner, R. D., and G. T. Marsischky, 1999. Eukaryotic DNA mismatch repair. Curr. Opin. Genet. Dev. 9 89–96. [DOI] [PubMed] [Google Scholar]
- Kroutil, L. C., M. W. Frey, B. F. Kaboord, T. A. Kunkel and S. J. Benkovic, 1998. Effect of accessory proteins on T4 DNA polymerase replication fidelity. J. Mol. Biol. 278 135–146. [DOI] [PubMed] [Google Scholar]
- Kunkel, T. A., S. S. Patel and K. A. Johnson, 1994. Error-prone replication of repeated DNA sequences by T7 DNA polymerase in the absence of its processivity subunit. Proc. Natl. Acad. Sci. USA 91 6830–6834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y., H. Asahara, V. S. Patel, S. Zhou and S. Linn, 1997. Purification, cDNA cloning, and gene mapping of the small subunit of human DNA polymerase epsilon. J. Biol. Chem. 272 32337–32344. [DOI] [PubMed] [Google Scholar]
- Li, Y., Z. F. Pursell and S. Linn, 2000. Identification and cloning of two histone fold motif-containing subunits of HeLa DNA polymerase epsilon. J. Biol. Chem. 275 31554. [PubMed] [Google Scholar]
- Loeb, L. A., 2001. A mutator phenotype in cancer. Cancer Res. 61 3230–3239. [PubMed] [Google Scholar]
- Loeb, L. A., K. Loeb and J. P. Anderson, 2003. Multiple mutations and cancer. Proc. Natl. Acad. Sci. USA 100 776–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longley, M. J., D. Nguyen, T. A. Kunkel and W. C. Copeland, 2001. The fidelity of human DNA polymerase gamma with and without exonucleolytic proofreading and the p55 accessory subunit. J. Biol. Chem. 276 38555–38562. [DOI] [PubMed] [Google Scholar]
- Lopez de Saro, F. J., R. E. Georgescu and M. O'Donnell, 2003. A peptide switch regulates DNA polymerase processivity. Proc. Natl. Acad. Sci. USA 100 14689–14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki, S., K. Hashimoto, T. Ohara and A. Sugino, 1998. DNA polymerase II (epsilon) of Saccharomyces cerevisiae dissociates from the DNA template by sensing single-stranded DNA. J. Biol. Chem. 273 21332–21341. [DOI] [PubMed] [Google Scholar]
- Makiniemi, M., H. Pospjech, S. Kilpelainen and M. Jokela, 1999. A novel family of DNA-polymerase-associated B subunits. Trends Biochem. Sci. 24 14–16. [DOI] [PubMed] [Google Scholar]
- Morrison, A., and A. Sugino, 1994. The 3′-5′ exonuclease of both DNA polymerases δ and ɛ participate in correcting errors of DNA replication in Saccharomyces cerevisiae. Mol. Gen. Genet. 242 289–296. [DOI] [PubMed] [Google Scholar]
- Morrison, A., H. Araki, A. B. Clark, R. K. Hamatake and A. Sugino, 1990. A third essential DNA polymerase in S. cerevisiae. Cell 62 1143–1151. [DOI] [PubMed] [Google Scholar]
- Mortimer, R. K., and J. R. Johnston, 1986. Genealogy of principal strains of the yeast genetic stock center. Genetics 113 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozzherin, D. J., M. McConnell, M. V. Jasko, A. A. Krayevsky, C. K. Tan et al., 1996. Proliferating cell nuclear antigen promotes misincorporation catalyzed by calf thymus DNA polymerase delta. J. Biol. Chem. 271 31711–31717. [DOI] [PubMed] [Google Scholar]
- Navas, T. A., Z. Zhou and and S. J. Elledge, 1995. DNA polymerase epsilon links the DNA replication machinery to the S phase checkpoint. Cell 13 29–39. [DOI] [PubMed] [Google Scholar]
- Navas, T. A., Y. Sanchez and S. J. Elledge, 1996. RAD9 and DNA polymerase epsilon form parallel sensory branches for transducing the DNA damage checkpoint signal in Saccharomyces cerevisiae. Genes Dev. 10 2632–2643. [DOI] [PubMed] [Google Scholar]
- Ohya, T. S., S. Maki, Y. Kawasaki and A. Sugino, 2000. Structure and function of fourth subunit (Dpb4p) of DNA polymerase ɛ in Saccharomyces cerevisiae. Nucleic Acids Res. 28 3846–3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov, Y. I., D. Nguyen and T. A. Kunkel, 2001. Mutator effects of overproducing DNA polymerase η (Rad30) and its catalytically inactive variant in yeast. Mutat. Res. 8 129–139. [DOI] [PubMed] [Google Scholar]
- Pavlov, Y. I., C. S. Newlon and T. A. Kunkel, 2002. Yeast origins establish a strand bias for replicational mutagenesis. Mol. Cell 10 207–213. [DOI] [PubMed] [Google Scholar]
- Pavlov, Y. I., S. Maki, H. Maki and T. A. Kunkel, 2004. Evidence for interplay among yeast replicative DNA polymerases alpha, delta and epsilon from studies of exonuclease and polymerase active site mutations. BMC Biol. 2 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham, P. T., M. W. Olson, C. S. McHenry and R. M. Schaaper, 1998. The base substitution and frameshift fidelity of Escherichia coli DNA polymerase III holoenzyme in vitro. J. Biol. Chem. 273 23575–23584. [DOI] [PubMed] [Google Scholar]
- Pham, P. T., M. W. Olson, C. S. McHenry and R. M. Schaaper, 1999. Mismatch extension by Escherichia coli DNA polymerase III holoenzyme. J. Biol. Chem. 274 3705–3710. [DOI] [PubMed] [Google Scholar]
- Pham, P. T., W. Zhao and R. M. Schaaper, 2006. Mutator mutants of Escherichia coli carrying a defect in the DNA polymerase III τ subunit. Mol. Microbiol. 59 1149–1161. [DOI] [PubMed] [Google Scholar]
- Pringle, J. R., and L. H. Hartwell, 1981. The Saccharomyces cerevisiae cell cycle, pp. 97–144 in The Molecular Biology of the Yeast Saccharomyces, edited by J. N. Strathern, E. W. Jones and J. R. Broach. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Pursell, Z. F., I. Isoz, E.-B. Lundstrom, E. Johansson and T. A. Kunkel, 2007. Yeast DNA polymerase e participates in leading-strand DNA replication. Science 317 127–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis, C. C., and J. L. Campbell, 2007. Contribution of Trf4/5 and the nuclear exosome to genome stability through regulation of histone mRNA levels in Saccharomyces cerevisiae. Genetics 175 993–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose, M., F. Winston and P. Hieter, 1990. Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Sambrook, J., E. F. Fritsch and T. Maniatis, 1989. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Schaaper, R. M., 1993. Base selection, proofreading, and mismatch repair during DNA replication in Escherichia coli. J. Biol. Chem. 268 23762–23765. [PubMed] [Google Scholar]
- Schaaper, R. M., and M. Radman, 1989. The extreme mutator effect of Escherichia coli mutD5 results from saturation of mismatch repair by excessive DNA replication errors. EMBO J. 8 3511–3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu, K., K. Hashimoto, J. M. Kirshner, W. Nakai, H. Nashikawa et al., 2002. Fidelity of DNA polymerase epsilon holoenzyme from budding yeast Saccharomyces cerevisiae. J. Biol. Chem. 277 37422–37429. [DOI] [PubMed] [Google Scholar]
- Sikorski, R. S., and P. Hieter, 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiga, M. G., and G. D'Urso, 2004. Identification and cloning of two putative subunits of DNA polymerase epsilon in fission yeast. Nucleic Acids Res. 32 4945–4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugino, A., 1995. Yeast DNA polymerases and their role at the replication fork. Trends Biochem. Sci. 20 319–323. [DOI] [PubMed] [Google Scholar]
- Tsubota, T., R. Tajima, K. Ode, H. Kubota, N. Fukuhara et al., 2006. Double-stranded DNA binding, an unusual property of DNA polymerase epsilon, promotes epigenetic silencing in Saccharomyces cerevisiae. J. Biol. Chem. 281 32898–32908. [DOI] [PubMed] [Google Scholar]
- Vojtek, A. B., M. Hollenberg and J. A. Cooper, 1993. Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell 74 205–214. [DOI] [PubMed] [Google Scholar]
- Wagner, J., H. Etienne, R. Janel-Bintz and R. P. Fuchs, 2002. Genetics of mutagenesis in E. coli: various combinations of translesion polymerases (Pol II, IV and V) deal with lesion/sequence context diversity. DNA Rep. 1 159–167. [DOI] [PubMed] [Google Scholar]
- Williamson, M. P., 1994. The structure and function of proline-rich regions in proteins. Biochem. J. 297 249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]