

Abstract
Neisseria meningitidis binds factor H (fH), a key regulator of the alternative complement pathway. A ~29 kD fH-binding protein expressed in the meningococcal outer membrane was identified by mass spectrometry as GNA1870, a lipoprotein currently under evaluation as a broad-spectrum meningococcal vaccine candidate. GNA1870 was confirmed as the fH ligand on intact bacteria by 1) abrogation of fH binding upon deleting GNA1870, and 2) blocking fH binding by anti-GNA1870 mAbs. fH bound to whole bacteria and purified rGNA1870 representing each of the three variant GNA1870 families. We showed that the amount of fH binding correlated with the level of bacterial GNA1870 expression. High levels of variant 1 GNA1870 expression (either by allelic replacement of gna1870 or by plasmid-driven high-level expression) in strains that otherwise were low-level GNA1870 expressers (and bound low amounts of fH by flow cytometry) restored high levels of fH binding. Diminished fH binding to the GNA1870 deletion mutants was accompanied by enhanced C3 binding and increased killing of the mutants. Conversely, high levels of GNA1870 expression and fH binding enhanced serum resistance. Our findings support the hypothesis that inhibiting the binding of a complement down-regulator protein to the neisserial surface by specific Ab may enhance intrinsic bactericidal activity of the Ab, resulting in two distinct mechanisms of Ab-mediated vaccine efficacy. These data provide further support for inclusion of this molecule in a meningococcal vaccine. To reflect the critical function of this molecule, we suggest calling it fH-binding protein.
Neisseria meningitidis is an important cause of meningitis and sepsis worldwide. Complement forms a critical arm of the innate immune defenses against meningococcal infections, evidenced by the epidemiological observation that persons deficient in components of the terminal complement pathway (C5–C9) or the alternative pathway (such as properdin and factor D) are at an increased risk for neisserial infections (1–3). The complement cascade can mediate direct bacterial killing by forming C5b–C9 complexes in the bacterial membrane, or can facilitate opsonophagocytic clearance of bacteria via complement receptors on phagocytic cells.
Effective capsular polysaccharide-based vaccines against serogroups A, C, W-135, and Y meningococcal strains have been developed (4), but an effective vaccine against serogroup B strains remains elusive. Serogroup B capsular polysaccharide is not effective as a vaccine in part because it is structurally similar to host neural-cell adhesion molecule and is therefore poorly immunogenic (5). Reverse vaccinology has identified a lipoprotein called GNA1870, also known as lipoprotein 2086 (6), that is surface expressed on all meningococcal strains tested and elicits bactericidal Abs that are protective in the infant rat model of meningococcal bacteremia (6–9). Based on amino acid sequence, two classification systems for GNA1870 have been developed. One system divides GNA1870 into three variant families called variant 1, variant 2, and variant 3 (9). The other classification system is based on sequence analysis of GNA1870 (also called 2086) predominantly derived from serogroup B strains and divides this protein into subfamilies A (divided further into 10 clusters, A1–A10) and B (divided into clusters B1–B9) (6). Despite detailed studies on the immunogenicity and vaccine potential of GNA1870, a function for this molecule has not yet been determined.
Factor H (fH)3 is a key fluid-phase regulator of the alternative complement pathway. It acts as a cofactor for the factor I-mediated cleavage of C3b to its hemolytically inactive fragment iC3b (10–12) and also promotes the decay of the alternative pathway C3, convertase C3bBb (13, 14). In the present study, we have identified GNA1870 to be a ligand for fH, a key regulator of the alternative complement pathway.
Materials and Methods
Bacterial strains
Strains of N. meningitidis used in this study and their relevant characteristics are listed in Table I. Bacteria were grown on chocolate agar plates supplemented with Isovitalex equivalent at 37°C in an atmosphere enriched with 5% CO2.
Table I.
Summary of Neisseria meningitidis strains
| Strain | Relevant Characteristics/Reference |
|---|---|
| H44/76 | B:15:P1.7,16: ST-32 (Norway, 1976); variant 1 GNA1870; invasive isolate (55, 56) |
| MC58 | B:15:P1.7,16: ST-74 (U.K., 1985); variant 1 GNA1870; invasive isolate (57, 58) |
| A2594 | A:4:P1.20,9a:ST-5 (Germany, 1991); GNA1870 not sequenced; invasive isolate (this study) |
| C2120 | C:NT:P1.5,2a:ST-11 (Germany, 1997); variant 3 GNA1870; invasive isolate (59) |
| W171b | W135:NT:P1.5-9,10a:ST-11 (U.S.); variant 2/3 hybrid GNA1870; site of isolation unknown (60) |
| Y2220 | Y:21:P1.19,15-1a (ST-172); variant 2/3 hybrid GNA1870; carrier isolate (16) |
| RM1090 | C:2a:P1.5,2:ST-unknown (U.S., 1985); variant 2 GNA1870; invasive isolate (8) |
| M1239 | B:14:P1.23,14:ST-437 (U.S., 1994); variant 3 GNA1870; invasive isolate (61) |
Denotes that PorA VR typing (62) was used to define the serosubtype.
W171 also is known as ATCC 35559, or M-603.
Construction of capsule-deficient mutants and mutants unable to sialylate lipooligosaccharide (LOS)
Mutant derivatives of strains representing serogroups B, C, W-135, and Y; H44/76, C2120, W171, and Y2220, respectively, lacking the ability to express capsular polysaccharide were constructed by inactivation of the polysialyltransferase gene (siaD), as described previously (15, 16). The serogroup A-representative strain A2594 was rendered unencapsulated by insertional inactivation of mynB, the gene that encodes the capsular polymerase of serogroup A meningococcal capsule (17). Using primers NT2 (5′-TACTACCATTACCCTTTTCTCA-3′) and NT4 (5′-ATACTTAA TAACAGAAAATGGCG-3′), a 1617-bp PCR product containing mynB was amplified and cloned into the vector pCR 2.1-TOPO (Invitrogen Life Technologies) to yield pNT3. Excision of a 267-bp fragment (base pairs 476–729) was accomplished by digestion of pNT3 with HincII and a blunt-ended chloramphenicol resistance cassette gene (cat) derived from pT-Nmax5 (18) was ligated into the HincII restriction sites, resulting in plasmid that was named pNT5. Strain A2594 was transformed with pNT5, and chloramphenicol-resistant clones were screened both by PCR and ELISA, the latter using anti-serogroup A capsular mAb932 (provided by D. Bitter-Suerbaumm, Medical School Hannover, Germany) to confirm the absence of capsule (data not shown). The ability to sialylate LOS in serogroups B, C, W-135, and Y strains was abrogated by insertional inactivation of the LOS sialyltransferase gene (lst) using a kanamycin-resistance marker as described previously (19). Mutants of strain H44/76 that lacked either porin A (PorA) or PorB3 were provided by P. van der Ley (Netherlands Vaccine Institute, Bilthoven, The Netherlands). The PorA and PorB3 mutants were selected using the Ab-dependent bactericidal activity of complement with mAbs Mn15A14H6 (against PorB3) and Mn5C11G (against PorA), respectively (20). siaD and lst mutations of these Por mutants were generated as described above.
Allelic replacement of Y2220 PorB2 and gna1870 with corresponding genes from H44/76
Using primers listed in Table II, overlap-extension PCR was used to generate a hybrid DNA fragment that contained (in 5′- to -3′ orientation) the H44/76 porB3 (1031 bp), the erythromycin-resistance cassette (ErmC; 870 bp) and 1095 bp of Y2220 DNA downstream of porB2. This hybrid amplicon was cloned into the TA cloning vector, pCR2.1-TOPO 2.1 (Invitrogen Life Technologies), to yield pH44/76PorB3-Erm. The sequence of the cloned DNA was confirmed, and pH44/76PorB3-Erm was used to transform strain Y2220 siaD lst. Erythromycin-resistant colonies from the Y2220 siaD lst transformation did not yield clones whose complete PorB2 was completely replaced. Consequently, we transformed strain H44/76 with pH44/76PorB3-Erm, and used chromosomal DNA from an erythromycin-resistant clone to then transform Y2220 siaD lst. Mutants (Y2220 siaD lst H44/76PorB3+) were screened by PCR using primers that were specific for H44/76 PorB3 loops 1 and 7. Selected mutants were sequenced to ensure that H44/76 PorB3 had entirely replaced Y2220 PorB2.
Table II.
Primers used for allelic replacement of Por and GNA1870
| Primer | Sequencea |
|---|---|
| H44/76 PorB3-F | 5′-TCTTAACCAAAAAAGGAATACAGC-3′ |
| H44/76 PorB3-R | 5′-tcctattttttgTCTTAGCAGATTAGAACTTGTGGCG-3′ |
| Erm-F | 5′-ctaatctgctaagaCAAAAAATAGGAACACGAAAAACAAG-3′ |
| Erm-R | 5′-ttttgttcataccaCTCATCTTGTTCATATTTATCAGAG-3′ |
| Y porB2 down-F | 5′-gaacaagatgagTGGTATGAACAAAAAGCCTGTCGC-3′ |
| Y porB2 down-R | 5′-GGGCAAGGAGGAAAAAGCGGTC-3′ |
| H44/76 1870-F | 5′-CCAAGGGCGAACTGAAC CAAATC-3′ |
| H44/76-1870-R | 5′-gactttaggtgGTGTTCGGACGGCATTTTCACA-3′ |
| Tet-F | 5′-gccgtccgaacacCACCTAAAGTCAGCCCCATACGA-3′ |
| Tet-R | 5′-tcagatggcattaTGGTGAATCCGTTAGCGAGGTG-3′ |
| 1870-down-F | 5′-aacggattcaccaTAATGCCATCTGAACCAACGAGA-3′ |
| 1870-down-R | 5′-GCATAAAACACTTCGCCCATACG-3′ |
Sequence overlapping with adjacent amplicon indicated in bold lower case.
Similarly, we constructed a 2800-bp hybrid DNA fragment that comprised (in 5′- to -3′ orientation) H44/76 gna1870 (1040 bp; this amplicon contained DNA upstream (5′) of gna1870 in addition to the gna1870 ORF), a 1347-bp fragment containing the tetracycline-resistance cassette (Tet) derived from pACYC184 (New England Biolabs), and 413-bp DNA downstream (3′) of gna1870. The hybrid amplicon was cloned into the TA cloning vector, pCR2.1-TOPO, to yield pH44/76-GNA1870-Tet, which was then used to transform Y2220 siaD. Tet-resistant clones were screened for expression of H44/76 GNA1870 by flow cytometry using mAb JAR1 (recognizes H44/76 GNA1870, but not Y2220 GNA1870). We sequenced gna1870 of one such JAR1-binding mutant, called Y2220 siaD H44/76GNA1870+, to confirm the presence of the entire H44/76 gna1870.
Insertional inactivation of gna1870
Inactivation of gna1870 in strains H44/76, RM1090, and M1239 using pBSUDGNA1870ERM (9) to yield H44/76 ΔGNA1870, RM1090 ΔGNA1870, and M1239 ΔGNA1870, respectively, was constructed as previously described (8). PCR was used to confirm interruption of gna1870 in all mutants.
Overexpression of variant 1 GNA1870 by N. meningitidis RM1090
Strain RM1090 naturally expresses low levels of variant 2 GNA1870 (8). To increase expression we used a shuttle vector, pFP12 (21) where the GFP gene was replaced with the variant 1 gna1870 derived from N. meningitidis MC58 (identical gna1870 sequence to that of H44/76; Ref. 8). RM1090 Δ1870 was transformed with pFP12-GNA1870 to yield RM1090 v.1 1870+, which expressed levels of variant 1 GNA1870 equivalent to that of MC58, also a high-level expresser of GNA1870 (8).
DNA sequencing and phylogenetic analysis of gna1870
The nucleotide sequences of gna1870 from N. meningitidis strains C2120, W171, and Y2220 were determined directly from purified PCR templates (Qiagen) amplified using the primers Up-1870-F (5′ CCAAGGGCGA ACTGAACCAAATC 3′) and Down-1870-R (5′ GATGGAACAGACG GGTTTCGCC 3′). These sequences have been deposited in GenBank (accession nos. DQ324737–DQ324739). The predicted GNA1870 amino acid sequence, beginning at the proposed GTG start codon, was aligned with published GNA1870 sequences obtained from GenBank and World Patent Application WO 2004/04840 A2 using clustalW software. To determine the GNA1870 variant class, a phylogenetic tree (branched length dendrogram) was constructed using clustalW software.
Sera and complement reagents
Sera from seven normal healthy adult volunteers with no history of meningococcal infections and who had not received meningococcal vaccine were pooled and stored at −70°C. fH was purchased from Advanced Research Technologies.
Antibodies
Affinity-purified goat anti-human fH was made by Bethyl Laboratories using purified fH (Advanced Research Technologies) both as the immunogen and the immunoadsorbant. mAbs against variant 1 GNA1870, called JAR1, JAR3, JAR4, and JAR5 have been described previously (7). Anti-PorA mAb 1.7, which recognizes PorA from H44/76 (22), was obtained from the National Institute for Biological Standards and Controls (Hertfordshire, U.K.). Polyclonal anti-GNA1870 antisera was raised by immunizing mice sequentially with His-tagged rGNA1870 variants 1, 2, and 3 proteins, as described previously (7). FITC-conjugated anti-human C3 and anti-human C4 (BioDesign) were used in flow cytometry assays and have been described previously (16, 23). Anti-goat IgG and anti-mouse IgG conjugated to alkaline phosphatase and anti-goat IgG-FITC (Sigma-Aldrich) were used as secondary disclosing Abs.
Outer membrane preparations
Outer membranes were isolated using a previously described method (24). Briefly, bacteria harvested from five plates after an overnight culture on chocolate agar were suspended in normal saline. Bacteria were washed, suspended in 5 ml of PBS containing 10 mM EDTA, and incubated at 60°C for 30 min. Bacterial suspensions were then sheared by sequential passage using syringes and progressively smaller-sized needles (18- to 25-gauge). The resultant suspension was centrifuged at 5000 × g for 10 min at 4°C to separate any intact cells and debris. Supernatants were ultracentrifuged at 80,000 × g for 90 min at 4°C to yield a pellet that was enriched in outer membranes.
Recombinant GNA1870 proteins
His-tagged rGNA1870 proteins representing variants 1, 2, and 3 strains MC58, 2996 and M1239, respectively, were expressed in Escherichia coli and purified as described previously (7). These rGNA1870 molecules lacked the N-terminal lipid substitution.
Flow cytometry
Detection by flow cytometry of fH, IgG, C3, and C4 to Neisseriae have all been described previously (23, 25, 26). In some experiments, mAbs specific for GNA1870 were used to attempt competitive inhibition of fH binding to bacteria. Relative expression of GNA1870 was measured using either mAbs against variant 1 GNA1870 at a concentration of 10 μg/ml, or with mouse polyclonal anti-GNA1870 antiserum (described above) at a 1/100 dilution. Binding of anti-GNA1870 Abs was disclosed with anti-mouse IgG-FITC (Sigma-Aldrich) at a 1/100 dilution.
Western blotting
Western blotting was used to assess fH binding to outer membrane preparations. Membrane proteins were separated on a 4–12% Bis-Tris gel (Invitrogen Life Technologies) using MOPS running buffer. Proteins were transferred to polyvinylidene difluoride membranes (Millipore) and blocked with PBS-1% dry milk for 30 min at room temperature. Membranes were then incubated overnight at 4°C with fH (1 μg/ml in PBS-0.05% Tween 20). fH-binding proteins (fHBPs) were detected using affinity-isolated anti-fH (1 μg/ml in PBS-0.05% Tween 20) and disclosed using anti-goat IgG-alkaline phosphatase as previously described (27).
Mass spectrometry (MS) identification of meningococcal fHBP
Outer membrane proteins were separated by electrophoresis as described above, and stained with colloidal Coomassie brilliant blue (Sigma-Aldrich). The band corresponding to the ~29-kDa band that bound fH was carefully excised, diced, washed extensively, and then digested overnight at 37°C with trypsin. Digested peptides were eluted and subjected to microreversed-phase chromatography (ZipTips; Millipore). Purified peptides were cocrystallized with the matrix 2,5-dihydroxybenzoic acid on target, and MALDI mass spectra were acquired using a Reflex IV MALDI-TOF mass spectrometer (Bruker). Spectra were analyzed using MoverZ software (Genomic Solutions), internally calibrated to within 30 ppm, and peak lists were submitted to the Web-based search engine, Mascot (Matrix Science), for peptide mass fingerprinting analysis.
ELISA
To assess direct binding of fH to rGNA1870 proteins, microtiter plates were coated with purified fH (5 μg/ml in PBS) overnight at 4°C. Subsequent steps were conducted at room temperature for 1 h each. Nonspecific binding sites were blocked with PBS-0.5% BSA, followed by the addition of 10 μg/ml each rGNA1870 protein in PBS-0.05% Tween 20. Bound rGNA1870 was detected with polyclonal mouse anti-GNA1870 antiserum, followed by anti-mouse IgG conjugated to alkaline phosphatase (both at a 1/1000 dilution in PBS-Tween).
Serum bactericidal assays
Bacteria from an overnight culture on chocolate agar plates were repassaged onto fresh chocolate agar and allowed to grow for ~6 h at 37°C in 5% CO2. Serum bactericidal assays were performed as described previously (28). Briefly, ~2000 CFUs of meningococci were incubated with serum (concentrations specified for each experiment) in a final reaction volume of 150 μl. Aliquots of 25 μl were plated in duplicate at the start of the assay (t0) and after incubating the reaction mixture at 37°C for 30 min (t30). Survival was calculated as the number of viable colonies at t30 relative to baseline colony counts at t0.
Results
Binding of fH to N. meningitidis strains
We screened five strains of N. meningitidis, representative of each of the five major meningococcal serogroups, and examined them for their ability to bind to fH by flow cytometry (Fig. 1, upper panel). We detected fH binding to all strains with the exception of Y2220. We have shown previously that sialylation of Neisseria gonorrhoeae LOS can enhance fH binding (26). We next determined whether expression of capsular polysaccharide or LOS sialic acid had an impact on meningococcal fH binding. As seen in Fig. 1 (lower panel), isogenic mutant derivatives of all these strains that lacked capsule (siaD mutants, or the mynB derivative of A2594) and LOS sialic acid (lst mutants of the B, C, W-135, and Y strains; serogroup A strains do not endogenously sialylate their LOS) bound fH. These data suggested that the receptor for fH is a somatic meningococcal Ag, and that neither capsular polysaccharide nor LOS sialylation have an impact on fH binding.
FIGURE 1.
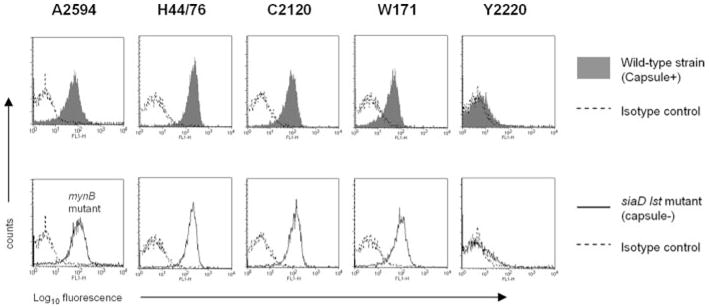
fH binding to N. meningitidis. A representative strain of N. meningitidis from each of the five major pathogenic serogroups (A–C, W-135, and Y) was chosen for study. Bacteria (~108 organisms) were incubated with 2 μg of pure fH, and bound fH was detected by flow cytometry using goat anti-human fH Ab. Upper panel, fH binding to encapsulated wild-type strains (shaded histogram). Lower panel, fH binding to unencapsulated mutant derivatives that also lacked the ability to sialylate their LOS (mynB mutant of A2594 and siaD lst mutants of the remaining four strains; solid line). Isotype controls (no fH added) are indicated by the broken lines. The x-axis represents fluorescence in arbitrary units on a log10 scale, and the y-axis represents the number of events.
PorA and PorB3 are not the receptors for fH on intact meningococci
We have shown previously that fH binds Por1A on certain strains of N. gonorrhoeae (25). The homologous protein in N. meningitidis is PorB3 (29). In addition, meningococci express a second porin molecule, called PorA (30). We speculated that either PorB3 or PorA may be the acceptor molecule for fH. Two lines of evidence showed that neither Por molecule bound fH.
In the first experiment, we examined fH binding to Por mutants of strain H44/76 that lacked either PorB3 (contained only PorA) or PorA (expressed PorB3 alone). Each of the mutants bound fH (Fig. 2A), which suggested that a receptor distinct from Por bound fH. An alternative explanation for this observation was that both Por molecules bound fH. Because porin is required for bacterial viability, it is not possible to delete both Por molecules simultaneously.
FIGURE 2.
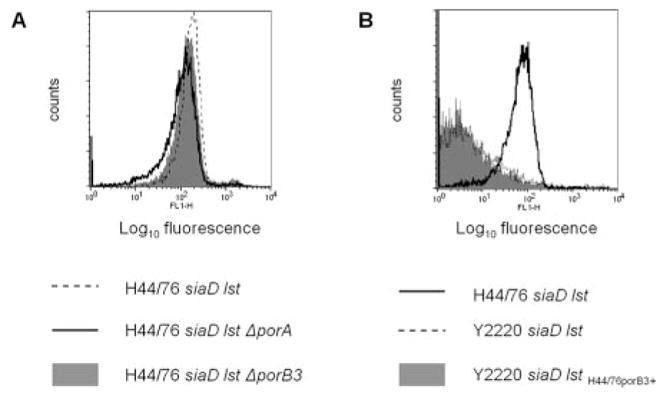
PorA and PorB3 on N. meningitidis strain H44/76 are not the ligands for fH. A, Binding of fH by flow cytometry to an unencapsulated mutant of H44/76 that lacked the ability to sialylate LOS (H44/76 siaD lst; broken line) and its two mutant derivatives that lacked either PorA (H44/76 siaD lst ΔporA; solid line) or PorB3 (H44/76 siaD lst ΔporB3; gray shaded histogram). B, fH binding to a mutant derivative of strain Y2220 siaD lst (fH nonbinder) whereby its PorB2 molecule was replaced with PorB3 of strain H44/76 (Y2220 siaD lst H44/76PorB3+; gray shaded histogram). fH binding to Y2220 siaD lst (negative control) is shown with the broken line, and the positive control strain, H44/76 siaD lst, by the solid line. Axes are as described for Fig. 1.
To address the possibility that both Por molecules bind fH, we replaced the PorB2 molecule of the fH nonbinding strain Y2220 siaD lst with the PorB3 molecule of strain H44/76. The resulting mutant Y2220 siaD lst H44/76porB3+, did not bind fH (Fig. 2B), suggesting that H44/76 PorB3 was not the ligand for fH on intact bacteria. This result made the possibility unlikely that both Por molecules on strain H44/76 bound fH, and we proceeded to identify the fH ligand by Western blotting as described below. As a control to demonstrate the ability of a Por molecule to bind fH in the background of N. meningitidis, we replaced Y2220 PorB2 with the fH-binding Por1A molecule from N. gonorrhoeae strain FA19 (25) and showed that the resultant mutant bound fH (data not shown).
fH binds to an ~29-kDa outer membrane protein identified as GNA1870
To identify the fH-binding molecule on N. meningitidis, we examined fH binding to an outer membrane protein preparation from strain H44/76 that was separated by electrophoresis on denaturing Bis-Tris gels followed by Western blotting. The membrane was incubated with fH, and bound fH was detected using affinity-isolated anti-fH. A discrete fH-binding band was seen at ~29 kDa (Fig. 3A, left lane; H44/76 OM). Bands were also evident at positions that corresponded to the expected migration velocity of PorB3 and PorA, but these had been excluded as fH ligands on intact bacteria as indicated above. The band that corresponded to the ~29-kDa band was identified on a Coomassie-stained gel and was analyzed by in-gel trypsin digestion followed by MALDI-TOF MS and peptide mass fingerprinting that was compared with the Neisseria proteome. The protein band was defined as lipoprotein GNA1870 using high probability-based Mowse scoring. The peptide ions covered 89% of the total protein sequence (Fig. 3B).
FIGURE 3.
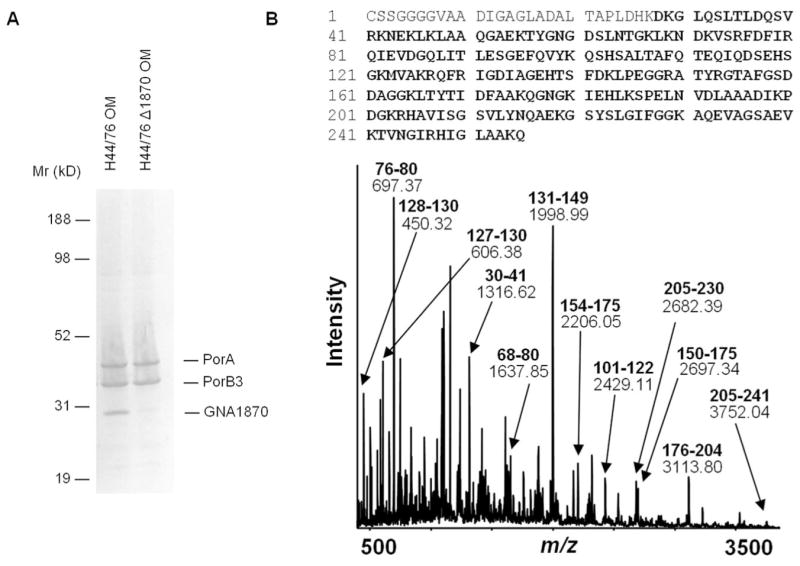
A, fH binding to outer membrane lysates prepared from strain H44/76 and its GNA1870 deletion mutant. B, Identification of GNA1870 by MS and peptide mass fingerprinting analysis. MALDI-TOF MS over the range m/z 400 to 3800 of peptides derived from in-gel trypsin digestion of the ~29 Kd protein band taken from a Bis-Tris gel. Major detected peptide ions assigned to GNA1870 are labeled with their experimental m/z value and their corresponding amino acid intervals (bold). The GNA1870 protein sequence is shown above, with the total peptide ion coverage (89%), including that represented by additional unlabeled minor peaks, shown in bold. Experimental m/z values were in agreement with theoretical values to within 50 ppm.
To validate the specificity of fH binding to GNA1870 in our Western blot binding assay, we prepared outer membranes from a GNA1870 deletion mutant of strain H44/76, and analyzed fH binding as detailed above. Fig. 3A, right lane; (H44/76 Δ1870 OM) shows that deleting GNA1870 results in loss of fH binding at the corresponding site on the blot.
GNA1870 is the ligand for fH on intact bacteria
Binding of a molecule to denatured proteins on Western blots could result from exposure of regions in the molecule that are otherwise cryptic on intact bacteria as was demonstrated in Fig. 3A, where fH had artifactually bound to PorA and PorB2 in Western blots. We used the following independent lines of evidence to confirm GNA1870 as the target for fH on live meningococci.
Deleting GNA1870 abrogates fH binding
We created isogenic mutant strains by insertional inactivation of gna1870 in the background of strains H44/76 and H44/76 siaD lst (unencapsulated and no LOS sialic acid). The resultant deletion mutants did not bind fH (Fig. 4A). These data indicated that GNA1870 is the acceptor for fH on live, whole meningococci.
FIGURE 4.
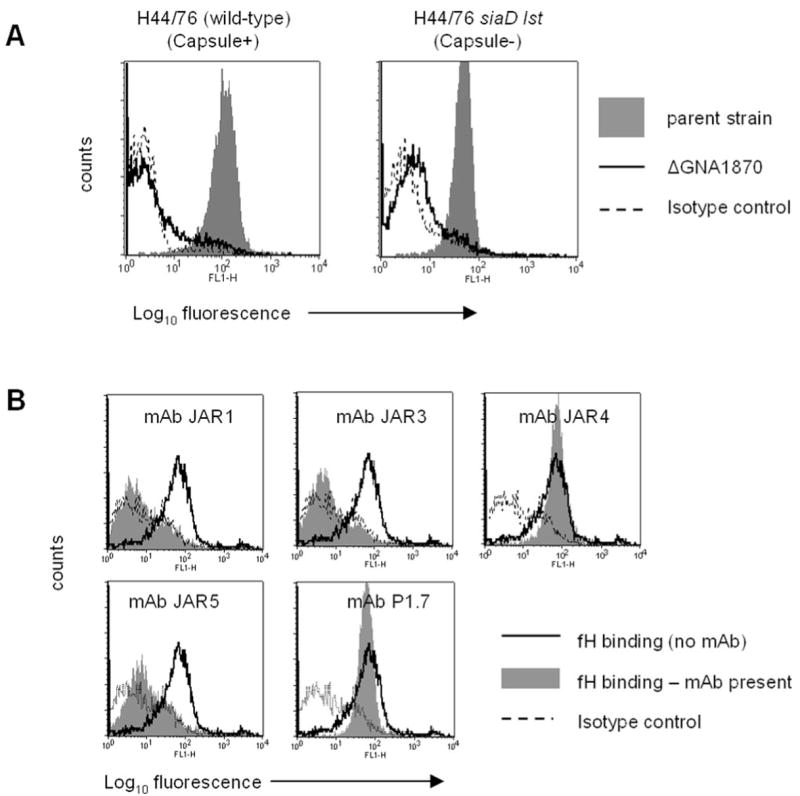
A, Deleting GNA1870 from wild-type strain H44/76 (graph on left) and its unencapsulated siaD lst mutant derivative (graph on right) abrogates fH binding. The shaded area indicates binding to the parent strain in both graphs, while binding to the isogenic mutant ΔGNA1870 derivative is indicated by the solid line. Broken lines indicate isotype controls (no fH added). B, fH binding to strain H44/76 can be blocked by anti-GNA1870 mAbs JAR1, JAR3, and JAR5, but not by JAR4. Bacteria were incubated with 5 μg of each mAb, followed by the addition of 1 μg of purified fH. Anti-PorA mAb 1.7 was used a negative control. Binding of fH in the absence of mAbs is shown by the solid line and is the same for each histogram, and fH binding in the presence of mAbs is the gray shaded histogram. Isotype controls are indicated by the broken line.
Monoclonal Abs directed against GNA1870 block fH binding
We have previously described a series of mAbs (JAR1, JAR3, JAR4, and JAR5) that are directed against the H44/76 GNA1870 molecule (7). Although the fine specificity of binding of these mAbs is not known, preliminary results suggest that the region spanned by aa 100–255 of GNA1870, which has been shown to be important for eliciting bactericidal Abs (31), also may be the region that binds these mAbs. We tested the ability of these Abs to selectively inhibit binding of fH to intact meningococci using a competitive inhibition assay. We observed that all anti-GNA1870 mAbs, with the exception of JAR4, blocked binding of fH to intact bacteria (Fig. 4B). All anti-GNA1870 mAbs bound live bacteria to a similar extent (7). As a negative control, we used an anti-PorA mAb, 1.7, which did not block fH binding to strain H44/76, but did bind to the strain.
fH can bind to all three GNA1870 variants
One system of GNA1870 classification divides the molecule into three variant families (variants 1–3) based on amino acid sequence analysis (9). Having thus far identified GNA1870 on strain H44/76 (variant 1 protein) as a ligand for fH, we sought to determine whether fH could bind to strains bearing variant 2 or variant 3 molecules. We detected binding of fH to strains RM1090 (variant 2) and M1239 (variant 3) (Fig. 5A). To confirm that the variant 2 and variant 3 proteins of strains RM1090 and M1239, respectively, were the ligands for fH, we constructed GNA1870 deletion mutants in these strains and noted that fH binding to each of the mutants was abrogated (Fig. 5A).
FIGURE 5.
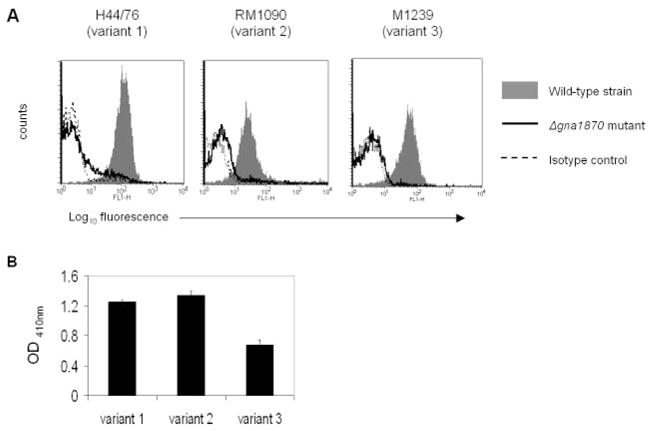
A, fH binds to all 3 GNA1870 variant strains. fH binding to variant 1 strain H44/76, variant 2 strain RM1090, and variant 3 strain M1239 (denoted by gray shaded histograms) and their Δgna1870 mutants (solid line) was examined by flow cytometry. B, Direct binding of fH to rGNA1870 by ELISA. One representative experiment of two experiments is shown. Each bar shows the mean (±SD) of quadruple wells. Control wells (rGNA1870 excluded from fH-coated wells, and binding to wells not coated with fH) all yielded OD410 nm of ≤0.008 (data not shown).
We confirmed a direct interaction between fH and GNA1870 from all three variant families by ELISA showing binding of purified recombinant GNA1870 (rGNA1870) representing strains MC58 (variant 1), 2996 (variant 2), and M1239 (variant 3) to fH that was immobilized on a microtiter plate (Fig. 5B).
Levels of GNA1870 expression correlate with fH binding
The data thus far indicated that fH could bind to GNA1870 from all three variant families. During the course of our investigations, we sequenced gna1870 from strains C2120, and W171, and Y2220 (high, low, and nonbinders of fH by flow cytometry, respectively; Fig. 1). A clustalW software (〈http://align.genome.jp/〉)-generated multisequence alignment of the predicted amino acid sequence of GNA1870 from these strains with representative variant 1, 2 and 3 sequences indicated that C2120 produces a variant 3 GNA1870, while Y2220 and W171 appear to produce a hybrid variant 2/3 protein. A dendrogram illustrating the protein sequence diversity is shown in Fig. 6A. Rather surprisingly, we found that the deduced amino acid sequences of Y2220 and W171 were identical, strongly suggesting that the primary amino acid sequence of GNA1870 did not determine fH binding. We confirmed that GNA1870 was the sole fH binding molecule on W171, by demonstrating abrogation of fH binding upon deleting GNA1870 (data not shown).
FIGURE 6.
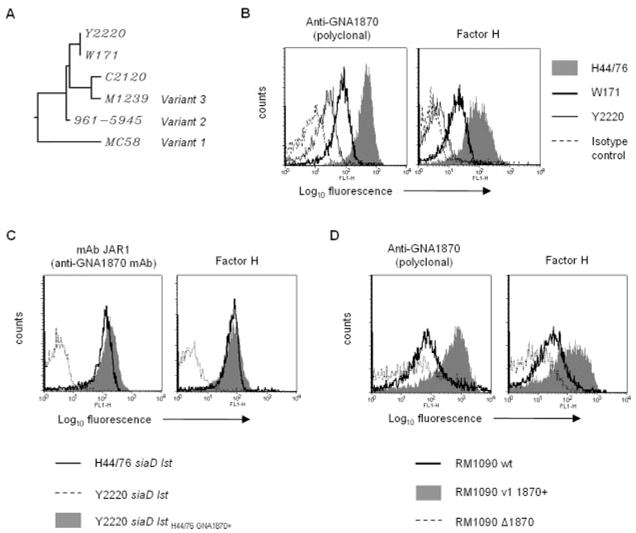
A, Phylogenetic tree showing the clustering of GNA1870 from strains Y2220, W171, and C2120 with GNA 1870 from variant 1, 2, and 3 type strains. B, Relative expression of GNA1870 on strains H44/76, W171, and Y2220, and correlation with fH binding. GNA1870 expression was measured by flow cytometry using polyclonal anti-GNA1870 antiserum (left graph). fH binding to the three strains is shown in the right histogram graph. In both histogram plots, H44/76 is represented by the shaded area, W171 by the thick solid line and Y2220 by the thin solid line. Isotype controls (anti-GNA1870 or fH excluded) are depicted by the broken line. C, High-level variant 1 GNA1870 expression in Y2220 correlates with increased fH binding. Left graph, Confirmation of phenotypic expression of H44/76 GNA1870 in the background of Y2220 siaD lst. Binding of JAR1 (specific for variant 1 GNA1870) to Y2220 siaD lst GNA1870H44/76+ (shaded area) by flow cytometry. Binding of JAR1 to the recipient strain Y2220 siaD lst is indicated by the broken line, and binding to the positive control strain H44/76 siaD lst by the solid line. Right graph, fH binding to Y2220 siaD lst GNA1870H44/76+, Y2220 siaD lst (negative control), and H44/76 siaD lst (positive control) are shown by the shaded area, broken line, and solid line, respectively. D, High levels of plasmid-mediated variant 1 GNA1870 expression in RM1090 results in enhanced fH binding. Left graph, Expression of GNA1870 in RM1090 (solid line), RM1090 overexpressing variant 1 GNA1870 (shaded area), and negative control strain lacking GNA1870 (RM1090 Δ1870, broken line) using polyclonal anti-GNA1870 antiserum. Right graph, fH binding to the three strains. Axes are as described in Fig. 1
Because GNA1870 expression levels have been shown to vary among different meningococcal strains (9), we hypothesized that levels of GNA1870 expression on the bacterial surface correlated with fH binding. We compared GNA1870 expression on strains H44/76 (high fH binder), W171 (low fH binder) and Y2220 (no detectable fH binding). fH binding also was measured in parallel. As seen in Fig. 6B, fH binding (right graph) was proportional to the amount of GNA1870 expressed on the bacterial surface (left graph). Expression of GNA1870 on these strains was also evaluated by western blotting, and confirmed the flow cytometry data (data not shown). We identified two additional strains, both belonging to serogroup B, that also express very low levels of GNA1870 on their surface, called NMB (6) and 2996 (J. Welsch and D. M. Granoff, unpublished observations). Consistent with the hypothesis that fH binding correlated with levels of GNA1870 expression, neither strain showed any detectable fH binding by flow cytometry (data not shown).
To demonstrate that high levels of GNA1870 expression enhanced fH binding in an isogenic background, we replaced the GNA1870 molecule of fH-nonbinding carrier strain Y2220 siaD lst, with the corresponding molecule from fH-binding strain 44/76. Surface expression of the H44/76 GNA1870 molecule on the resultant mutant (called Y2220 siaD lst H44/76GNA1870+) was confirmed by binding of the H44/76 GNA1870-specific mAbs JAR1, JAR3, JAR4 and JAR 5 to the mutant strain (representative data with mAb JAR1 shown in Fig. 6C, left graph). As expected, the Y2220 mutant strain with the replaced GNA1870 molecule bound fH to the same extent as strain H44/76 siaD lst (Fig. 6C, right graph).
As additional proof for correlation between levels of GNA1870 expression and fH binding, we examined fH binding to strain RM1090 (a low-level expresser of variant 2 GNA1870; Ref. 8) and RM1090 v.1 GNA1870+ (Fig. 6D, right graph). RM1090 ΔGNA1870 was used as a negative control. The level of GNA1870 expression was also assessed in parallel (Fig. 6D, left graph). Taken together, these observations suggest that the amount of fH binding correlates with the level of GNA1870 expression on the bacterial surface.
GNA1870 enhances serum resistance of meningococci
fH functions to down-regulate the alternative pathway of complement. Bacteria that bind fH would be expected to be more serum resistant than their isogenic mutant derivatives that do not bind fH. We compared the ability of strains H44/76, MC58, and M1239 and their isogenic GNA1870 deletion mutant derivatives to resist the effects of direct complement-mediated killing. Different serum concentrations were used because each strain differed in its baseline susceptibility to NHS. As seen in Fig. 7A, the wild-type strains were more serum resistant than their Δ1870 derivatives.
FIGURE 7.

Deleting GNA1870 enhances complement-mediated killing. A, Serum bactericidal assays were performed on encapsulated strains H44/76, MC58, and M1239 and their Δgna1870 mutants. Serum concentrations used are indicated on the x-axis. B, Unencapsulated strains lacking GNA1870 are more serum sensitive. H44/76 siaD (unencapsulated; LOS sialylated) and its GNA1870 deletion mutant were assessed for their ability to resist 5% NHS. Survival at 15 and 30 min was measured. C, Replacing the GNA1870 molecule of strain Y2220 siaD lst with that of H44/76 enhances serum resistance. Serum bactericidal assays were performed using 5% NHS.
We also examined the effects of deleting GNA1870 in the background of an unencapsulated derivative of strain H44/76 that possessed sialylated LOS (siaD mutant), a situation that may mimic bacteria at the mucosal surfaces. Again, the GNA1870-bearing strain was more serum resistant (Fig. 7B).
As further proof for the complement regulatory function of GNA1870, we compared the ability of strain Y2220 siaD lst H44/76GNA1870+ (fH binder) to resist complement compared with strain Y2220 siaD lst (fH nonbinder). As expected, the ability to bind fH was associated with enhanced serum resistance (Fig. 7C).
Complement regulation by GNA1870
We examined IgG, C3, and C4 binding to H44/76 siaD lst and its GNA1870 deletion derivative (Fig. 8). Of note, the GNA1870 deletion mutant bound less IgG than did the siaD lst mutant, which resulted in less C4 binding. Despite less classical pathway activation, the GNA1870 mutant bound more C3. These results demonstrate uninhibited activation of the alternative pathway positive feedback loop when the ability to bind fH is lost.
FIGURE 8.
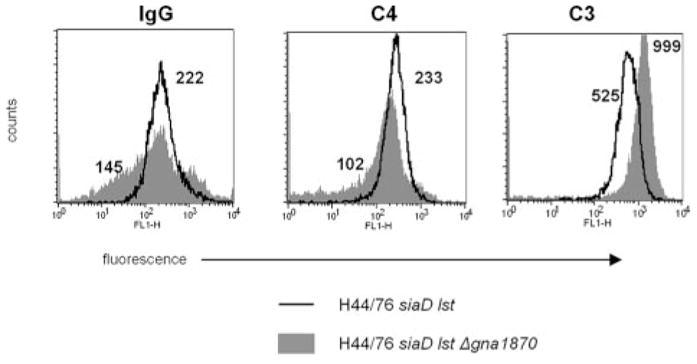
fH binding to meningococci via GNA1870 regulates the alternative pathway of complement. Strain H44/76 siaD lst and its isogenic mutant that lacked GNA1870 were incubated with NHS (15% v/v) and binding of IgG, C4, and C3 were measured by flow cytometry. Numbers beside each histogram indicate the geometric mean fluorescence of the entire population. Axes are as described in Fig. 1.
Discussion
N. meningitidis must survive complement-mediated killing to cause invasive disease. The classical pathway is required to initiate bactericidal activity on Neisseriae (32). The importance of Abs in protection against invasive disease has been detailed in elegant studies by Goldschneider et al. (33, 34). Once the classical pathway is set into motion, the positive feedback loop of the alternative pathway augments C3b deposition on bacteria. Persons deficient in alternative pathway activators such as properdin and factor D (1, 35, 36) are predisposed to invasive meningococcal disease.
Down-regulation of the alternative pathway may be beneficial for meningococci to survive in the human host. Almost all isolates recovered from the bloodstream or cerebrospinal fluid are encapsulated and capsular polysaccharide is required for high-level serum resistance (37, 38). However, isolates recovered from the nasopharynx are often shown to be unencapsulated (39–42). Encapsulation may actually hinder bacterial invasion of epithelial cells (43), and it has been suggested that meningococci may need to down-regulate capsule expression to traverse the epithelial barrier (44, 45). Meningococci, therefore, use redundant mechanisms to evade complement at various stages of disease pathogenesis. Binding of the alternative pathway down-regulatory molecule, fH to microbial surfaces is used by several bacterial species to regulate the alternative pathway of complement (reviewed in Ref. 46). The major classical pathway regulator, C4b-binding protein (C4bp) may also be important and we have recently demonstrated binding of C4bp to meningococcal PorA under hypotonic conditions, which may constitute a separate mechanism of complement evasion by N. meningitidis (47). The other pathogenic Neisseria species, N. gonorrhoeae, uses both C4bp and fH to its advantage to regulate complement activation on its surface. Sialylation of the lacto-N-neotetraose LOS species enhances fH binding to N. gonorrhoeae (26). In addition, several gonococcal strains that express the porin 1A (Por1A) molecule bind fH, which confers serum resistance in the absence of LOS sialylation (25). In this study, we report binding of fH to the meningococcal lipoprotein GNA1870. All meningococcal isolates that have been tested express GNA1870 (6, 9).
fH binds to selected polyanions, such as heparin, dextran sulfate, chondroitin sulfate A, and carrageenan (types III and IV), but not others such as chondroitin sulfate C, keratan sulfate, hyaluronic acid, colominic acid (bacterial polysialic acid), or polyaspartic acid (48, 49). An important observation we made was that fH did not bind to the polyanionic capsular polysaccharides expressed by any of the five major meningococcal serogroups, based on the finding that fH bound equally well to wild-type strains and their unencapsulated (siaD lst) mutants (Fig. 1). Serogroup B capsular polysaccharide regulates the alternative complement pathway (50), but our observations suggest that this effect is not attributable to enhanced fH binding.
We previously reported an association between expression of meningococcal PorB3 and the ability to bind to fH (51). We speculated that PorB3 might be the acceptor for fH, based on sequence similarities between meningococcal PorB3 and gonococcal Por1A (52), the latter having been shown to bind fH (25). In this study, we have identified a ligand for fH on N. meningitidis. Of the five diverse strains of N. meningitidis that we examined for fH binding, strains C2120 and W171 do not express PorB3 (they express PorB2), yet they still bind fH, suggesting that fH binds to a target(s) other than PorB3. Using Por deletion mutants, as well as allelic replacement (Fig. 2), we confirmed that fH did not bind to either Por molecule on intact N. meningitidis strain H44/76. Although binding of fH to PorB3 purified from strain H44/76 has been reported previously using ELISA (53), our results indicate that a fH-PorB3 interaction may not occur on live bacteria.
The ~29 kDa molecule in outer membrane preparations of H44/76 that bound fH in Western blots was identified as GNA1870 by MALDI-TOF MS and peptide mass fingerprinting analysis. Attempts at N-terminal Edman sequencing of this protein failed, consistent with the protein N terminus being modified or blocked, in this instance by palmitoylation (9). A limitation in the definitive identification of ligands using Western blotting is that surface molecules that are out of context of the membrane and/or denatured could bind fH via regions that may otherwise be cryptic on intact bacteria, thereby yielding false positive results. In addition, binding sites that require a conformational or three-dimensional structure will not be identified by this method. Therefore, we used two independent approaches (Fig. 4) to confirm that GNA1870 was the receptor for fH on intact meningococci.
We observed fH binding to all three GNA1870 variant classes, despite sequence diversity among these molecules (Fig. 5). This observation suggests that retaining the ability to bind fH may be beneficial to bacteria in vivo. The lower OD410 nm reading seen with the variant 3 protein may reflect differences in recognition of the proteins by the polyclonal variant 1–3 anti-GNA1870 Ab (8), and does not necessarily imply decreased binding of this protein to fH (Fig. 5). The N-terminal ~100 aa of all sequenced GNA1870 molecules bears maximum sequence homology, raising the possibility that the fH binding motif may reside in this region.
Differences in fH binding were attributable to differences in expression levels of GNA1870, and not to differences in amino acid sequences. A comparison of GNA1870 expression and fH binding among strains H44/76, W171, and Y2220 showed a correlation between levels of GNA1870 expression and fH binding (Fig. 6B). The polyclonal anti-GNA1870 antiserum used in this assay binds better to variant 2 GNA1870 than variant 1 proteins (8), thereby making it unlikely that there was preferential detection of H44/76 GNA1870 (variant 1). Furthermore, W171 and Y2220 were chosen for this experiment because their gna1870 genes are identical, allowing for a symmetric and unbiased comparison of GNA1870 expression. Y2220 expresses low amounts of GNA1870, and may bind fH but in amounts below the threshold of detection by flow cytometry. Low levels of GNA1870 expression and concomitant low or no fH binding are not unique to serogroup Y strains; we identified two other serogroup B strains of N. meningitidis (2996 and NMB) that express low levels of GNA1870 (6) and did not bind fH. We constructed a GNA1870 deletion mutant in strain 2996 and observed an increase in serum sensitivity (data not shown). This result suggests that even low levels of factor H binding (below the threshold of detection by flow cytometry) may diminish serum killing. This finding may have implications for the use of GNA1870 as a vaccine candidate because curtailing fH binding by specific immune Ab (Fig. 4B) will enhance killing, even by normal serum.
It is noteworthy that the DNA sequence spanning the region between the gna1870 ORF and the gene 3′ to gna1870 in Y2220 and W171 are identical, which suggests that differential protein expression is not because of differences in promoter sequences identified previously (9). Y2220 is capable of high-level GNA1870 expression, as evidenced by the observation that Y2220 siaD lst H44/76GNA1870+ expressed similar amounts of GNA1870 as H44/76, the latter having been shown to express high levels of GNA1870 (7, 9). The factor(s) responsible for variable GNA1870 expression among meningococcal strains is not clear.
Both capsulated and unencapsulated bacteria showed heightened serum resistance when they expressed GNA1870 and bound fH. Capsular polysaccharide is a key determinant of high-level serum resistance in meningococci (38). The ability to bind fH provides an additional level of protection to encapsulated bacteria against complement-mediated killing. The boosted level of serum resistance seen with encapsulated strains that bind fH could be critical for bacterial survival in the bloodstream, where high levels of complement are present. Although bacteria at mucosal surfaces encounter lower levels of complement (54), colonizing meningococci (which are often unencapsulated and potentially more serum sensitive) need to evade complement to successfully inhabit the nasopharynx.
The ability of bacterial surface-bound fH to regulate the C3-amplification loop of the alternative pathway of complement was illustrated by increased C3 binding to an isogenic mutant of strain H44/76 that lacked GNA1870 (Fig. 8). It is noteworthy that the GNA1870-deletion mutant bound slightly less IgG and C4 than its parent, suggesting that normal human serum may contain Abs directed against this protein. Despite less C4 binding to the mutant strain (and consequently diminished activation of C3 via the classical pathway of complement), we observed more C3 activation on this strain, which is consistent with uninhibited activation of C3 by the alternative pathway in the absence of fH binding.
In conclusion, we have demonstrated an important function for GNA1870, an immunogenic protein under investigation as a broadly effective meningococcal vaccine candidate. In addition to activating the classical pathway of complement in their own right, Abs directed against GNA1870 could promote bacterial killing and opsonophagocytosis by diverting fH away from the bacterial surface. This potential dual mechanism of bacterial killing makes GNA1870 an attractive vaccine candidate. One reason for vaccine failures is the selection of bacteria that lack the target Ag. Escape mutants that do not express GNA1870 because of selection by Ab pressure would be at a significant disadvantage because they would be more susceptible to complement-dependent killing. Our findings provide a strong rationale for the use of GNA1870 as one component of a meningococcal vaccine. We suggest referring to GNA1870 as fHBP to reflect the important function of this protein.
Acknowledgments
We thank Dr. Peter Rice for invaluable suggestions and critically reading the manuscript, and Maohua Lei for expert technical assistance. We thank Dr. Peter van der Ley for providing H44/76 PorA and PorB3 deletion mutant strains.
This work was supported by Public Health Service Grants R01-AI054544 and R01-AI32725 (to S.R.), R01-AI46464, R01-AI58122, and R21-AI061533 (to D.M.G.) from the National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH). L.A.L. and J.W. were supported by NIH Training Grants T32-AI052070 and T32-HL007951, respectively. C.E.C. was supported by NIH Grants P41-RR10888 and S10-RR15942. G.M. and J.W. contributed equally to this work.
Footnotes
Abbreviations used in this paper: fH, factor H; siaD, polysialyltransferase; cat, chloramphenicol resistance cassette; LOS, lipooligosaccharide; Por, porin; Erm, erythromycin-resistance cassette; Tet, tetracycline-resistance cassette; fHBP, fH-binding protein; MS, mass spectrometry; C4bp, C4b-binding protein.
Disclosures
Dan M. Granoff is a part-time consultant for Chiron Vaccines; grant support from Sanofi and Chiron Vaccines.
References
- 1.Ross SC, Densen P. Complement deficiency states and infection: epidemiology, pathogenesis, and consequences of neisserial and other infections in an immune deficiency. Medicine. 1984;63:243–273. [PubMed] [Google Scholar]
- 2.Fijen CA, Kuijper EJ, te Bulte MT, Daha MR, Dankert J. Assessment of complement deficiency in patients with meningococcal disease in The Netherlands. Clin Infect Dis. 1999;28:98–105. doi: 10.1086/515075. [DOI] [PubMed] [Google Scholar]
- 3.Figueroa J, Andreoni J, Densen P. Complement deficiency states and meningococcal disease. Immunol Res. 1993;12:295–311. doi: 10.1007/BF02918259. [DOI] [PubMed] [Google Scholar]
- 4.Bilukha OO, Rosenstein N. Prevention and control of meningococcal disease. Recommendations of the Advisory Committee on Immunization Practices (ACIP) Morbid Mortal Weekly Rep. 2005;54:1–21. [PubMed] [Google Scholar]
- 5.Weisgerber C, Husmann M, Frosch M, Rheinheimer C, Peuckert W, Gorgen I, Bitter-Suermann D. Embryonic neural cell adhesion molecule in cerebrospinal fluid of younger children: age-dependent decrease during the first year. J Neurochem. 1990;55:2063–2071. doi: 10.1111/j.1471-4159.1990.tb05796.x. [DOI] [PubMed] [Google Scholar]
- 6.Fletcher LD, Bernfield L, Barniak V, Farley JE, Howell A, Knauf M, Ooi P, Smith RP, Weise P, Wetherell M, et al. Vaccine potential of the Neisseria meningitidis 2086 lipoprotein. Infect Immun. 2004;72:2088–2100. doi: 10.1128/IAI.72.4.2088-2100.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Welsch JA, Rossi R, Comanducci M, Granoff DM. Protective activity of monoclonal antibodies to genome-derived neisserial antigen 1870, a Neisseria meningitidis candidate vaccine. J Immunol. 2004;172:5606–5615. doi: 10.4049/jimmunol.172.9.5606. [DOI] [PubMed] [Google Scholar]
- 8.Hou VC, Koeberling O, Welsch JA, Granoff DM. Protective antibody responses elicited by a meningococcal outer membrane vesicle vaccine with overexpressed genome-derived neisserial antigen 1870. J Infect Dis. 2005;192:580–590. doi: 10.1086/432102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Masignani V, Comanducci M, Giuliani MM, Bambini S, Adu-Bobie J, Arico B, Brunelli B, Pieri A, Santini L, Savino S, et al. Vaccination against Neisseria meningitidis using three variants of the lipoprotein GNA1870. J Exp Med. 2003;197:789–799. doi: 10.1084/jem.20021911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Whaley K, Ruddy S. Modulation of the alternative complement pathways by β1H globulin. J Exp Med. 1976;144:1147–1163. doi: 10.1084/jem.144.5.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sim E, Wood AB, Hsiung LM, Sim RB. Pattern of degradation of human complement fragment, C3b. FEBS Lett. 1981;132:55–60. doi: 10.1016/0014-5793(81)80426-7. [DOI] [PubMed] [Google Scholar]
- 12.Pangburn MK, Schreiber RD, Muller-Eberhard HJ. Human complement C3b inactivator: isolation, characterization, and demonstration of an absolute requirement for the serum protein β1H for cleavage of C3b and C4b in solution. J Exp Med. 1977;146:257–270. doi: 10.1084/jem.146.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weiler JM, Daha MR, Austen KF, Fearon DT. Control of the amplification convertase of complement by the plasma protein β1H. Proc Natl Acad Sci USA. 1976;73:3268–3272. doi: 10.1073/pnas.73.9.3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fearon DT, Austen KF. Activation of the alternative complement pathway due to resistance of zymosan-bound amplification convertase to endogenous regulatory mechanisms. Proc Natl Acad Sci USA. 1977;74:1683–1687. doi: 10.1073/pnas.74.4.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frosch M, Weisgerber C, Meyer TF. Molecular characterization and expression in Escherichia coli of the gene complex encoding the polysaccharide capsule of Neisseria meningitidis group B. Proc Natl Acad Sci USA. 1989;86:1669–1673. doi: 10.1073/pnas.86.5.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ram S, Cox AD, Wright JC, Vogel U, Getzlaff S, Boden R, Li J, Plested JS, Meri S, Gulati S, et al. Neisserial lipooligosaccharide is a target for complement component C4b: inner core phosphoethanolamine residues define C4b linkage specificity. J Biol Chem. 2003;278:50853–50862. doi: 10.1074/jbc.M308364200. [DOI] [PubMed] [Google Scholar]
- 17.Swartley JS, Liu LJ, Miller YK, Martin LE, Edupuganti S, Stephens DS. Characterization of the gene cassette required for biosynthesis of the (α1→6)-linked N-acetyl-D-mannosamine-1-phosphate capsule of serogroup A Neisseria meningitidis. J Bacteriol. 1998;180:1533–1539. doi: 10.1128/jb.180.6.1533-1539.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kahrs AF, Odenbreit S, Schmitt W, Heuermann D, Meyer TF, Haas R. An improved TnMax mini-transposon system suitable for sequencing, shuttle mutagenesis and gene fusions. Gene. 1995;167:53–57. doi: 10.1016/0378-1119(95)00671-0. [DOI] [PubMed] [Google Scholar]
- 19.Vogel U, Claus H, Heinze G, Frosch M. Functional characterization of an isogenic meningococcal α-2,3-sialyltransferase mutant: the role of lipooligosaccharide sialylation for serum resistance in serogroup B meningococci. Med Microbiol Immunol. 1997;186:159–166. doi: 10.1007/s004300050059. [DOI] [PubMed] [Google Scholar]
- 20.Tommassen J, Vermeij P, Struyve M, Benz R, Poolman JT. Isolation of Neisseria meningitidis mutants deficient in class 1 (porA) and class 3 (porB) outer membrane proteins. Infect Immun. 1990;58:1355–1359. doi: 10.1128/iai.58.5.1355-1359.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pagotto FJ, Salimnia H, Totten PA, Dillon JR. Stable shuttle vectors for Neisseria gonorrhoeae, Haemophilus spp. and other bacteria based on a single origin of replication. Gene. 2000;244:13–19. doi: 10.1016/s0378-1119(99)00557-0. [DOI] [PubMed] [Google Scholar]
- 22.Poolman JT, Kriz-Kuzemenska P, Ashton F, Bibb W, Dankert J, Demina A, Froholm LO, Hassan-King M, Jones DM, Lind I, et al. Serotypes and subtypes of Neisseria meningitidis: results of an international study comparing sensitivities and specificities of monoclonal antibodies. Clin Diagn Lab Immunol. 1995:69–72. doi: 10.1128/cdli.2.1.69-72.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ram S, Cullinane M, Blom A, Gulati S, McQuillen D, Monks B, O’Connell C, Boden R, Elkins C, Pangburn M, et al. Binding of C4b-binding protein to porin: a molecular mechanism of serum resistance of Neisseria gonorrhoeae. J Exp Med. 2001;193:281–296. doi: 10.1084/jem.193.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gnehm HE, Pelton SI, Gulati S, Rice PA. Characterization of antigens from nontypable Haemophilus influenzae recognized by human bactericidal antibodies: role of Haemophilus outer membrane proteins. J Clin Invest. 1985;75:1645–1658. doi: 10.1172/JCI111872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ram S, McQuillen DP, Gulati S, Elkins C, Pangburn MK, Rice PA. Binding of complement factor H to loop 5 of porin protein 1A: a molecular mechanism of serum resistance of nonsialylated Neisseria gonorrhoeae. J Exp Med. 1998;188:671–680. doi: 10.1084/jem.188.4.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ram S, Sharma AK, Simpson SD, Gulati S, McQuillen DP, Pangburn MK, Rice PA. A novel sialic acid binding site on factor H mediates serum resistance of sialylated Neisseria gonorrhoeae. J Exp Med. 1998;187:743–752. doi: 10.1084/jem.187.5.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blake MS, Johnston KH, Russell-Jones GJ, Gotschlich EC. A rapid, sensitive method for detection of alkaline phosphatase-conjugated anti-antibody on Western blots. Anal Biochem. 1984;136:175–179. doi: 10.1016/0003-2697(84)90320-8. [DOI] [PubMed] [Google Scholar]
- 28.McQuillen DP, Gulati S, Rice PA. Complement-mediated bacterial killing assays. Methods Enzymol. 1994;236:137–147. doi: 10.1016/0076-6879(94)36013-8. [DOI] [PubMed] [Google Scholar]
- 29.Wolff K, Stern A. The class 3 outer membrane protein (PorB) of Neisseria meningitidis: gene sequence and homology to the gonococcal porin PIA. FEMS Microb Lett. 1991;67:179–185. doi: 10.1016/0378-1097(91)90351-a. [DOI] [PubMed] [Google Scholar]
- 30.Poolman JT, Timmermans HA, Hopman CT, Teerlink T, Van Vught PA, Witvliet MH, Beuvery EC. Comparison of meningococcal outer membrane protein vaccines solubilized with detergent or C polysaccharide. Antonie Van Leeuwenhoek. 1987;53:413–419. doi: 10.1007/BF00415495. [DOI] [PubMed] [Google Scholar]
- 31.Giuliani MM, Santini L, Brunelli B, Biolchi A, Arico B, Di Marcello F, Cartocci E, Comanducci M, Masignani V, Lozzi L, et al. The region comprising amino acids 100 to 255 of Neisseria meningitidis lipoprotein GNA 1870 elicits bactericidal antibodies. Infect Immun. 2005;73:1151–1160. doi: 10.1128/IAI.73.2.1151-1160.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ingwer I, Petersen BH, Brooks G. Serum bactericidal action and activation of the classic and alternate complement pathways by Neisseria gonorrhoeae. J Lab Clin Med. 1978;92:211–220. [PubMed] [Google Scholar]
- 33.Goldschneider I, Gotschlich EC, Artenstein MS. Human immunity to the meningococcus. I. The role of humoral antibodies. J Exp Med. 1969;129:1307–1326. doi: 10.1084/jem.129.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goldschneider I, Gotschlich EC, Artenstein MS. Human immunity to the meningococcus. II. Development of natural immunity. J Exp Med. 1969;129:1327–1348. doi: 10.1084/jem.129.6.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sjoholm AG, Braconier JH, Soderstrom C. Properdin deficiency in a family with fulminant meningococcal infections. Clin Exp Immunol. 1982;50:291–297. [PMC free article] [PubMed] [Google Scholar]
- 36.Biesma DH, Hannema AJ, van Velzen-Blad H, Mulder L, van Zwieten R, Kluijt I, Roos D. A family with complement factor D deficiency. J Clin Invest. 2001;108:233–240. doi: 10.1172/JCI12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vogel U, Claus H, Heinze G, Frosch M. Role of lipopolysaccharide sialylation in serum resistance of serogroup B and C meningococcal disease isolates. Infect Immun. 1999;67:954–957. doi: 10.1128/iai.67.2.954-957.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vogel U, Frosch M. Mechanisms of neisserial serum resistance. Mol Microbiol. 1999;32:1133–1139. doi: 10.1046/j.1365-2958.1999.01469.x. [DOI] [PubMed] [Google Scholar]
- 39.Claus H, Maiden MC, Maag R, Frosch M, Vogel U. Many carried meningococci lack the genes required for capsule synthesis and transport. Microbiology. 2002;148:1813–1819. doi: 10.1099/00221287-148-6-1813. [DOI] [PubMed] [Google Scholar]
- 40.Claus H, Maiden MC, Wilson DJ, McCarthy ND, Jolley KA, Urwin R, Hessler F, Frosch M, Vogel U. Genetic analysis of meningococci carried by children and young adults. J Infect Dis. 2005;191:1263–1271. doi: 10.1086/428590. [DOI] [PubMed] [Google Scholar]
- 41.Patrick DM, Champagne S, Goh SH, Arsenault G, Thomas E, Shaw C, Rahim T, Taha F, Bigham M, Dubenko V, et al. Neisseria meningitides carriage during an outbreak of serogroup C disease. Clin Infect Dis. 2003;37:1183–1188. doi: 10.1086/378743. [DOI] [PubMed] [Google Scholar]
- 42.Sadler F, Fox A, Neal K, Dawson M, Cartwright K, Borrow R. Genetic analysis of capsular status of meningococcal carrier isolates. Epidemiol Infect. 2003;130:59–70. doi: 10.1017/s0950268802007987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stephens DS, Spellman PA, Swartley JS. Effect of the (α2→8)-linked polysialic acid capsule on adherence of Neisseria meningitidis to human mucosal cells. J Infect Dis. 1993;167:475–479. doi: 10.1093/infdis/167.2.475. [DOI] [PubMed] [Google Scholar]
- 44.Deghmane AE, Giorgini D, Larribe M, Alonso JM, Taha MK. Down-regulation of pili and capsule of Neisseria meningitidis upon contact with epithelial cells is mediated by CrgA regulatory protein. Mol Microbiol. 2002;43:1555–1564. doi: 10.1046/j.1365-2958.2002.02838.x. [DOI] [PubMed] [Google Scholar]
- 45.Hammerschmidt S, Muller A, Sillmann H, Muhlenhoff M, Borrow R, Fox A, van Putten J, Zollinger WD, Gerardy-Schahn R, Frosch M. Capsule phase variation in Neisseria meningitidis serogroup B by slipped-strand mispairing in the polysialyltransferase gene (siaD): correlation with bacterial invasion and the outbreak of meningococcal disease. Mol Microbiol. 1996;20:1211–1220. doi: 10.1111/j.1365-2958.1996.tb02641.x. [DOI] [PubMed] [Google Scholar]
- 46.Kraiczy P, Wurzner R. Complement escape of human pathogenic bacteria by acquisition of complement regulators. Mol Immunol. 2006;43:31–44. doi: 10.1016/j.molimm.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 47.Jarva H, Ram S, Vogel U, Blom AM, Meri S. Binding of the complement inhibitor C4bp to serogroup B Neisseria meningitidis. J Immunol. 2005;174:6299–6307. doi: 10.4049/jimmunol.174.10.6299. [DOI] [PubMed] [Google Scholar]
- 48.Pangburn MK, Atkinson MA, Meri S. Localization of the heparin-binding site on complement factor H. J Biol Chem. 1991;266:16847–16853. [PubMed] [Google Scholar]
- 49.Meri S, Pangburn MK. Discrimination between activators and non-activators of the alternative pathway of complement: regulation via a sialic acid/polyanion binding site on factor H. Proc Natl Acad Sci USA. 1990;87:3982–3986. doi: 10.1073/pnas.87.10.3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jarvis GA, Vedros NA. Sialic acid of group B Neisseria meningitidis regulates alternative complement pathway activation. Infect Immun. 1987;55:174–180. doi: 10.1128/iai.55.1.174-180.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ram S, Mackinnon FG, Gulati S, McQuillen DP, Vogel U, Frosch M, Elkins C, Guttormsen HK, Wetzler LM, Oppermann M, et al. The contrasting mechanisms of serum resistance of Neisseria gonorrhoeae and Neisseria meningitidis. Mol Immunol. 1999;4–5:915–928. doi: 10.1016/s0161-5890(99)00114-5. [DOI] [PubMed] [Google Scholar]
- 52.Derrick JP, Urwin R, Suker J, Feavers IM, Maiden MC. Structural and evolutionary inference from molecular variation in Neisseria porins. Infect Immun. 1999;67:2406–2413. doi: 10.1128/iai.67.5.2406-2413.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Estabrook MM, Jack DL, Klein NJ, Jarvis GA. Mannose-binding lectin binds to two major outer membrane proteins, opacity protein and porin, of Neisseria meningitidis. J Immunol. 2004;172:3784–3792. doi: 10.4049/jimmunol.172.6.3784. [DOI] [PubMed] [Google Scholar]
- 54.Mezei G, Varga L, Veres A, Fust G, Cserhati E. Complement activation in the nasal mucosa following nasal ragweed-allergen challenge. Pediatr Allergy Immunol. 2001;12:201–207. doi: 10.1034/j.1399-3038.2001.012004201.x. [DOI] [PubMed] [Google Scholar]
- 55.Frasch CE, Zollinger WD, Poolman JT. Serotype antigens of Neisseria meningitidis and a proposed scheme for designation of serotypes. Rev Infect Dis. 1985;7:504–510. doi: 10.1093/clinids/7.4.504. [DOI] [PubMed] [Google Scholar]
- 56.Seiler A, Reinhardt R, Sarkari J, Caugant DA, Achtman M. Allelic polymorphism and site-specific recombination in the opc locus of Neisseria meningitidis. Mol Microbiol. 1996;19:841–856. doi: 10.1046/j.1365-2958.1996.437970.x. [DOI] [PubMed] [Google Scholar]
- 57.McGuinness BT, I, Clarke N, Lambden PR, Barlow AK, Poolman JT, Jones DM, Heckels JE. Point mutation in meningococcal por A gene associated with increased endemic disease. Lancet. 1991;337:514–517. doi: 10.1016/0140-6736(91)91297-8. [DOI] [PubMed] [Google Scholar]
- 58.Tettelin H, Saunders NJ, Heidelberg J, Jeffries AC, Nelson KE, Eisen JA, Ketchum KA, Hood DW, Peden JF, Dodson RJ, et al. Complete genome sequence of Neisseria meningitidis serogroup B strain MC58. Science. 2000;287:1809–1815. doi: 10.1126/science.287.5459.1809. [DOI] [PubMed] [Google Scholar]
- 59.Vogel U, Morelli G, Zurth K, Claus H, Kriener E, Achtman M, Frosch M. Necessity of molecular techniques to distinguish between Neisseria meningitidis strains isolated from patients with meningococcal disease and from their healthy contacts. J Clin Microbiol. 1998;36:2465–2470. doi: 10.1128/jcm.36.9.2465-2470.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Claus H, Borrow R, Achtman M, Morelli G, Kantelberg C, Longworth E, Frosch M, Vogel U. Genetics of capsule O-acetylation in serogroup C, W-135 and Y meningococci. Mol Microbiol. 2004;51:227–239. doi: 10.1046/j.1365-2958.2003.03819.x. [DOI] [PubMed] [Google Scholar]
- 61.Comanducci M, Bambini S, Brunelli B, Adu-Bobie J, Arico B, Capecchi B, Giuliani MM, Masignani V, Santini L, Savino S, et al. NadA, a novel vaccine candidate of Neisseria meningitidis. J Exp Med. 2002;195:1445–1454. doi: 10.1084/jem.20020407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sacchi CT, Lemos AP, Brandt ME, Whitney AM, Melles CE, Solari CA, Frasch CE, Mayer LW. Proposed standardization of Neisseria meningitidis PorA variable-region typing nomenclature. Clin Diagn Lab Immunol. 1998;5:845–855. doi: 10.1128/cdli.5.6.845-855.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]