

Abstract
Background:
In a subset of patients with metastatic melanoma, T lymphocytes bearing the cell-surface marker CD8 (CD8+ T cells) can cause the regression of even large tumors. These antitumor CD8+ T cells recognize peptide antigens presented on the surface of tumor cells by major histocompatibility complex (MHC) class I molecules. The MHC class I molecule is a heterodimer composed of an integral membrane glycoprotein designated the α chain and a noncovalently associated, soluble protein called beta2-microglobulin (β2m). Loss of β2m generally eliminates antigen recognition by antitumor CD8+ T cells.
Purpose:
We studied the loss of β2m as a potential means of tumor escape from immune recognition in a cohort of patients receiving immunotherapy.
Methods:
We successfully grew 13 independent tumor cell cultures from tumor specimens obtained from 13 patients in a cohort of 40 consecutive patients undergoing immunotherapy for metastatic melanoma and for whom tumor specimens were available. These cell lines, as well as another melanoma cell line (called 1074mel) that had been derived from tumor obtained from a patient in a cytokine–gene therapy study, were characterized in vitro cytofluorometrically for MHC class I expression and by northern and western blot analyses for messenger RNA (mRNA) and protein expression, respectively, and ex vivo by immunohistochemistry.
Results:
After one melanoma cell line (1074mel) was found not to express functional β2m by cytofluorometric analysis, four (31%) of the 13 newly established melanoma cell lines were found to have an absolute lack of functional MHC class I expression. Northern blot analysis of RNA extracted from the five cell lines exhibiting no functional MHC class I expression showed that these cells contained normal levels of α-chain mRNA but variable levels of β2m mRNA. In addition, no immunoreactive β2m protein was detected by western blot analysis. When human β2m was transiently expressed with the use of a recombinant vaccinia virus, cell-surface MHC class I expression was reconstituted and the ability of these five cell lines to present endogenous antigens was restored. Immunohistochemical staining of tumor sections revealed a lack of immunoreactive MHC class I in vivo, supporting the notion that the in vitro observations were not artifactual. Furthermore, archival tumor sections obtained from patients prior to immunotherapy were available from three patients and were found to be β2m positive. This result was consistent with the hypothesis that loss of β2m resulted from immunotherapy.
Conclusions:
These data suggest that the loss of β2m may be a mechanism whereby tumor cells can acquire immunoresistance. This study represents the first characterization of a molecular route of escape of tumors from immune recognition in a cohort of patients being treated with immunotherapy.
A subset of T lymphocytes bearing the cell-surface marker CD8 can be shown to directly lyse tumor cells in vitro (1,2). These T cells, designated CD8+ T cells, can be expanded to large numbers ex vivo and adoptively transferred back to patients together with interleukin 2 (IL-2) where they can, in some cases, effect the regression of even large tumors (3-5). In patients with a number of different human malignancies, antitumor T cells can be elicited that are capable of recognizing autologous tumor cells as measured by cytolytic- and cytokine-release assays (6-10). Tumor deposits from patients with metastatic melanoma lesions yield tumor-infiltrating lymphocytes (TILs), with antitumor specificity in approximately 30% of the cases, making melanoma the most tractable of human cancers to T-cell-based immunotherapy. Objective clinical responses are observed in approximately 35% of patients with metastatic melanoma who are treated with TIL-based therapy; some responses are complete and long lasting [reviewed in (11)].
The reasons why the metastatic lesions of some patients yield successful. TIL cultures ex vivo while others do not are unknown. It is also unknown why some patients with apparently specific and lytic TILs often fail to respond. Even more perplexing is why some tumors escape after an initial response to therapy and why only some, but not all, of the lesions in an individual respond to treatment.
Abnormalities in T-cell signal transduction induced by tumor cells are potential mechanisms for tumor escape from immune recognition (12). However, since most patients with melanoma do not have measurable immunosuppression, the tumor cell has become the target of investigation to determine the mechanisms of escape from immunologic recognition (13). The steps required for the processing and presentation of antigens for recognition by CD8+ T cells, in particular, may be involved in tumor escape, since CD8+ T cells do not recognize intact antigens on the surfaces of tumor cells but generally recognize peptide fragments of protein antigens presented by major histocompatibility complex (MHC) class I molecules (14-16).
MHC molecules are known as human leukocyte antigens (HLAs) in humans and H-2 antigens in mice (17). MHC class I molecules are heterodimers composed of a 44- to 46-kd integral membrane glycoprotein designated the α chain and a noncovalently associated, soluble 12-kd protein called beta2-microglobulin (β2m). All nucleated cells in the body, with the exception of germline cells and some neurons (18), express class I–peptide complexes, the ligands for CD8+ T cells. A molecular understanding of the structure of MHC molecules has made clear their true function with respect to antigen recognition by T cells: MHC molecules are receptors for peptide antigens (19). MHC molecules are physically associated with peptide antigens, and x-ray crystallographic data have indicated precisely how such an interaction occurs. The solution of the crystal structure of MHC class I and II molecules has clarified the molecular structure of MHC molecules and specifically the way MHC molecules bind antigen (20-22). MHC molecules have peptide-binding domains, consisting of a deep groove that runs between two long α helices found on the outward-facing surface of the MHC molecule. In the case of class I molecules, x-ray crystallographic findings have since been refined and extended to include x-ray images of particular peptides lying in the cleft of MHC molecules (23-26). These findings reveal that the peptides are bound in an extended conformation. Antigenic peptides bound by MHC class I molecules are generally eight to 10 amino acids in length, usually resulting from proteolytic activity in the cytoplasm.
Peptides are transported into the endoplasmic reticulum by a specialized adenosine triphosphate-dependent transporter called TAP, which is related to the multidrug resistance protein, Mdrl. Peptides then assemble together with α chains and β2m to form a trimolecular complex that is then transported out of the endoplasmic reticulum, through the Golgi apparatus, to the cell surface for potential recognition by CD8+ T cells. With few exceptions (27), recognition by CD8+ T cells does not occur in the absence of β2m (28,29), since MHC class I α chains not associated with β2m are retained in the endoplasmic reticulum (30).
Bicknell et al. (31), Bodmer et al. (32), and Momburg and Koch (33) have shown that human cancer cells, especially cells of adenocarcinomas of the colon, may fail to express β2m; Wang et al. (34) and D'Urso et al. (35) have shown that two human melanoma cell lines do not express MHC class I as a result of mutations in the genes encoding β2m. The loss or mutation of the genes encoding β2m is a highly efficient mechanism for tumor escape from immune recognition, since stable presentation of peptide antigen by MHC class I molecules does not occur in the absence of β2m. The purpose of our study was to investigate the loss of β2m as a potential means of tumor escape from immune recognition in a cohort of patients undergoing immunotherapy.
Materials and Methods
Patients
Tumors were obtained according to institutional guidelines, and National Institutes of Health (NIH) Clinical Center standards for informed consent were adhered to for all patients. Thirteen independent tumor cell cultures were successfully grown from a cohort of 40 consecutive patients receiving immunotherapy for whom tumor specimens were available. These patients were enrolled in clinical trials at the National Cancer Institute (NCI), Bethesda, MD; the results of one of these trials was recently published (4).
Tumor Cells
Melanoma cell lines (1074mel, 1106mel, 1180mel, 1174mel, 1259mel, 526mel, 888mel, 397mel, and 624mel) were established in our laboratory, and the last four of these cell lines have been previously described (36). One tumor cell culture (1074mel; i.e., melanoma cell culture from patient No. 1074) was not part of the cohort of patients undergoing immunotherapy for metastatic melanoma and was derived for a different purpose [a cytokine–gene-therapy study (37)]. The β2m-deficient melanoma cell line FO-1 has been previously described (35). Daudi (a B-lymphoblastoid cell line derived from a patient with Burkitt's lymphoma), H82sclc [a small-cell lung cancer cell line (14)], and K562 (human chronic myelogenous leukemia) were obtained from the American Type Culture Collection (ATCC), Rockville, MD. Tumor cells were maintained as a monolayer culture in complete medium containing RPMI-1640, 10% heat-inactivated fetal calf serum (both from Biofluids, Rockville, MD), and 0.03% (100 mM) glutamine (NIH Media Unit, Bethesda, MD).
Cytofluorography
Cultured tumor cell lines were harvested with 0.05% trypsin–0.02% versene without calcium and magnesium (Biofluids), washed, then incubated with hybridoma culture supernatant containing the W6/32 monoclonal antibody (MAb) for 30 minutes (Sera Labs, Westbury, NY) (mouse immunoglobulin G2a [IgG2a] isotype). W6/32 reacts with monomorphic determinants on the HLA-A, -B, and -C molecules. In all cases, cells were stained with the appropriate isotype-matched control antibody, nonspecific mouse IgG2a (Becton Dickinson Immunocytometry Systems, San Jose, CA). MAb binding to cells was followed by binding with goat anti-mouse fluorescein isothiocyanate-conjugated antibody (Boehringer Mannheim Biochemicals, Indianapolis, IN) and detected by fluorescence-activated cell sorting (FACS) using a FACScan 440 (Becton Dickinson, Mountain View, CA).
Cytotoxic T Lymphocytes
Cytotoxic T lymphocytes (CTLs) were generated from excised tumor specimens by culturing suspension cells in complete medium containing IL-2 (6000 U/mL) (Chiron Therapeutics, Emeryville, CA) for 30-70 days as previously described (38).
Experiments that use mouse CTLs to study antigen processing in human tumors have been described elsewhere (14). In brief, polyclonal CD8+ T-cell populations were generated from 6- to 8-week-old female BALB/c mice by intravenous injection of 5 × 106 plaque-forming units of vaccinia virus. After at least 2 weeks, spleens were removed, dispersed to single-cell suspensions, and stimulated in vitro with vaccinia-infected BALB/c splenocytes at a ratio of 2:1. Cells were then cultured in complete medium to generate CD8+ T cells. The care of animals was in accord with NIH guidelines.
Microcytotoxicity Assays
5lCr release assays were performed as previously described (39). Briefly, 5000 target cells labeled with 200 μCi of Na5lCrO4 (Du Pont NEN, Boston, MA) were mixed with various numbers of effector cells and incubated for 6 hours. In experiments involving infection of cells with vaccinia virus, target cells were infected with 10 plaque-forming units per cell of the designated recombinant vaccinia virus(es) for 60-90 minutes, then labeled with 5lCr for 1 hour. Target cells were then washed three times and mixed with CD8+ T cells. In all micro-cytotoxicity assays, the amount of released 51Cr was determined by γ-photon counting of aliquots of supernatants of cells after lysis experiments. Percent specific lysis was calculated as follows: ([experimental cpm–spontaneous cpm]/[maximal cpm–spontaneous cpm]) × 100.
Immunohistochemical Staining of Archival Tissue Specimens From Patients
Immunohistochemistry was performed on archival tissue sections where available. Formalin-fixed, paraffin-embedded sections, 5 μm thick, were deparaffinized in xylene and stained as previously described (40). Murine MAb, ATCC HB159 (anti-human β2m, also known as SF1-1.1.1), supernatant was brought to pH 7 by the addition of HC1 and used at a 1:5 dilution in phosphate-buffered saline (PBS) (Biofluids). Biotinylated horse anti-mouse IgG at 1:200 was used as the secondary antibody (Boehringer Mannheim Biochemicals). Immunoperoxidase staining was completed with the use of the avidin–peroxidase complex (Vector Laboratories, Inc., Burlingame, CA). Diaminobenzidine-hydrogen peroxide (Sigma Chemical Co., St. Louis, MO) was used as the developing solution, and the sections were counterstained with hematoxylin–eosin.
Western Blot Analyses
Tumor cells were lysed by adding a buffer consisting of PBS containing 2 mM EDTA, 2 mM phenylmethylsulfonyl fluoride, and 1 mM dithiothreitol (Sigma Chemical Co.) at 4 °C. The cells were then sonicated for 10-15 seconds, and the lysate was centrifuged at 3000 rpm for 5 minutes at 4 °C. The concentration of protein in the cell homogenate supernatant was determined by the use of the Bio-Rad protein assay (Bio-Rad Laboratories, Richmond, CA). Equal amounts of protein (20 μg) from each cell lysate were electrophoretically resolved on a 16% sodium dodecyl sulfate–polyacrylamide gel and transferred to a nitrocellulose membrane with the use of a Bio-Rad Transblott Mini cell (Bio-Rad) at 250 mA for 1 hour. The nitrocellulose sheet was blocked overnight with Blocking Buffer (5 Prime → 3 Prime, Inc., West Chester, PA) and first incubated with the anti-β2m MAb HB159 (ATCC) for 2 hours and then with horseradish peroxidase-labeled secondary goat–anti-mouse antibody for 2 hours. Immunofluorescence was detected with the use of the Enhanced Chemi Luminescence (Amersham Life Science Inc., Arlington Heights, IL). Protein sizes were estimated with the use of low-molecular-weight standards (14 000-70 000; Sigma Chemical Co.).
Northern Blot Analysis
Specific probes for β2m were generated by the use of the reverse transcription-polymerase chain reaction (PCR) method. The total RNA was isolated with the use of guanidine isothiocyanate (Fluka Chemical Corp., Ronkonkoma, NY) then purified with the use of the cesium chloride (Serva Biochemicals) centrifugation method, as previously described (14) from the Epstein-Barr virus-transformed B-cell line designated 501 that was established in our laboratory (41). First-strand complementary DNA (cDNA) was synthesized from 10 μg total RNA using an oligo dT primer and murine leukemia virus reverse transcriptase (Life Technologies [GIBCO BRL], Gaithersburg, MD). The specific 5′-side oligonucleotide primer for β2m that was used had the sequence 5′ CAC GTC ATC CAG AGA ATG G 3′, and the 3′-side oligonucleotide primer had the sequence 5′ CGA TCC CAC TTA ACT ATC TTG G 3′. For PCR amplification, samples were initially denatured for 2 minutes at 94 °C, then 30 cycles were performed with the use of the following conditions: 94 °C for 30 seconds, 60 °C for 30 seconds, and 72 °C for 1 minute followed by an extension cycle of 10 minutes at 72 °C. PCR products were electrophoresed on 1.5% agarose gels with an acetate buffer, and the predicted fragment sizes of bands (260 bp) were isolated, purified by the glass-powder method (GeneClean, La Jolla, CA), and used as hybridization probes for northern blots. The HLA-A2.1 full-length cDNA was used as a probe template for MHC class I (42). Human β-actin cDNA was purchased from Clontech Laboratories, Inc., Palo Alto, CA. The probes were labeled with [32P]-deoxycytidine triphosphate (Amersham Life Science Inc.) by the random-priming method as previously described (14). For the northern blot analysis, 10 μg of total RNA from each tumor was electrophoresed in a 1% agarose–formaldehyde gel, transferred to a nylon membrane (Zeta-Probe, Bio-Rad Laboratories), and fixed to the membrane by UV cross-linking. Prehybridization was done for 30 minutes with the use of the 5 Prime → 3 Prime Northern Hybridization buffer. Hybridization was done in a 40% formamide hybridization solution (Northern hybridization buffer, 5 Prime → 3 Prime, Inc.) at 42 °C overnight. Membranes were washed three to four times in 2× standard saline citrate at 60 °C for 30 minutes for each wash, and autoradiography was then performed.
Gene Replacement Studies
The production of a recombinant vaccinia virus expressing the Kd gene (Kd-Vac) has been described (14), as has the production of the recombinant vaccinia virus containing the β2m gene (β2m-Vac) (42).
Results
Steady-state cell surface expression of MHC class I was studied in a group of patients with metastatic melanoma at the NCI. All of the patients entered in the T-cell-based immunotherapy protocol had either failed standard therapy or had disease for which no effective therapy was available. All patients had clinically assessable disease and received no other therapy during the 30 days before the protocol treatment or during treatment. Patient characteristics, including the immunotherapy received, are shown in Table 1.
Table 1.
Patient characteristics
| Patient No. |
Age, y; sex | Specimen site* |
Comments† | Tumor-infiltrating lymphocyte growth |
Class I, histology |
Response to immunotherapy‡ |
|---|---|---|---|---|---|---|
| 1106 | 46; male | SC | Previous response to treatment with IL-2 and IFN α; recurred and did not rerespond | No | No | — |
| SC | Same as above | Yes | Yes§ | Mixed | ||
| SC∥ | Treatment with TILs from above, no response | No | No¶,# | — | ||
| SC | Additional biopsies | No attempt | No | — | ||
| 1180 | 46; female | SC | No previous treatment of metastasis | Yes | Yes | No |
| SC | New nodule after no response to treatment with TILs/IL-2/IFN α | Yes | Low** | No | ||
| SC∥ | New nodules after no response to TILs/IL-2/IFN α | No | No††,‡‡ | — | ||
| 1074§§ | 26; female | SC∥ | Previous mixed response to IL-2 | Yes | No | No |
| 1174 | 53; male | LN∥ | No response to previous IL-2 | No | No | — |
| 1259 | 30; male | SC | No previous immunotherapy | Yes | Yes | Yes |
| SC∥ | Previous response to TILs; given cryopreserved TILs | No | No | No |
SC = subcutaneous; LN = lymph node.
IL-2 = interleukin 2; IFN α = interferon alpha; TILs = tumor-infiltrating lymphocytes.
For definitions of clinical responses, see (4).
Immunohistochemical staining, shown in Fig. 6, panel B.
Tumor from which melanoma cell line was derived.
Hematoxylin–eosin stain, shown in Fig. 6, panel A.
Immunohistochemical staining, shown in Fig. 6, panel C.
Immunohistochemical staining, shown in Fig. 6, panel E.
Hematoxylin–eosin stain, shown in Fig. 6, panel D.
Immunohistochemical staining, shown in Fig. 6, panel F.
From another study (42).
Of the 40 attempts to grow tumor cells obtained from patients from whom melanoma deposits were surgically resected, 18 tumor cell lines could not be grown at all and nine could be derived, but did not grow adequately, and therefore were not studied for their expression of cell surface HLA. Of the 13 tumor cell lines that could be derived into tumor cell cultures that could be grown to quantities sufficient for in vitro evaluation, four were found to be devoid of MHC class I and were included in the present study for the characterization of their MHC class I deficits. One additional tumor cell culture (from patient No. 1074) was not part of this group of 40 and was derived for a different purpose [a cytokine gene-therapy study (37)], but is nevertheless included here because it was observed to be MHC class I deficient.
From this screening analysis, five independently established melanoma cell cultures from patients with metastatic melanoma were devoid of cell-surface HLA class I molecules as measured cytofluorometrically (Fig. 1). The loss of MHC class I expression on these cell lines was complete, even when assessed by the MAb W6/32 (that can recognize virtually all human HLA class I alleles).
Fig. 1.
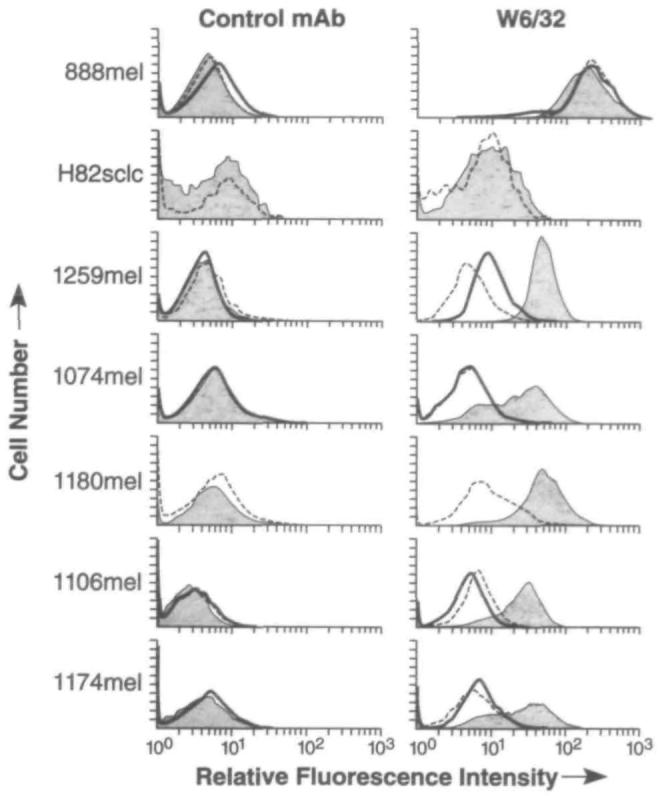
Absence of major histocompatibility complex class I on the surfaces of metastatic human melanoma cell lines and restoration by transient expression of beta2-microglobulin (β2m). Fluorescence-activated cell sorter analysis was performed on tumor cell lines alone (heavy solid line), infected with wild-type vaccinia virus (dashed line), and infected with the recombinant vaccinia virus expressing β2m (light solid line, shaded gray under the curve); cell lines were sequentially incubated with either the control immunoglobulin G2a antibody (left panels) or the W6/32 monoclonal antibody (right panels) and then with goat anti-mouse fluorescein isothiocyanate-conjugated Fab fragments. The cell lines analyzed were the positive control cell line 888mel (that is normal in its antigen-presenting capacity), the negative-control H82sclc (a small-cell lung cancer cell line that does not process antigens), and the five melanoma cell lines (1259mel, 1074mel, 1180mel, 1106mel, and 1174mel) that have lost functional β2m. For two cell lines (H82sclc and 1180mel), only two curves are shown in this experiment because the tumor-cell-alone control was not performed because of inadequate numbers of cells.
To characterize the mechanisms underlying the lack of MHC class I expression, we considered the possibility that HLA α chains were poorly loaded with peptides in the endoplasmic reticulum because of down-regulation or mutation of the TAP peptide transporters or proteasome components, since we have observed this mechanism to be functioning in small-cell lung cancer (14). However, defects in the above-named components of the antigen-processing machinery have generally resulted in MHC class I expression that is decreased, but not absent, since some peptides can originate from peptides or proteins translocated across the endoplasmic reticulum membrane by TAP-independent signal sequence mechanisms, exposing the proteins to proteolytic activities in the endoplasmic reticulum (43,44).
The completeness of the loss observed in the melanoma cell lines suggested the structural loss or mutation of either all of the heavy-chain MHC class I molecules or of β2m. The former would require disabling mutations of six independent genomic transcripts (two alleles each of HLA-A, -B, and -C) or the complete loss of approximately 1500 bp of the MHC class I region on the short arm of both copies of chromosome 6 (since MHC class I molecules are codominantly expressed). Another, more likely, possibility was the loss or mutation of both copies of β2m, a structural component of cell-surface MHC class I.
To explore the possibility that the deficit in functional β2m occurred at the level of decreased gene transcription or increased degradation of messenger RNA (mRNA), northern blot analysis of the melanoma tumor cell lines was performed. The α-chain mRNA levels were found to be normal (data not shown). However, expression of β2m mRNA was variable and lower than that seen in control cell lines (Fig. 2). The controls used in this experiment were the antigen-presentation normal melanoma cell lines 624 and 397 and the FO-1 cell line, which is known to have a defect in the expression of functional β2m, due to a lack of β2m gene expression (35). A western blot analysis, however, failed to detect any immunoreactive protein (Fig. 3).
Fig. 2.
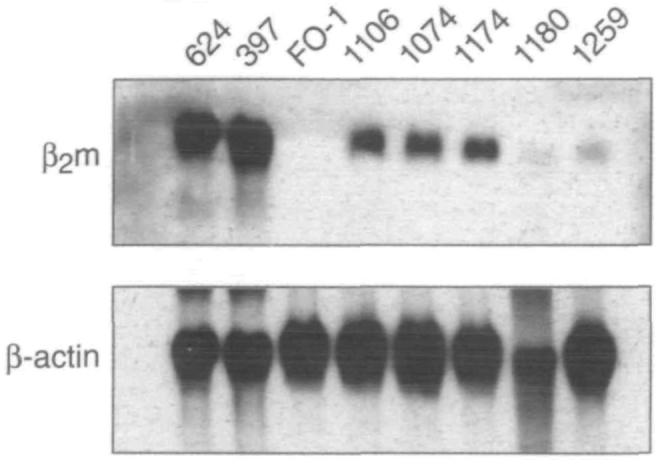
Northern blot analysis of total RNA extracted from the indicated cells (624[mel], 397[mel], FO-1, 1106[mel], 1074[mel], 1l74[mel], 1180[mel], 1259[mel]) and probed for the expression of beta2-microglobulin (β2m) messenger RNA. The northern blot probed for β2m messenger RNA (top panel) was stripped and probed for β-actin to ensure sufficient loading of all lanes (bottom panel).
Fig. 3.

Western blot analysis of total protein extracts for expression of immunoreactive beta2-microglobulin (β2m). Lysates of melanoma cell lines 1074mel, 1106mel, 1180mel, 1174mel, and 1259mel do not express any measurable immunoreactive β2m. Line l074mel is shown after infection with recombinant vaccinia virus expressing β2m (left-most panel) or after infection with wild-type control vaccinia virus (second from left).
To test the hypothesis that β2m was responsible for the absence of MHC class I on the cell surfaces, we sought to replace functional β2m within the cells. Since MHC class I molecules are generally retained in the endoplasmic reticulum if they are not complexed with β2m, the addition of functional β2m would likely reconstitute cell-membrane expression of MHC class I molecules. One rapid, although transient, method of β2m gene expression involved infection of the cells with a recombinant vaccinia virus that expresses β2m. As shown in Fig. 1, expression of β2m mRNA by a recombinant vaccinia virus elicited MHC class I expression on the surfaces of the five human melanoma cell lines under study. Indeed, there may be some suppression of endogenous MHC class I expression by control vaccinia virus in line 1259M, a phenomenon that has been described by others (45). The melanoma cell line 888 was used as a positive control, since it exhibits normal antigen presentation; the small-cell lung cancer cell line H82 was used as a negative control, since it is defective in antigen processing, as previously reported (14).
The results of the western blot analysis, together with the cytofluorographic evidence, suggested that the genetic insertion of functional β2m would fully restore the ability of the cell lines to process and present endogenous antigens for recognition by CD8+ T cells. On the other hand, if there were additional abnormalities in antigen processing, a full restoration of function in the antigen-presenting capabilities of the cell lines being studied would not be seen with the replacement of β2m. To test this question directly, we employed a method for the study of antigen processing that was independent of both the HLA type of the tumor and the presence or absence of specific cellular proteins. With the use of a recombinant vaccinia virus encoding the mouse H-2 Kd MHC class I molecule (Kd-Vac), we tested human tumor cell lines for presentation of viral antigens to mouse Kd-restricted, vaccinia-specific CD8+ T-cell populations and thus studied antigen-processing capabilities of human tumor cells per se. As shown in Fig. 4, restoration of the β2m molecule in the tumor cells completely restored the antigen-processing capabilities of all five of the melanoma cell lines tested. The expression of β2m had no effect on H82, the negative control in this experiment (14).
Fig. 4.

Microcytotoxicity assay evaluating the antigen-presenting capacities of beta2-microglobulin (β2m)-deficient cell lines. Cell lines were screened for their capacities to present vaccinia virus antigens in the context of a transiently expressed H-2 Kd molecule. Effector cells (E) were vaccinia virus-specific, H-2 Kd-restricted murine cytotoxic T lymphocytes. Target cells (T) were either uninfectcd (▲), infected with vaccinia virus expressing H-2 Kd alone (●), or in combination with recombinant vaccinia virus expressing β2m (△). The E:T ratios for the six points in each curve, reading from left to right, were 100:1, 50:1, 25:1, 12.5:1, 6.25:1, and 3.125:1. The melanoma cell line 526 (the positive control) is an antigen-processing intact cell line that processes vaccinia antigens efficiently in the absence of β2m. The small-cell lung cancer cell line H82sclc fails to process antigens even in the presence of β2m, since the antigen-processing lesions in this cell line are elsewhere (14). Note that 1259mel, 1180mel, 1106mel, 1174mel, and 1074mel all fail to present vaccinia virus antigens but can do so efficiently when functional β2m is expressed endogenously.
To test whether lack of functional β2m was the sole mechanism of escape of these tumors from recognition by CD8+ T cells, we studied whether restoration of β2m in these cell lines was associated with the restoration of killing of these cell lines by antitumor CD8+ T cells. This was a critical question because the loss of β2m could be accompanied by the loss of tumor-associated antigens, making the restoration of β2m in these cell lines a less relevant aspect of therapy. The problem with testing this question was that autologous TILs were not available from patients whose tumors gave rise to these cell lines. Since one of the five patients lacking functional β2m expression was HLA-A2.1 positive (No. 1074) and since we have generated CD8+ T cells that were capable of recognizing ubiquitously expressed melanoma tumor-associated antigens in the context of HLA-A2.1 (46), we could directly test whether restoration of β2m led to immune recognition by CD8+ T cells. As shown in Fig. 5, expression of functional β2m in the 1074mel cell line restored its susceptibility to lysis by HLA-A2.1-restricted antimelanoma CD8+ T cells.
Fig. 5.
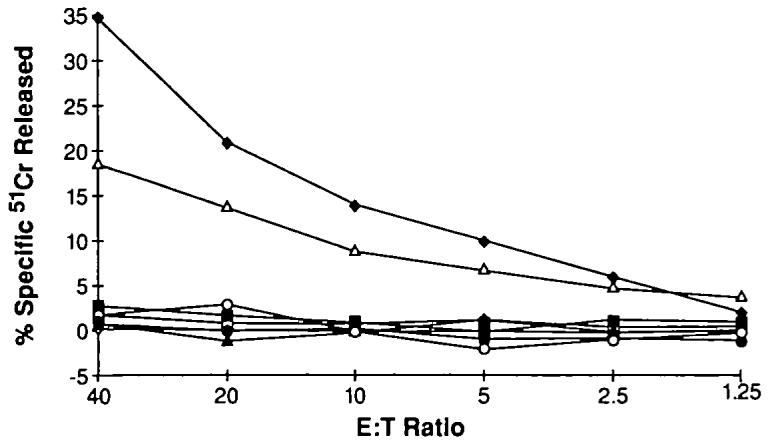
Microcytotoxicity assay evaluating the capacity of a beta2-microblobulin (β2m)-deficient cell line to present tumor-associated antigen after restoration of β2m. Melanoma cell lines were tested for recognition by HLA-A2.1-restricted antimelanoma effector (E) cells. Target cells (T) were the HLA-A2.1-positive, β2m-positive 526 cell line (◆); the HLA-A2.1-positive but β2m-negative 1074 cell line infected with control vaccinia virus (▲) or infected with recombinant vaccinia virus-expressing β2m (△); the HLA-A2.1-negative but β2m-positive 888 melanoma cell line with control (□) or β2m vaccinia virus (■); and the HLA-A2.1-negative and β2m- negative 1259 cell line with control (●) or β2m vaccinia virus (○).
Despite the lack of expression of cell-surface MHC class I molecules, all five of the β2m-deficient melanoma cell lines were capable of being lysed by lymphokine-activated killer cells (data not shown).
To test the possibility that these observations were due simply to an artifact of the culture of the melanoma cells, we sought to assess the status of MHC class I expression of these tumors in vivo. Since a portion of each of the fresh melanoma specimens from which the melanoma cultures were developed was sent for pathologic analysis, we procured the paraffin-embedded tissue blocks and performed immunohistochemical analysis for the presence of β2m. Tissue sections in all five of the cases in question were found to be uniformly devoid of β2m (Fig. 6 and data not shown). Note that each tumor cell line contains the positive control of infiltrating cells that make up the vasculature of the tumor. Thus, the cells of the lines under scrutiny did not express β2m in vivo that was capable of complexing with MHC class I α chains and stabilizing them on the tumor cell surfaces.
Fig. 6.
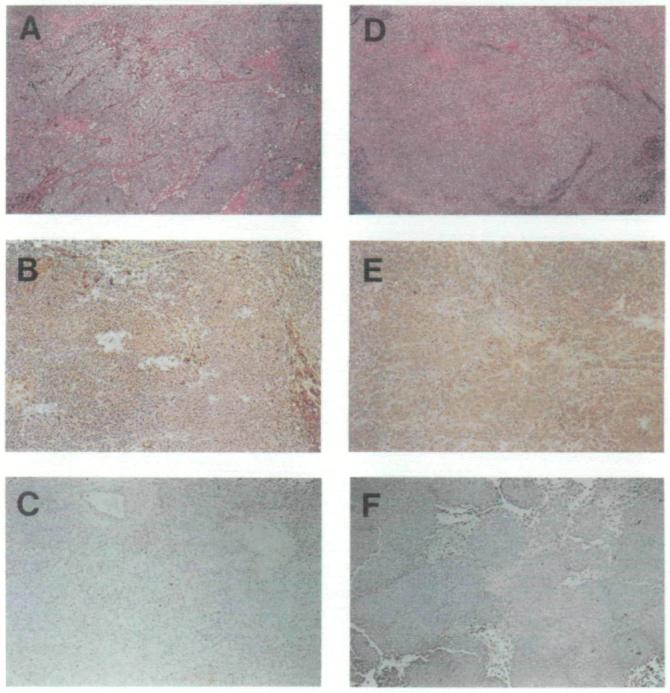
Immunohistochemical staining of sections of melanoma in situ. Sections from tumor specimens described in Table 1 from patient Nos. 1106 (panels A-C) and 1180 (panels D-F) are shown. Immunohistochemical staining in panels B, C, E, and F was done at the same time, under exactly the same conditions with anti-human beta2-microglobulin (β2m) monoclonal antibody followed by immunoperoxidase staining (see “Materials and Methods” section for details). Strongly positive and weakly positive staining for β2m is shown in panels B and E, respectively. Panels C and F are examples of specimens that stain negative for β2m. Hematoxylin–eosin stains are shown in panels A and D, where melanin deposits that appear dark brown can be seen.
Finally, archival tumor sections obtained from patients prior to immunotherapy were available for three patients (Nos. 1106, 1180, and 1259) and were found to be β2m positive (Table 1, Fig. 6, and data not shown). This result was consistent with the hypothesis that the loss of β2m resulted from immunotherapy.
Discussion
The five cell lines described in this article were independently derived from metastatic melanomas obtained from five patients who had undergone immunotherapy. The tumors from which these five cell lines were derived may escape immune recognition by CD8+ T cells via the loss of expression of functional β2m, since replacement of β2m fully restores the antigen-processing capabilities of all of these cell lines (Fig. 4) and can restore the ability of antitumor CD8+ T cells to recognize the cell line in the tumor (1074mel) that was matched for HLA-A2.1. Importantly, the loss of β2m by these tumors was also observed in vivo by immunohistochemical analysis.
The loss of β2m may be an important mechanism for the escape of some melanoma cells from recognition by CD8+ T cells and could explain why some patients experience a recrudescence of tumor growth, often in aggressive and ultimately lethal forms, after excellent responses to T-cell-based therapy. Indeed, none of the patients responded to immunotherapy subsequent to the observed loss of functional β2m, and all of these five patients (Table 1) have died of disease that was noted to be clinically very aggressive. Perhaps most importantly, antitumor CD8+ T cells could not be grown from any of these lesions once there was loss of β2m. However, the sample is too small to enable us to definitively draw the statistical conclusion that patients bearing tumors deficient in functional β2m have poor outcomes compared with patients whose tumors do not suffer such a loss. Furthermore, we do not have enough illustrative examples to conclude that immunotherapy, or T-cell-based immunotherapy, was responsible for the loss of β2m. Many patients whose tumors do express MHC class I fail to respond, and thus tumors are likely to use multiple different mechanisms to evade immune destruction. Nevertheless, the findings presented here could be used as the basis for prospective studies of much larger cohorts of patients receiving immunotherapy that could provide more definitive answers to these questions.
The incidence of the loss of functional μ2m in this study, four (31%) of 13, is strikingly high. However, loss of MHC class I expression in a cohort of patients undergoing immunotherapy with IL-2 has not yet been performed. Abnormalities in MHC class I expression in the absence of immunotherapy may be much higher than has been described in the earlier literature, as indicated by more recent studies examining particular allospecificities (36,47). Furthermore, the loss of β2m has recently been associated with a mutator phenotype, frequently an early event in the development of carcinoma. The molecular basis for the loss of β2m in these patients, particularly as it relates to microsatellite instability, is now being explored (48).
We have previously reported poor expression of MHC class I molecules by human small-cell lung cancers and an experimental mouse sarcoma (14,38,49). In our previous studies of deficient MHC class I expression, all of the small-cell lung cancer cell lines could be induced to express MHC class I by the addition of interferon gamma (IFN γ). We could not induce MHC class I expression with the use of IFN γ in the five human melanoma cell lines reported here (data not shown). Thus, there appears to be histologic specificity in the mechanisms underlying a failure of recognition by CD8+ T cells: Six of six small-cell lung cancers previously studied presented antigen poorly because of a down-regulation of molecules important in antigen processing, while loss of functional β2m was responsible for this effect in five of five melanomas in the present study.
The loss of β2m is a highly effective mechanism of tumor escape from recognition by CD8+ T cells, since MHC class I molecules are not stable in its absence. The loss of expression of both copies of β2m is also an efficient mechanism of escape, since loss of expression of any two other genes involved in antigen processing, such as TAP or proteasome components, would cause a much less profound loss of antigen-presenting capacity (13,50). Loss of β2m may result in increased susceptibility to lysis by natural killer (NK) cells (51-53). However, the aggressive lethality of each of the five β2m-deficient tumors reported in this study suggests either that lysis of these tumors by NK cells is ineffective or that the tumor cells have additional mechanisms of escape from immune recognition by NK cells.
Positive expression of β2m mRNA by northern blot analysis suggested that the deletion of both copies of the gene was not responsible for the loss of functional ν2m and that mutation of one or both copies was probably responsible for the complete loss of functional β2m. The absence of β2m shown by western blot analysis indicated that the epitopes recognized by the MAb were not present to any measurable extent. Since western blot analysis measures the steady-state level of antigen, either the epitope was not produced because of mutation or premature termination or the protein produced had a vastly reduced half-life because of enhanced proteolysis due to a change in sequence or to a misfolding of the protein.
Studies in experimental murine models indicate that loss of β2m expression by germline transmission of a homozygous disruption of the β2m gene is not lethal (54). The β2m-deficient mice express little or no functional class I antigen, and while other T-cell subsets were found in their normal distribution, the mice were found to have no mature CD8+ T cells (29). It is interesting that these mice were still capable of rejecting skin grafts (55) and were able to clear some viral infections (56,57). These findings are consistent with a view of the immune system as highly redundant. Indeed, all of the β2m-loss mutants remained sensitive to lysis by lymphokine-activated killer cells, suggesting that immunotherapy based on non-MHC-restricted effector cells may be more effective in selected patients.
Acknowledgments
We thank Akash Taggarse, Kevin Heller, Jonathan W. Yewdell, and Jack R. Bennink for their helpful advice and assistance.
References
- 1.Topalian SL. Cell transfer therapy: preclinical studies. In: DeVita VT Jr, Hellman S, Rosenberg SA, editors. Biologic therapy of cancer. 2nd ed. Lippincott; Philadelphia: 1995. pp. 467–86. [Google Scholar]
- 2.Peoples GE, Goedegebuure PS, Smith R, Linehan DC, Yoshino I, Everlein TJ. Breast and ovarian cancer-specific cytotoxic T lymphocytes recognize the same HER2/neu-derived peptide. Proc Natl Acad Sci U S A. 1995;92:432–6. doi: 10.1073/pnas.92.2.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoppe RT, Medeiros LJ, Warnke RA, Wood GS. CD8-positive tumor-infiltrating lymphocytes influence the long-term survival of patients with mycosis fungoides. J Am Acad Dermatol. 1995;32:448–53. doi: 10.1016/0190-9622(95)90067-5. [DOI] [PubMed] [Google Scholar]
- 4.Rosenberg SA, Yannelli JR, Yang JC, Topalian SL, Schwartzentruber DF, Weber JS, et al. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2 [see comment citation in Medline] J Natl Cancer Inst. 1994;86:1159–66. doi: 10.1093/jnci/86.15.1159. [DOI] [PubMed] [Google Scholar]
- 5.Ikarashi H, Fujita K, Takakuwa K, Kodama S, Tokunaga A, Takahashi T, et al. Immunomodulation in patients with epithelial ovarian cancer after adoptive transfer of tumor-infiltrating lymphocytes. Cancer Res. 1994;54:190–6. [PubMed] [Google Scholar]
- 6.Peoples GE, Blotnick S, Takahashi K, Freeman MR, Klagsbrun M, Eberlein TJ. T lymphocytes that infiltrate tumors and atherosclerotic plaques produce heparin-binding epidermal growth factor-like growth factor and basic fibroblast growth factor: a potential pathologic role. Proc Natl Acad Sci U S A. 1995;92:6547–51. doi: 10.1073/pnas.92.14.6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schoof DD, Terashima Y, Peoples GE, Goedegebuure PS, Andrews JV. CD4+ T cell clones isolated from human renal cell carcinoma possess the functional characteristics of Th2 helper cells. Cell Immunol. 1993;150:114–23. doi: 10.1006/cimm.1993.1183. [DOI] [PubMed] [Google Scholar]
- 8.Yoshino I, Peoples GE, Goedegebuure PS, Maziarz R, Eberlein TJ. Association of HER2/neu expression with sensitivity to tumor-specific CTL in human ovarian cancer. J Immunol. 1994;152:2393–400. [PubMed] [Google Scholar]
- 9.Qin H, Chen W, Takahashi M, Disis ML, Byrd DR, McCahill L, et al. CD4+ T-cell immunity to mutated ras protein in pancreatic and colon cancer patients. Cancer Res. 1995;55:2984–7. [PubMed] [Google Scholar]
- 10.Fisk B, Blevins TL, Wharton JT, Ioannides CG. Identification of an immunodominant peptide of HER-2/neu protooncogene recognized by ovarian tumor-specific cytotoxic T lymphocyte lines. J Exp Med. 1995;181:2109–17. doi: 10.1084/jem.181.6.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosenberg SA. Cell transfer therapy: clinical applications. In: DeVita VT Jr, Hellman S, Rosenberg SA, editors. Biologic therapy of cancer. 2nd ed. Lippincott; Philadelphia: 1995. pp. 487–506. [Google Scholar]
- 12.Mizoguchi H, O'Shea JJ, Longo DL, Loeffler CM, McVicar DW, Ochoa AC. Alterations in signal transduction molecules in T lymphocytes from tumor-bearing mice [see comment citation in Medline] Science. 1992;258:1795–8. doi: 10.1126/science.1465616. [DOI] [PubMed] [Google Scholar]
- 13.Restifo NP, Kawakami Y, Marincola F, Shamamian P, Taggarse A, Esquivel F, et al. Molecular mechanisms used by tumors to escape immune recognition: immunogenetherapy and the cell biology of major histocompatibility complex class I. J Immunother. 1993;14:182–90. doi: 10.1097/00002371-199310000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Restifo NP, Esquivel F, Kawakami Y, Yewdell JW, Mulé JJ, Rosenberg SA, et al. Identification of human cancers deficient in antigen processing. J Exp Med. l993;177:265–72. doi: 10.1084/jem.177.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cromme FV, Airey J, Heemels MT, Ploegh HL, Keating PJ, Stem PL, et al. Loss of transporter protein, encoded by the TAP-1 gene, is highly correlated with loss of HLA expression in cervical carcinomas. J Exp Med. 1994;179:335–40. doi: 10.1084/jem.179.1.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fenton RG, Longo DL. Genetic instability and tumor cell variation: implications for immunotherapy [editorial; see comment citation in Medline] J Natl Cancer Inst. 1995;87:241–3. doi: 10.1093/jnci/87.4.241. [DOI] [PubMed] [Google Scholar]
- 17.Hansen TH, Carreno BM, Sachs DH. The major histocompatibility complex. In: Paul W, editor. Fundamental immunology. 3rd ed. Raven Press; New York: 1993. pp. 577–628. [Google Scholar]
- 18.Neumann H, Cavalie A, Jenne DE, Wekerle H. Induction of MHC class I genes in neurons. Science. 1995;269:549–52. doi: 10.1126/science.7624779. [DOI] [PubMed] [Google Scholar]
- 19.Rammensee HG, Falk K, Rotzschke O. MHC molecules as peptide receptors. Curr Opin Immunol. 1993;5:35–44. doi: 10.1016/0952-7915(93)90078-7. [DOI] [PubMed] [Google Scholar]
- 20.Brown JH, Jardetzky TS, Gorga JC, Stem JL, Urban RG, Strominger JL, et al. Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1 [see comment citation in Medline] Nature. 1993;364:33–9. doi: 10.1038/364033a0. [DOI] [PubMed] [Google Scholar]
- 21.Bjorkman PJ, Saper MA, Samraoui B, Bennett WS, Strominger JL, Wiley DC. Structure of the human class I histocompatibility antigen, HLA-A2. Nature. 1987;329:506–12. doi: 10.1038/329506a0. [DOI] [PubMed] [Google Scholar]
- 22.Bjorkman PJ, Saper MA, Samraoui B, Bennett WS, Strominger JL, Wiley DC. The foreign antigen binding site and T cell recognition regions of class I histocompatibility antigens. Nature. 1987;329:512–8. doi: 10.1038/329512a0. [DOI] [PubMed] [Google Scholar]
- 23.Wilson IA, Fremont DH. Structural analysis of MHC class I molecules with bound peptide antigens. Semin Immunol. 1993;5:75–80. doi: 10.1006/smim.1993.1011. [DOI] [PubMed] [Google Scholar]
- 24.Stura EA, Matsumura M, Fremont DH, Saito Y, Peterson PA, Wilson IA. Crystallization of murine major histocompatibility complex class I H-2Kb with single peptides. J Mol Biol. 1992;228:975–82. doi: 10.1016/0022-2836(92)90881-j. [DOI] [PubMed] [Google Scholar]
- 25.Matsumura M, Fremont DH, Peterson PA, Wilson IA. Emerging principles for the recognition of peptide antigens by MHC class I molecules [see comment citation in Medline] Science. 1992;257:927–34. doi: 10.1126/science.1323878. [DOI] [PubMed] [Google Scholar]
- 26.Fremont DH, Matsumura M, Stura EA, Peterson PA, Wilson IA. Crystal structures of two viral peptides in complex with murine MHC class I H-2Kb [see comment citation in Medline] Science. 1992;257:919–27. doi: 10.1126/science.1323877. [DOI] [PubMed] [Google Scholar]
- 27.Zugel U, Schoel B, Kaufmann SH. Beta 2-microglobulin independent presentation of exogenously added foreign peptide and endogenous self-epitope by MHC class I alpha-chain to a cross-reactive CD8+ CTL clone. J Immunol. 1994;153:4070–80. [PubMed] [Google Scholar]
- 28.Sumida T, Furukawa M, Sakamoto A, Namekawa T, Maeda T, Zijlstra M, et al. Prevention of insulitis and diabetes in beta 2-microglobulin-deficient non-obese diabetic mice. Int Immunol. 1994;6:1445–9. doi: 10.1093/intimm/6.9.1445. [DOI] [PubMed] [Google Scholar]
- 29.Zijlstra M, Bix M, Simister NE, Loring JM, Raulet DH, Jaenisch R. β2-microglobulin deficient mice lack CD4− CD8+ cytolytic T cells [see comment citation in Medline] Nature. 1990;344:742–6. doi: 10.1038/344742a0. [DOI] [PubMed] [Google Scholar]
- 30.Sugita M, Brenner MB. An unstable beta 2-microglobulin: major histocompatibiity complex class I heavy chain intermediate dissociates from calnexin and then is stabilized by binding peptide. J Exp Med. 1994;180:2163–71. doi: 10.1084/jem.180.6.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bicknell DC, Rowan A, Bodmer WF. Beta 2-microglobulin gene mutations: a study of established colorectal cell lines and fresh tumors. Proc Natl Acad Sci U S A. 1994;91:4751–6. doi: 10.1073/pnas.91.11.4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bodmer WF, Browning MJ, Krausa P, Rowan A, Bicknell DC, Bodmer JG. Tumor escape from immune response by variation in HLA expression and other mechanisms. Ann N Y Acad Sci. 1993;690:42–9. doi: 10.1111/j.1749-6632.1993.tb43994.x. [DOI] [PubMed] [Google Scholar]
- 33.Momburg F, Koch S. Selective loss of beta 2-microglobulin mRNA in human colon carcinoma. J Exp Med. 1989;169:309–14. doi: 10.1084/jem.169.1.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Z, Cao Y, Albino AP, Zeff RA, Houghton A, Ferrone S. Lack of HLA class I antigen expression by melanoma cells SK-MEL-33 caused by a reading frameshift in beta 2-microglobulin messenger RNA. J Clin Invest. 1993;91:684–92. doi: 10.1172/JCI116249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.D'Urso CM, Wang ZG, Cao Y, Tatake R, Zeff RA, Ferrone S. Lack of HLA class I antigen expression by cultured melanoma cells FO-1 due to a defect in β2m gene expression. J Clin Invest. 1991;87:284–92. doi: 10.1172/JCI114984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marincola FM, Shamamian P, Alexander RB, Gnarra JR, Turetskaya RL, Nedospasov SA, et al. Loss of HLA haplotype and B locus down-regulation in melanoma cells lines. J Immunol. 1994;153:1225–37. [PubMed] [Google Scholar]
- 37.Rosenberg SA. The gene therapy of cancer. Prev Med. 1994;23:624–6. doi: 10.1006/pmed.1994.1102. [DOI] [PubMed] [Google Scholar]
- 38.Topalian SL, Solomon D, Rosenberg SA. Tumor-specific cytolysis by lymphocytes infiltrating human melanomas. J Immunol. 1989;142:3714–25. [PubMed] [Google Scholar]
- 39.Restifo NP, Esquivel F, Asher AL, Stotter H, Barth RJ, Bennink JR, et al. Defective presentation of endogenous antigens by a murine sarcoma. Implications for the failure of an anti-tumor immune response. J Immunol. 1991;147:1453–9. [PMC free article] [PubMed] [Google Scholar]
- 40.Cole DJ, Taubenberger JK, Pockaj BA, Yannelli JR, Carter C, Carrasquillo J, et al. Histopathological analysis of metastatic melanoma deposits in patients receiving adoptive immunotherapy with tumor-infiltrating lymphocytes. Cancer Immunol Immunother. 1994;38:299–303. doi: 10.1007/BF01525507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Topalian SL, Rivoltini L, Mancini M, Ng J, Hartzman RJ, Rosenberg SA. Melanoma-specific CD4+ T lymphocytes recognize human melanoma antigens processed and presented by Epstein-Barr virus-transformed B cells. Int J Cancer. 1994;58:69–79. doi: 10.1002/ijc.2910580113. [DOI] [PubMed] [Google Scholar]
- 42.O'Neil BH, Kawakami Y, Restifo NP, Bennink JR, Yewdell JW, Rosenberg SA. Detection of shared MHC-restricted human melanoma antigens after vaccinia virus-mediated transduction of genes coding for HLA. J Immunol. 1993;151:1410–8. [PMC free article] [PubMed] [Google Scholar]
- 43.Hammond SA, Bollinger RC, Tobery TW, Silliciano RF. Transporter-independent processing of HIV-1 envelope protein for recognition by CD8+ T cells. Nature. 1993;364:158–61. doi: 10.1038/364158a0. [DOI] [PubMed] [Google Scholar]
- 44.Henderson RA, Michel H, Sakaguchi K, Shabanowitz J, Appella E, Hunt DF, et al. HLA-A2.1-associated peptides from a mutant cell line: a second pathway of antigen presentation [see comment citations in Medline] Science. 1992;255:1264–6. doi: 10.1126/science.1546329. [DOI] [PubMed] [Google Scholar]
- 45.Townsend A, Bastin J, Gould K, Brownlee G, Andrew M, Coupar B, et al. Defective presentation to class I-restricted cytotoxic T lymphocytes in vaccinia-infected cells is overcome by enhanced degradation of antigen. J Exp Med. 1988;168:1211–24. doi: 10.1084/jem.168.4.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kawakami Y, Eliyahu S, Sakaguchi K, Robbins PF, Rivoltini L, Yannelli JR, et al. Identification of the immunodominant peptides of the MART-1 human melanoma antigen recognized by the majority of HLA-A2-restricted tumor infiltrating lymphocytes. J Exp Med. 1994;180:347–52. doi: 10.1084/jem.180.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kageshita T, Wang Z, Calorini L, Yoshii A, Kimura T, Ono T, et al. Selective loss of human leukocyte class I allospecificities and staining of melanoma cells by monoclonal antibodies recognizing monomorphic determinants of class I human leukocyte antigens. Cancer Res. 1993;53:3349–54. [PubMed] [Google Scholar]
- 48.Branch P, Bicknell DC, Rowan A, Bodmer WF, Karran P. Immune surveillance in colorectal carcinoma [letter] Nat Genet. 1995;9:231–2. doi: 10.1038/ng0395-231. [DOI] [PubMed] [Google Scholar]
- 49.Restifo NP, Spiess PJ, Karp SE, Mulé JJ, Rosenberg SA. A nonimmunogenic sarcoma transduced with the cDNA for interferon gamma elicits CD8+ T cells against the wild-type tumor: correlation with antigen presentation capability. J Exp Med. 1992;175:1423–31. doi: 10.1084/jem.175.6.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Esquivel F, Yewdell J, Bennink J. RMA/S cells present endogenously synthesized cytosolic proteins to class I-restricted cytotoxic T lymphocytes. J Exp Med. 1992;175:163–8. doi: 10.1084/jem.175.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maio M, Altomonte M, Tatake R, Zeff RA, Ferrone S. Reduction in susceptibility to natural killer cell-mediated lysis of human FO-1 melanoma cells after induction of HLA class I antigen expression by transfection with β2m gene. J Clin Invest. 1991;88:282–9. doi: 10.1172/JCI115289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Karre K. Express yourself or die: peptides, MHC molecules, and NK cells [see comment citation in Medline] Science. 1995;267:978–9. doi: 10.1126/science.7863341. [DOI] [PubMed] [Google Scholar]
- 53.Malnati MS, Peruzzi M, Parker KC, Biddison WE, Ciccone E, Moretta A, et al. Peptide specificity in the recognition of MHC class I by natural killer cell clones [see comment citation in Medline] Science. 1995;267:1016–8. doi: 10.1126/science.7863326. [DOI] [PubMed] [Google Scholar]
- 54.Zijlstra M, Li E, Sajjadi F, Subramani S, Jaenisch R. Germ-line transmission of a distrupted β2-microglobulin gene produced by homologous recombination in embryonic stem cells. Nature. 1989;342:435–8. doi: 10.1038/342435a0. [DOI] [PubMed] [Google Scholar]
- 55.Zijlstra M, Auchincloss H, Jr, Loring JM, Chase CM, Russell PS, Jaenisch R. Skin graft rejection by beta 2-microglobulin-deficient mice. J Exp Med. 1992;175:885–93. doi: 10.1084/jem.175.4.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eichelberger M, Allan W, Zijlstra M, Jaenisch R, Doherty PC. Clearance of influenza virus respiratory infection in mice lacking class I major histocompatibihty complex-restricted CD8+ T cells. J Exp Med. 1991;174:875–80. doi: 10.1084/jem.174.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hou S, Doherty PC, Zijlstra M, Jaenisch R, Katz JM. Delayed clearance of Sendai virus in mice lacking class I MHC-restricted CD8+ T cells. J Immunol. 1992;149:1319–25. [PubMed] [Google Scholar]