

Abstract
The problem of predicting the enzymes and non-enzymes from the protein sequence information is still an open problem in bioinformatics. It is further becoming more important as the number of sequenced information grows exponentially over time. We describe a novel approach for predicting the enzymes and non-enzymes from its amino-acid sequence using artificial neural network (ANN). Using 61 sequence derived features alone we have been able to achieve 79 percent correct prediction of enzymes/non-enzymes (in the set of 660 proteins). For the complete set of 61 parameters using 5-fold cross-validated classification, ANN model reveal a superior model (accuracy = 78.79 plus or minus 6.86 percent, Q(pred) = 74.734 plus or minus 17.08 percent, sensitivity = 84.48 plus or minus 6.73 percent, specificity = 77.13 plus or minus 13.39 percent). The second module of ANN is based on PSSM matrix. Using the same 5-fold cross-validation set, this ANN model predicts enzymes/non-enzymes with more accuracy (accuracy = 80.37 plus or minus 6.59 percent, Q(pred) = 67.466 plus or minus 12.41 percent, sensitivity = 0.9070 plus or minus 3.37 percent, specificity = 74.66 plus or minus 7.17 percent).
Keywords: enzymes, non enzymes, neural network, sequence derived features, PSSM
Background
It is generally accepted that protein structure is determined by its amino acid sequence [1] and that the knowledge of protein structures plays an important role in understanding their functions. To understand the rules relating amino acid sequence to three-dimensional protein structure is one of the major goals of contemporary molecular biology. A priori knowledge of protein as enzymes and non-enzymes has become quite useful from both an experimental and theoretical point of view.
One of the fundamental problems in post-genome era is the prediction and classification of proteins given only their primary sequence. [2] The number of proteins that are being made available to public and private databases is growing exponentially, and new methods must be found to understand and classify that information. The enormous task of function determination for every entry in GenBank has prompted the development of more sophisticated methods for protein automatic classification. [3,4] A computational method allowing for the automatic determination of protein function from its sequence alone is one of the prevailing problems in bioinformatics. [5] Determination of three-dimensional structure is the traditional approach to functional classification of proteins. This is a very time-consuming process, and the need for a faster method of classification is obvious. [6]
It has been reported that structural classes of proteins correlate strongly with amino acid composition, marked the onset of algorithm developments aimed at predicting the structural class of a protein from its amino acid composition alone. [7] In addition to amino acid composition, considering the sequence order along the primary structure of a protein into account would result in the improvement of prediction accuracy. [8] Hence, in this study we have develop two different neural networks which extract valuable information from protein sequence only for prediction into enzymes/non-enzymes. The first network used sequence derived features derived from PEPSTAT (EMBOSS suite) and the second network used PSSM profile obtained from PSI-BLAST, which would be useful for the systematic analysis of small or medium size protein sequences. Results are discussed, assessing the benefits of using this methodology in binary prediction of enzymes / non-enzymes. The preliminary results suggest that sequence derived feature can be used as a fast and effective classification methodology for proteins.
Methodology
Training data
A data set of 660 proteins, consisting of 330 non redundant enzymes and the same number of non redundant non-enzymes, were used for training and testing. The enzyme data set used in this study is obtained from the BRENDA database http://www.brenda.uni-koeln.de. [9] The pairwise sequence identities in the datasets are less than 54 percent for enzyme class and 45 perent for non-enzyme class.
Sequence derived parameters calculation and selection
To build a binary ANN model enabling effective prediction of enzymes/non-enzymes we initially calculated 61 parameters (Table 1 in supplementary material) from the protein sequence alone using PEPSTAT (EMBOSS suite) ftp://emboss.open-bio.org/pub/EMBOSS [10] for all 660 protein sequences. The normalized values (varying from 0 to 1) have been then used to generate ANN models for binary prediction.
Fivefold cross-validation
Fivefold cross-validation technique has been used for training and testing the ANN model, in which the dataset is randomly divided into five subsets, each containing equal number of enzyme sequences. Each set is a balanced set that consist of 50 percent of enzymes and 50 percent non-enzymes. The data set has been divided into training and testing set. The training set consists of five subsets. The network is validated for minimum error on testing set to calculate the performance measure for each fold of validation. This has been done five times to test for each subset. The final prediction results have been averaged over five testing sets.
ANN model for prediction of enzyme/non-enzyme using sequence derived features
Stuttgart neural network simulator package (SNNS version 4.2) [11] with standard back propagation was used to implement the ANN model. ANN configuration consists of 61 inputs and 1 output node. Whereas the number of nodes in the hidden layer was varied from 0 to 6 in order to find the optimal network that allows most accurate separation of enzymes/non-enzymes in the training sets (Figure 1a). During the learning phase, a value of 1 was assigned for the enzyme sequence and 0 for non-enzyme. The corresponding counts of the false/true positive and negative predictions were estimated using 0.1 and 0.9 cut-off values for non-enzymes and enzymes respectively. Thus, an enzyme from the testing set was considered correctly predicted by the ANN only when its output value ranged from 0.9 to 1.0. For each non-enzyme of the testing set the correct prediction was assumed if the corresponding ANN output lies between 0 and 0.1.
Figure 1.
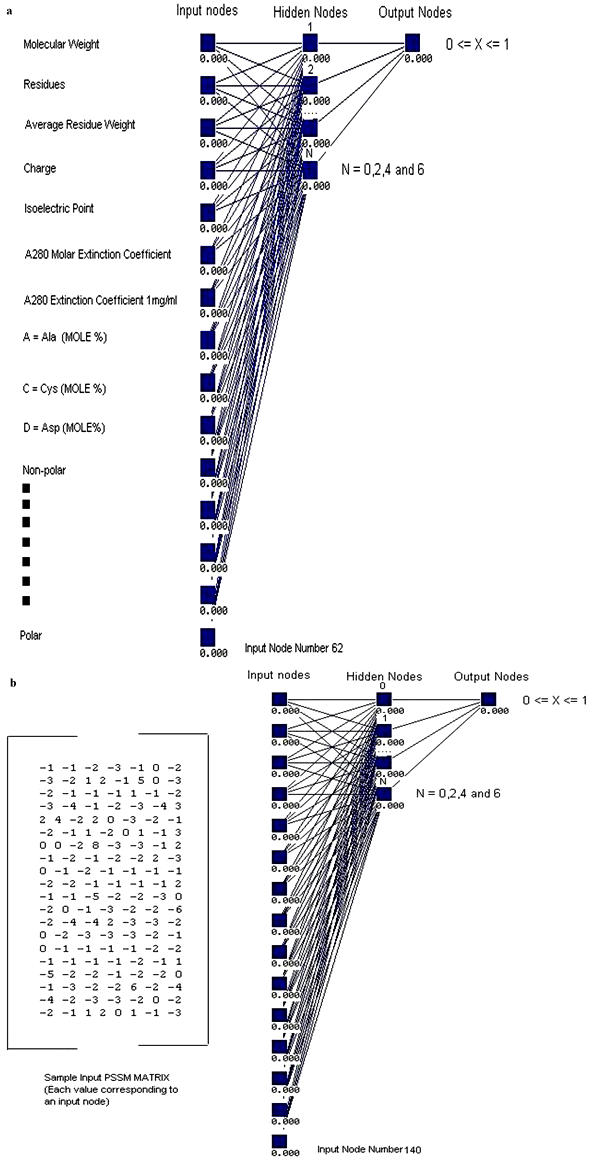
Configuration of artificial neural network used to develop binary primary sequence descriptor model for enzyme or non-enzyme proteins based on (a) sequence derived features and (b) PSSM matrix
ANN model for prediction of enzyme/non-enzyme using PSSM matrix
In this module of the developed tool, the position-specific scoring matrix generated by PSI-BLAST has been used as input to the neural network. The matrix has 20 x M real-number elements, where M is the length of the sliding window (M = 7). Each element represents the likelihood of that particular residue substitution at that position. Thus 20 real numbers rather than binary bits encode each residue. A standard back-propagation ANN configuration consisting of 140 inputs and 1 output node was used. The number of nodes in the hidden layer was varied from 0 to 6 in order to find the optimal network that allows most accurate separation of enzymes/non-enzymes in the training sets (Figure 1b). The training and validation methods are similar as mentioned above. The corresponding counts of the false/true positive and negative predictions were estimated using 0.4 and 0.9 cut-off values for non-enzymes and enzymes respectively. Thus, an enzyme from the testing set was considered correctly predicted by the ANN only when its output value ranged from 0.9 to 1.0. For each non-enzyme of the testing set the correct prediction was assumed if the corresponding ANN output lies between 0.1 and 0.4.
Performance measures
The prediction results of ANN model developed in the study were evaluated using the equations given in the supplementary material.
Results and discussion
The two different ANN models developed in this study are based on sequence derived features and PSSM matrix method. Applying a fivefold cross-validation using five testing subdata sets, we found that the network reached an overall accuracy of 78.79 ± 6.86% based on sequence derived features. The network has achieved an MCC of 0.596 ± 0.135. The other performance measures are: Qpred = 67.466 ± 17.084%, sensitivity = 90.70 ± 6.73% and specificity = 74.66 ± 13.39%. The vast majority of the predictions have been contained within 0.0 to 0.1 for non-enzymes and 0.9 to 1.0 for enzymes in case of sequence derived module. This illustrates that 0.1 and 0.9 cut-off values provide very adequate separation of two bioactive classes using ANN. To further enhance the prediction performance, the PSSM matrix is used for prediction. The network 7(20)-4-1 is trained on PSI-BLAST generated position-specific matrices (PSSM). The prediction results for both the networks are presented in Table 2 (supplementary material). It is clear from the results that the performance is improved slightly when PSI-BLAST-generated scoring matrices are used as input, compared with sequence derived features. The prediction accuracy is improved from 78.79% to 80.37%. However, most dramatic improvement is achieved in other parameters like Qpred, sensitivity and specificity. This is because it uses improved searching tool for multiple sequence alignment such as PSI-BLAST. PSI-BLAST searches the homologs against a larger database such as a nonredundant database and use multiple sequence information to generate PSSM matrix. From this study, it is clear that a combination of neural network and evolutionary information contained in multiple sequence alignment has improved the performance of prediction method.
The results demonstrate that the developed ANN-based binary prediction of enzymes/non-enzymes is adequate and can be considered an effective tool for in silico screening. The results also demonstrate that the sequence derived parameters as well as PSSM matrix readily accessible from the protein sequences only, can produce a variety of useful information to be used in silico; clearly demonstrates an adequacy and good predictive power of the developed ANN model. There is strong evidence that the introduced sequence features do adequately reflect the structural properties of proteins. The structure of a protein is an important determinant for the detailed molecular function of proteins, and would consequently also be useful for prediction of enzymes and non-enzymes. Based on the analysis of limited sequence features from protein sequences, differences in the parameters between enzymes and non-enzymes have previously been shown to exist and used for prediction of enzymes/non-enzymes in archaeal. [12] This agrees well with our result that sequence derived features can be used for predicting enzymes.
Presumably, accuracy of the approach operating by the sequence derived features can be improved even further by expanding the parameters or by applying more powerful classification techniques such as Support Vector Machines or Bayesian Neural Networks. Use of merely statistical techniques in conjunction with the sequence parameters would also be beneficial, as they will allow interpreting individual parameter contributions into “enzymes/non-enzymes-likeness”.
The results of the present work demonstrate that both the sequence derived features and PSSM matrix with ANN appear to be a very fast protein classification mechanism providing good results, comparable to some of the current efforts in the literature.
Availability
The program is implemented on the web server EnzymePred, available at http://www.juit.ac.in/enzyme/tool.html by using CGI/Perl script. The SNNS-generated network is converted into C program and is used as an interface. Users can enter primary amino acid sequence in fasta or free format. The protein sequence can be predicted as enzyme or non-enzyme.
Conclusion
We have demonstrated the feasibility of combining ANN with sequence derived features and PSSM matrix for prediction of enzymes/non-enzymes from protein sequence only. Even as prototype, both the ANNs we implemented have shown practical performance. With appraisal tests, we have found clues to improve prediction accuracy of ANN further. Expanding the sequence derived features, use of merely statistical techniques in conjunction with the sequence parameters and an adequate and low-noise training set, are critical to the success of ANN. Apparently, the more specifically an enzyme is to predict, thus the more definite a training set can be assembled, and the higher predicting power the corresponding ANN can acquire. In the future, we envisage an array of ANNs being trained to predict different classes and sub-classes of enzymes and to parse genomic sequence data in parallel, complementing current methods to achieve more reliable, high-throughput gene function prediction.
Supplementary materials
References
- 1.Anfinsen CBN. Science. 1973;181:223. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 2.Eisenberg D, et al. Nature. 2000;405:823. doi: 10.1038/35015694. [DOI] [PubMed] [Google Scholar]
- 3.Cai CZ, et al. Math Biosci. 2003;185:111. doi: 10.1016/s0025-5564(03)00096-8. [DOI] [PubMed] [Google Scholar]
- 4.King RD, et al. Bioinformatics. 2004;20:1110. doi: 10.1093/bioinformatics/bth047. [DOI] [PubMed] [Google Scholar]
- 5.Bork P, Koonin EV. Nat Genet. 1998;18:313. doi: 10.1038/ng0498-313. [DOI] [PubMed] [Google Scholar]
- 6.Baker EN, et al. Applied Bioinformatics. 2003;2:s3. [PubMed] [Google Scholar]
- 7.Nakashima H, et al. J Biochem. 1986;99:152. doi: 10.1093/oxfordjournals.jbchem.a135454. [DOI] [PubMed] [Google Scholar]
- 8.Bu WS, et al. Eur J Biochem. 1999;266:1043. doi: 10.1046/j.1432-1327.1999.00947.x. [DOI] [PubMed] [Google Scholar]
- 9.Schomburg I, et al. Nucleic Acids Res. 2004;32:D431. doi: 10.1093/nar/gkh081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rice P, et al. Trends in Genetics. 2000;16:276. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 11. http://www-ra.informatik.uni-tuebingen.de/SNNS/
- 12.Jensen LJ, et al. Protein Sci. 2002;11:2894. doi: 10.1110/ps.0225102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.