

Abstract
Nicotinamide, the amide form of niacin (vitamin B3), is the precursor for the coenzyme β-nicotinamide adenine dinucleotide (NAD+) and plays a significant role during the enhancement of cell survival as well as cell longevity. Yet, these abilities of nicotinamide appear to be diametrically opposed. Here we describe the development of nicotinamide as a novel agent that is critical for modulating cellular metabolism, plasticity, longevity, and inflammatory microglial function as well as for influencing cellular life span. The capacity of nicotinamide to govern not only intrinsic cellular integrity, but also extrinsic cellular inflammation rests with the modulation of a host of cellular targets that involve mitochondrial membrane potential, poly(ADP-ribose) polymerase, protein kinase B (Akt), Forkhead transcription factors, Bad, caspases, and microglial activation. Further knowledge acquired in regards to the ability of nicotinamide to foster cellular survival and regulate cellular lifespan should significantly promote the development of therapies against a host of disorders, such as aging, Alzheimer’s disease, diabetes, cerebral ischemia, Parkinson’s disease, and cancer.
Keywords: Akt, Alzheimer’s disease, apoptosis, caspases, diabetes, erythropoietin, Huntington’s disease, microglia, NAD+, Parkinson’s disease, stroke, vitamin B3
NICOTINAMIDE, NAD+ PRECURSOR, AND CELLULAR METABOLISM
As the amide form of niacin or vitamin B3, nicotinamide is the precursor for the coenzyme β-nicotinamide adenine dinucleotide (NAD+) and is essential for the synthesis of nicotinamide adenine dinucleotide phosphate (NADP+) [1, 2]. Nicotinamide and nicotinic acid can be obtained either through synthesis in the body or through a dietary source [3]. The predominant form of niacin in dietary plant sources is nicotinic acid that is rapidly absorbed through the gastrointestinal epithelium. Nicotinamide is formed through the conversion of nicotinic acid in the liver or through the hydrolysis of NAD+. Once nicotinamide is obtained in the body, it is utilized to synthesize NAD+ [4].
Nicotinamide through NAD+ plays a critical physiological role in cellular metabolism and can be directly utilized by cells to synthesize NAD+ [4]. Nicotinamide also participates in energy metabolism through the tricarboxylic acid cycle by utilizing NAD+ in the mitochondrial respiratory electron transport chain for the production of ATP, DNA synthesis, and DNA repair [5–7]. Furthermore, nicotinamide can significantly increase NAD+ levels in vulnerable regions of the ischemic brain, suggesting that nicotinamide may offer cytoprotection of injured tissue through the maintenance of NAD+ levels [8]. Administration of nicotinamide can significantly improved glucose utilization, prevent excessive lactate production and improve electrophysiologic capacity in ischemic animal models [9]. During axonal degeneration, nicotinamide also may trigger neuronal protection through NAD+-dependent mechanisms [10].
The maintenance of cellular energy reserves and cellular function by nicotinamide are closely tied to the function of mitochondria. Oxidative stress can trigger the opening of the mitochondrial membrane permeability transition pore [11–14] and lead to a significant loss of mitochondrial NAD+ stores and subsequent apoptotic cell injury [15]. For example, in animal models of Parkinson’s disease that use 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine to elicit central nigrostriatal dopamine neurotoxicity, subsequent depletion of NAD+ and adenine triphosphate have been associated with neuronal loss [16]. Restoration of NAD+ content in mitochondria with liposomal NAD+ prevents neuronal injury [17].
Cellular function in the presence of nicotinamide also relies upon the maintenance of mitochondrial membrane potential. Mitochondria are a significant source of superoxide radicals that are associated with oxidative stress [18]. Blockade of the electron transfer chain at the flavin mononucleotide group of complex I (NADPH ubiquinone oxidoreductase) or at the ubiquinone site of complex III (ubiquinone-cytochrome c reductase) results in the active generation of free radicals which can impair mitochondrial electron transport and enhance free radical production [19, 20]. Furthermore, mutations in the mitochondrial genome have been associated with the potential development of a host of disorders, such as hypertension, hypercholesterolemia, and hypomagnesemia [21]. Loss of mitochondrial membrane potential through the opening of the mitochondrial permeability transition pore represents a significant determinant for cell injury and the subsequent induction of the apoptotic cascade [12, 13, 22].
Nicotinamide can preserve mitochondrial NAD-linked respiration [23] and block the depolarization of the mitochondrial membrane [2, 24, 25] (Fig. 1). Nicotinamide appears to function directly at the level of mitochondrial membrane pore formation to prevent the release of cytochrome c [26]. Pretreatment of cells with either nicotinamide alone or in combination with the mitochondrial permeability transition pore inhibitor cyclosporin A [27, 28] prior to an injury paradigm can equally prevent mitochondrial membrane depolarization. Other studies that involve oxygen-glucose deprivation demonstrate that nicotinamide maintains mitochondrial membrane potential and prevents the release of cytochrome c [29]. Interestingly, nicotinamide appears to act directly at the level of mitochondrial membrane pore formation to prevent cytochrome c release. Nicotinamide can prevent the chemical induction of mitochondrial membrane depolarization during exposure to either tert-butylhydorperoxide or atractyloside [29].
Fig. 1.
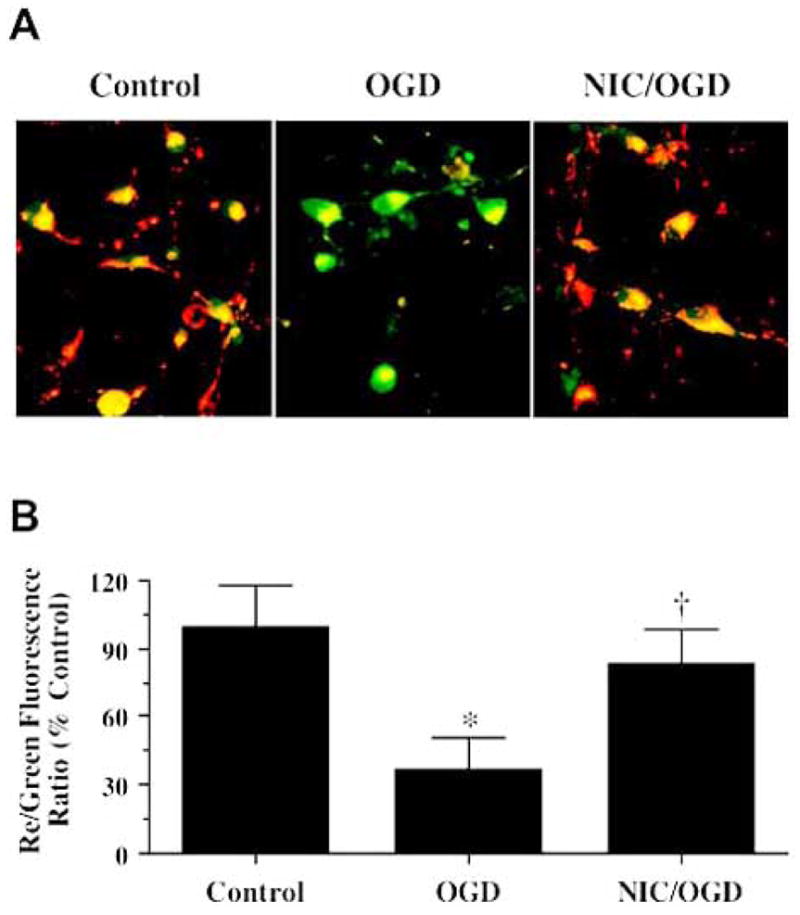
Nicotinamide (NIC) prevents the loss of mitochondrial membrane potential during oxygen glucose deprivation (OGD). (A) Representative pictures demonstrate that exposure to a 3 hour period of OGD produced a significant decrease in the red/green fluorescence intensity ratio in cultured rat hippocampal neurons using a cationic membrane potential indicator JC-1 within 3 hours when compared with untreated control cultures, suggesting that OGD results in mitochondrial membrane depolarization. Application of NIC (12.5 mM) 1 hour prior to OGD exposure significantly increased the red/green fluorescence intensity of neurons, indicating that mitochondrial membrane potential was restored. (B) The relative ratio of red/green fluorescent intensity of mitochondrial staining in both untreated (control) neurons and neurons exposed to OGD or NIC (12.5 mM) plus OGD 3 hour following the initial insult was measured in 4 independent experiments with analysis performed using the public domain NIH Image program (developed at the U.S. National Institutes of Health and available on the Internet at http://rsb.info.nih.gov/nih-image/) (Control vs. OGD, *p<0.01; OGD vs. NIC/OGD, †p<0.01).
Reactive oxygen species also have been postulated as a potential mechanism for the induction of acidosis-induced cellular toxicity [30] and subsequent mitochondrial failure [31]. In the nervous system, toxic insults, such as hypercapnia [32], hypoxia [33], glutamate toxicity [34], and free radicals [35–37] can result in the disturbance of intracellular pH. In addition, modulation of intracellular pH is physiologically relevant for endonuclease activities during apoptosis [35, 36, 38]. Yet, nicotinamide cannot prevent cellular injury during intracellular acidification paradigms [13]. As a result, nicotinamide may use other mechanisms to preserve cellular energy metabolism that could depend upon glycolytic metabolism with glyceraldehydes-3-phosphate dehydrogenase [39].
Furthermore, there exist additional pathways that nicotinamide may use to maintain cellular metabolic homeostasis through the maintenance of mitochondrial membrane potential. Nicotinamide can phosphorylate Bad [26] to prevent mitochondrial membrane depolarization and subsequent cytochrome c release. In addition, nicotinamide may inhibit the assembly of the mitochondrial permeability transition pore complex similar to the action of cyclosporin A [40] as well as stabilize cellular energy metabolism since the maintenance of mitochondrial membrane potential is an ATP facilitated process [41]. Another pathway that will be discussed later in more detail involves nicotinamide increasing the activation of Akt [26], since Akt is closely linked to the maintenance of mitochondrial membrane potential [42, 43].
NICOTINAMIDE, POLY (ADP-RIBOSE) POLYMERASE (PARP), AND CELLULAR FUNCTION
Nicotinamide has an intimate relationship with poly (ADP-ribose) polymerase (PARP). PARP is a nuclear protein that binds to DNA strand breaks and cleaves NAD+ into nicotinamide and ADP-ribose [44]. During DNA repair, ADP-ribose is polymerized onto nuclear proteins that include histones and transcription factors at DNA strand breaks [45]. Nicotinamide may provide cellular protection through the maintenance of PARP integrity and the preservation of cellular energy reserves. PARP catalyses the synthesis of poly (ADP-ribose) from its substrate NAD+, which stimulates the process of DNA repair [46]. Nicotinamide concentrations of at least 1 mM have been shown to provide sufficient stores of NAD+ for PARP activation [47]. Nicotinamide can prevent PARP degradation and allow for DNA repair through the direct inhibition of caspase 3 – like activity [13, 24, 26]. In contrast, elevated concentrations of nicotinamide can lead to PARP degradation and apoptotic injury [48].
Yet, excessive PARP activity may be detrimental to cellular function. Augmented PARP activation leads to a rapid depletion of its sole substrate NAD+ and lowered ATP production. As a cell consumes ATP in an effort to replenish NAD+, this results in a cellular energy crisis that precipitates cell death. For example, reactive oxygen injury in pulmonary artery endothelial cells can result in DNA strand breaks followed by the loss of membrane integrity with enhanced PARP activity and the depletion of NAD+ [49]. Exposure to bleomycin, a DNA-cleaving antitumor antibiotic, causes sustained PARP activation and NAD+ depletion and subsequent tissue injury [50]. In experimental diabetic rats, NAD+ and ATP levels are lowered with increased DNA breakage and malondialdehyde production following enhanced PARP activity. A significant increase in PARP activity also can promote NF-κB-driven transcription and microglial activation leading to the overexpression of pro-inflammatory and adhesion molecules [51].
Given the potential detrimental effects of excessive PARP activity, it may be necessary for nicotinamide to reduce PARP activity to block apoptotic injury [52, 53]. Several studies confirm that a reduction in PARP activity can enhance cell survival, such as during injury paradigms with photoreceptor cells [54], during homocysteine toxicity [55], during cerebral ischemia [56], or during free radical injury [57, 58]. Prevention of NAD+ depletion during enhanced PARP activity also has been demonstrated to prevent cellular lysis during oxidative stress [49].
In relation to clinical disorders, PARP activity has been associated with cell injury during neurodegenerative disorders. During acute neurodegenerative injury, increased PARP activity with subsequent consumption of NAD+ can exacerbate acute cerebral ischemia [59]. Even during periods of free radical injury in human blood lymphocytes, nicotinamide may block necrotic death through pathways that limit excessive PARP activity that can consume essential NAD+ stores [60]. Under some circumstances such as diabetic neuropathy, nicotinamide has the capacity to not only significantly reduce PARP activity, but also partially restore the NAD+ and ATP contents [61].
In a similar vein, inhibition of PARP activity by nicotinamide may be critical for disorders such as Alzheimer’s disease. Alzheimer’s disease has two pathologic hallmarks that consist of extracellular plaques of amyloid-β peptide aggregates and intracellular neurofibrillary tangles composed of hyperphosphorylated microtubular protein tau [30, 62]. The β-amyloid deposition that constitutes the plaques is composed of a 39-42 amino acid peptide (Aβ), which is the proteolytic product of the amyloid precursor protein (APP) [30]. Overexpression of APP can increase Aβ secretion which can form insoluble amyloid aggregates contributing to the development of Alzheimer’s disease [63, 64]. A limited pilot study has suggested that administration of nicotinamide adenine dinucleotide (NADH) in patients with Alzheimer type dementia may show improvement in their cognitive function [65]. More recently, sufficient dietary niacin intake examined in a series of patients aged 65 and older has been implicated as a potential factor to protect against the development or progression of Alzheimer’s disease [66]. Although the mechanisms responsible for these protective effects of NADH and niacin are not well established, it has been shown that in patients with Alzheimer’s disease, both PARP and poly(ADP-ribose) is present in the frontal and temporal cortex more frequently than in controls, suggesting that increased levels of functional PARP enzyme are present to result in a significant consumption of NAD+ stores [67]. In addition, amyloid toxicity may require increased PARP activity [68]. It is conceivable that modulation of niacin or NADPH can sufficiently control PARP activity to slow or prevent the progression of Alzheimer’s disease and cognitive loss.
It is worth mentioning that unexpected effects or toxicity with nicotinamide also may result through mechanisms that can be exclusive of PARP activity. Some studies have observed that toxicity with increased concentrations of nicotinamide may be a result of a secondary metabolic acidosis generated by nicotinamide [69]. Nicotinamide also may alter the metabolism of other exogenously administered agents through inhibition of P450 enzymes [70]. Exposure to elevated concentrations of nicotinamide can inhibit the function of rat pancreatic beta-cells, decrease DNA content of adult rat pancreatic islet cells, and induce cell death in fetal rat pancreatic islet cells [71]. Nicotinamide also has been shown to result in the release of choline that may precipitate neuronal injury when levels of choline become excessive [72]. Furthermore, excessive exposure to nicotinamide may lead to autointoxication and precipitate disorders such as Parkinson’s disease [73].
NICOTINAMIDE, SIRTUINS, AND CELLULAR LONGEVITY
Recently, nicotinamide has been reported to have diverse roles during cell biology that involve potential lifespan reduction in addition to modulating cellular energy reserves and metabolism [1, 74]. As a result, one must approach therapy with nicotinamide with caution, since nicotinamide may have detrimental effects with cellular aging that appear to be concentration dependent [26]. Nicotinamide offers protection in millimole concentrations against free radicals [13], anoxia [75], and oxygen glucose deprivation [29]. Nicotinamide also antagonizes cell injury during free radical generating toxins such as tertiary butylhydroperoxide [76]. Yet in relation to cell longevity, nicotinamide can function as an inhibitor of sirtuins in concentrations that range from 50–100 μM [74]. Increased longevity in yeast has been shown to be dependent upon silent information regulator 2 (Sir2) protein expression and is associated with nicotinamide and pyrazinamidase/nicotinamidase 1 (PNC1), an enzyme that deaminates nicotinamide to convert nicotinamide into nicotinic acid. In the absence of nicotinamide, Sir2 is activated and PNC1 expression is increased to lead to yeast lifespan extension during calorie restriction.
The Sir2 gene belongs to a family of genes which is a highly conserved group in the genomes of organisms ranging from archaebacteria to eukaryotes [77, 78]. Nicotinamide can block cellular Sir2 by intercepting an ADP-ribosyl-enzyme-acetyl peptide intermediate with the regeneration of NAD+ (transglycosidation) [79]. Physiological concentrations of nicotinamide noncompetitively inhibit Sir2, suggesting that nicotinamide is a physiologically relevant regulator of Sir2 enzymes [80].
Interestingly, it has been postulated that sirtuins may prevent nicotinamide and PARP to foster DNA repair by altering the accessibility of DNA damaged sites for repair enzymes. Homologues of the yeast gene Sir2 in mammalian cells code type III histone deacetylases that are dependent upon NAD+ and inhibited by nicotinamide. In yeast cells, Sir2 prevents transcriptional activity and DNA double strand break repair. It is possible that a similar mechanism exists in mammalian cells that will alter chromatin structure and prevent accessible sites for DNA repair. As a result, activation of PARP at the sites of DNA strand breaks may result in a local increase in nicotinamide concentration to block sirtuin activity and allow DNA repair to proceed [81, 82].
Given the intimate and inverse relationship between nicotinamide and sirtuins in controlling cell longevity and survival, alternative approaches for the treatment of neurodegenerative diseases should be considered that may involve the prevention of intracellular nicotinamide accumulation. During nicotinamide depletion, Sir2 is activated and employs PNC1 to regulate cell longevity. Increased expression of PNC1 has been found to be necessary for lifespan extension during calorie restriction in Saccharomyces cerevisiae [83] as well as cytoprotective in neuronal cell lines [84]. PNC1 can stimulate Sir2 histone deactylase activity by preventing the accumulation of nicotinamide through its conversion to nicotinic acid in the NAD+ salvage pathway. Overexpression of PNC1 has been demonstrated to suppress the inhibitory effect of exogenous nicotinamide on decreasing life span and promoting transcriptional repression of Sir2. As a result, PNC1 can positively regulate Sir2-mediated silencing and longevity by preventing the accumulation of intracellular nicotinamide [85]. Although some emerging work suggests that sirtuins may offer protection against neurodegenerative disorders, such as Huntington’s disease [86], further work is clearly required to link these studies to higher organisms and neurodegenerative disorders.
NICOTINAMIDE AND CELLULAR INFLAMMATION
During inflammation of brain tissue, microglial cells can proliferate and become active for the removal of injured as well as non-injured cells [87, 88]. A number of factors can modulate microglial activation. Secreted factors by either apoptotic or phagocytic cells, such as milk fat globule-EGF-factor 8 [89], fractalkine [90], and lipid lysophosphosphatidylcholine [91] have been shown to assist with the phagocytic removal of injured cells. However, the translocation of membrane phosphatidylserine (PS) residues from the inner cellular membrane to the outer surface is a necessary feature for the removal of apoptotic cells [92–94]. The phospholipids of the plasma membrane are normally in an asymmetric pattern with the outer leaflet of the plasma membrane consisting primarily of choline-containing lipids, such as phosphatidylcholine and sphingomyelin, and the inner leaflets consisting of aminophospholipids that include phosphatidylethanolamine and PS. The loss of membrane phospholipid asymmetry leads to the externalization of membrane PS residues and serves to identify cells for phagocytosis [2, 14, 95, 96].
The expression of the phosphatidylserine receptor (PSR) on microglia during oxidative stress also functions in concert with cellular membrane PS externalization to activate microglia [97]. Treatment with an anti-PSR neutralizing antibody in microglia prevents this microglial activation [43, 98] and application of PS directly results in microglial activation that can be blocked by a PSR neutralizing antibody [14, 98], suggesting that both PS exposure in target cells and PSR expression in microglia are necessary for microglial recognition of apoptotic cells. Recognition of cellular membrane PS by the PS-specific receptors on microglia may require cofactors, such as Gas6 [99] or other agents, such as integrin and lectin [100].
Yet, microglia cause cellular damage in disease entities through the generation of reactive oxygen species [101, 102] and through the production of cytokines [103, 104]. In neurodegenerative disorders, microglial activation has been reported in regions of the nervous system that are specific for diseases such as Alzheimer’s and Huntington’s disease [105, 106]. Microglial cells also co-localize with the perivascular deposits of Aβ and microglial activation correlates with the development of amyloid plaques [107]. Ultrastructural three-dimensional reconstruction of human amyloid plaques in different stages of development illustrates that the number of microglia parallels a progressive increase in fibrillar deposition and the size of fibrillar plaque [108]. During ischemic injury to cells, activation of microglia can parallel the induction of cellular apoptosis and correlate well with the severity of the ischemic insult [14, 109]. Microglia promote the production of pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α) and interleukin-1β, free radicals such as nitric oxide and superoxide [102], and fatty acid metabolites such as eicosanoids that can precipitate cell death [110]. TNF-α production by microglia may be linked to neurodegeneration by increasing the sensitivity of neurons to free radical exposure [111].
The efficacy of several cytoprotective agents is closely aligned with the ability to modulate microglial inflammatory activation. For example, erythropoietin (EPO) can prevent microglial cell activation and proliferation to block phagocytosis of injured cells through pathways that involve cellular membrane PS exposure [112, 113], Akt [109], and the regulation of caspases [98, 114]. EPO also can modulate cellular inflammation by inhibiting several pro-inflammatory cytokines, such as IL-6, TNF-α, and monocyte chemoattractant protein 1 [115, 116].
Nicotinamide is no exception to this rule. Nicotinamide can prevent inflammatory cell demise through the maintenance of membrane asymmetry and the inhibition of cytokine release. Nicotinamide can prevent cellular membrane PS externalization during a variety of insults that involve anoxia, free radical exposure, and oxygen-glucose deprivation [2, 13, 24, 25]. Nicotinamide may regulate membrane PS exposure and microglial activation through activation of Akt [29]. The protein Akt can modulate the spatial regulation of actin assembly [117], suggesting a relationship between Akt and the coordination of cytoskeletal organization [118]. In addition, Akt appears to be a necessary component for the modulation of membrane PS externalization [109] and prevent microglial activation [14]. Activation of Akt can prevent membrane PS exposure on injured cells and block the activation of microglia that are exposed to media taken from cells that overexpress active, phosphorylated Akt during cellular injury [14, 43]. In addition to targeting the activity of membrane PS exposure and microglial activation, nicotinamide inhibits several pro-inflammatory cytokines, such as interleukin-1β, interleukin-6, interleukin-8, tissue factor, and TNF-α [119–122]. Nicotinamide also can alter major histocompatibility complexes [123], inhibit intracellular adhesion molecule expression [124], and modulate the production of TNF in vascular cells [123]. In systems that involve hepatic cells, nicotinamide can reduce expression of several cytokines that include transforming growth factor (TGF) β2, IL-1β, TNF-α, and macrophage chemotactic protein-1 [125]. Nicotinamide also may be a viable option for the treatment of inflammatory mechanisms that lead to arthritis, such as the inhibition of collagen II expression [126]. However translation of these experimental studies to clinical efficacy appears to require further work, since investigations that examined the ability of oral nicotinamide administration to reduce cytokine production following endotoxin challenge in healthy volunteers did not demonstrate a significant effect upon serum cytokine levels [127].
NICOTINAMIDE, PROGENITOR CELLS, AND CELLULAR PROLIFERATION
Nicotinamide influences several cellular pathways in a number of cell systems. For example, nicotinamide can function as an anxiolytic [128], increase brain choline levels [72], and act as an endogenous ligand for benzodiazepine receptors [129]. Additional work has indicated that nicotinamide can increase cell tolerance against vascular thermal injury and increase capillary density [130]. Yet what may be more significant is the capacity of nicotinamide to foster cellular proliferation and Development, Nicotinamide can promote the maturation of fetal cells [131], the proliferation and differentiation of embryonic stem cells to yield insulin-producing cells [132], assist with trophic regulation to generate insulin secreting cells from adult pancreatic exocrine cells [133], and enhance an adaptive response to physical and chemical agents in mouse bone marrow cells that consists of an error-free DNA repair mechanism [134]. Nicotinamide also appears to be a critical requirement to promote the effective biological function of some embryonic stem cells. In one study, only nicotinamide treated embryonic stem cells that replenished dopamine reserves in animals receiving 6-hydroxdopamine could correct rotational behavior [135]. In addition, nicotinamide has been shown to positively foster the long-term survival and the subsequent generation of neuronal phenotypes from embryonic cells in some avian cell systems [136].
NICOTINAMIDE AND CELLULAR LIFE
The ability of nicotinamide to function as an essential cellular nutrient as well as assist and guide the development of progenitor cells appears to provide nicotinamide with essential qualities also necessary to protect cells against toxic insults (Table 1). Nicotinamide offers significant protection for neurons and can provide protection against free radical injury [13], anoxia [75], and oxygen-glucose deprivation [29, 137]. In cortical neurons, nicotinamide antagonizes cell injury during free radical generating toxins such as tertiary butylhydroperoxide [76]. Nicotinamide also can protect both rod and cone photoreceptor cells against N-methyl-N-nitrosourea toxicity [138, 139] as well as against glycation end products in all layers of the retina [140]. In animal studies, nicotinamide prevents neuronal degeneration against trauma [141], axonal degeneration [10], oxidative stress [13, 24, 26, 29, 39, 75], cerebral ischemia [142–144], and spinal cord injury [145, 146]. In one analysis of the literature, nicotinamide appeared to be more effective when administered intravenously during transient cerebral ischemia models that were absent of comorbidities [147].
Table 1.
Examples of Cytoprotection by Nicotinamide During Cell Injury
| Injury Model | Nicotinamide | Outcome | Ref. |
|---|---|---|---|
| Cerebral Ischemia | |||
| MCAO in Wistar rats | 500 mg/kg, ip, within 6 h after MCAO | Cerebral infarct volume decreased; neurological deficit scores decreased | [142] |
| MCAO in SHR or Fischer 344 rats | 500~750 mg/kg, iv, at 2 h after MCAO | Cerebral infarct volume decreased | [144] |
| MCAO in rats | 500 mg/kg, ip, with combination therapy | Cerebral infarct volume, edema, and neurological deficit scores decreased | [143] |
| Spinal cord injury | |||
| Injection of QUIS in Long-Evans rats | 500 mg/kg, ip, 30 min after QUIS injection | Grey matter damage reduced | [145] |
| Oxidative Stress | |||
| Glyoxal incubation (300 –800 μM) in rat retina for AGE accumulation | 10 mM simultaneous incubation with glyoxal | Apoptotic injury reduced in the outer nuclear layer | [140] |
| NO in hippocampal neurons and cerebral microvascular ECs | 12.5 mM, 1 h prior to or 2~ 6 h following NO | DNA fragmentation and PS exposure decreased, mitochondrial membrane potential preserved, caspase inhibition | [13, 24, 75] |
| OGD, anoxia in hippocampal neurons | 12.5 mM, 1 h prior to or 2~ 6 h following injury | Akt activation increased, phosphorylation of Bad and FOXO3a, PARP and FOXO3a integrity maintained, caspase inhibition | [29, 63] |
| OGD in rabbit retina | 10 mM, 35 min prior to OGD | Functional recovery and glucose utilization improved | [9] |
| MNU application in rats and mice | 100~1000 mg/kg, sc, immediately or 1000 mg/kg2~6 h after ip 60 mg/kg MNU | Photoreceptor cell loss decreased | [138, 139] |
| Hydrogen peroxide in rat ventricular myoblasts | 3 mM 10 min prior to oxidant stress | Mitochondrial respiration improved; cell necrosis reduced | [152] |
AGE: advanced glycation end products; EC: endothelial cell; ip: intraperitoneal injection; iv: inravenous injection; MCAO: middle cerebral artery occlusion; MNU: N-methyl-N-nitrosourea; NO: nitric oxide; OGD: oxygen-glucose deprivation; PARP: poly(ADP-ribose) polymerase; PS: phosphatidylserine; sc: subcutaneous injection; QUIS: quisqualic acid.
In regards to vascular cell protection, nicotinamide prevents vascular compromise during endotoxic shock [148]. Nicotinamide also may influence arteriolar dilatation and blood flow [149], although the effects on vascular flow may be tissue specific and pertain primarily to neoplastic disorders [150]. Nicotinamide also can maintain endothelial cell membrane integrity during oxygen radical exposure [2, 24, 75, 151]. Nicotinamide is believed to be responsible for the preservation of endocardial endothelial cell integrity during models of oxidative stress and to assist with left ventricular cardiac function [152, 153].
In disorders that affect both neuronal and vascular cell populations, nicotinamide also proves to be effective in preventing the loss of cellular function. Treatment with nicotinamide can maintain normal or nearly normal fasting blood glucose in animals with streptozotocin-induced diabetes [154, 155]. Yet, prolonged exposure to nicotinamide in some studies lead to impaired β-cell function and reduction in cell growth [71, 156]. Oral nicotinamide (1200mg/m2/day) protects β-cell function and prevents clinical disease in islet-cell antibody-positive first-degree relatives of type-1 diabetes [157]. In addition, treatment with nicotinamide (25mg/kg) in patients with recent onset type-1 diabetes mellitus combined with intensive insulin therapy for up to two years after diagnosis significantly reduced HbA1c levels [158]. Potentially relevant to diabetic patients with renal failure, nicotinamide also has been shown to reduce intestinal absorption of phosphate and prevent the development of hyperphosphatemia and progressive renal dysfunction [159].
Nicotinamide prevents apoptotic injury by impacting cellular pathways that involve DNA fragmentation and membrane PS exposure [2, 13, 24, 26, 29, 75]. Apoptosis, also termed programmed cell death, is considered to be an active component of cell death that contributes to the destruction of cells in multiple systems. Apoptosis contributes to a variety of disease states in the nervous system such as stroke, Alzheimer’s disease, and trauma [160–164]. Exclusive of the nervous system, apoptosis leads to cardiovascular injury [2, 165] and can result in cell death of cardiomyocytes during ischemic conditions [166].
Induction of apoptosis involves the onset of membrane PS exposure and DNA fragmentation [113]. As an early event in the dynamics of cellular apoptosis, PS exposure may be required for embryogenesis [167]. Yet, in mature tissues, membrane PS externalization can become a signal for early injury mechanisms [168] and the phagocytosis of cells [169]. As previously described in relation to inflammatory microglial activation, cells expressing externalized PS may be removed by microglia [1, 170]. An additional role for membrane PS externalization in the vascular cell system is the activation of coagulation cascades [109, 171]. Exposure of membrane PS residues in endothelial cells can promote the formation of a procoagulant surface [172]. Given the early onset of PS externalization during apoptosis, membrane PS exposure has become an attractive target to detect and follow the onset of cellular injury [94, 173–175].
In contrast to the early externalization of membrane PS residues, the cleavage of genomic DNA into fragments is considered to be a later event during cell injury[14, 94, 176, 177]. Several enzymes responsible for DNA degradation have been differentiated based on their ionic sensitivities to zinc [178] and magnesium [179]. Calcium, a critical independent component that can determine cell survival [180], also may determine endonuclease activity through calcium/magnesium - dependent endonucleases such as DNase I [181]. Other enzymes that may degrade DNA include the acidic, cation independent endonuclease (DNase II) [182], cyclophilins [183], and the 97 kDa magnesium - dependent endonuclease [184]. In the nervous system, three separate endonuclease activities are present. These include that a constitutive acidic cation-independent endonuclease, a constitutive calcium/magnesium-dependent endonuclease, and an inducible magnesium dependent endonuclease [30, 36, 38].
In many respects, nicotinamide can be considered a “broad spectrum” cytoprotectant that prevents both necrotic as well as apoptotic injury depending upon the experimental models examined. Irrespective of injuries that involve either clinical deficits, such as cognitive decline [185], or lead to cell destruction with DNA fragmentation [38, 98, 186, 187], nicotinamide can block the progression of injury and may, in some cases, reverse a prior insult. Yet, an ideal cytoprotectant would prevent not only DNA degradation, but also membrane PS exposure to provide greater overall protection against cellular dysfunction and demise from microglial phagocytosis [2, 165]. Studies with nicotinamide demonstrate a robust capacity for cytoprotection in addition to preventing passive cellular destruction during necrosis [142]. Nicotinamide yields immediate cytoprotection through the maintenance of an intact genomic DNA and can maintain membrane PS asymmetry to provide a more long-term protection by inhibiting the destruction of cells by phagocytes [1, 13, 24, 26]. Application of nicotinamide during anoxia, oxygen-glucose deprivation, and free radical exposure can prevent the early exposure of membrane PS residues and also inhibit the later stages of genomic DNA destruction [13, 24, 139]. Potentially more significant, nicotinamide prevents membrane PS exposure in vascular cells [2, 24], since exposure of membrane PS residues in vascular cells can lead to the loss of anticoagulant membrane components, the propagation of the coagulation process, antibody-dependent aggregations, and cellular Inflammation, Thus, nicotinamide, through the prevention of vascular membrane PS exposure, may enhance an organism’s ability to prevent a procoagulant state and lower the risk for diseases such as stroke and arteriosclerosis.
An important caveat to cell injury focuses upon the initial stages of apoptotic death, namely membrane PS residue exposure, and whether this is reversible in nature [13, 170, 188]. Investigations that examine the efficacy of cytoprotectants during cerebral ischemia have supported the premise that cellular apoptotic injury can be reversible. For example, the application of growth factors [189–191], benzothiazole compounds [192–194], metabotropic glutamate receptor agonists [92, 170, 195–197], cytokine modulation [112, 163, 198], and regulation of central signal transduction pathways [109, 117, 199] have been shown to reverse, or at least block, the progression of membrane and nuclear changes associated with apoptosis. The use of post-treatment strategies with nicotinamide in studies using reversible labeling of annexin V for PS exposure in living cells illustrate that apoptotic injury can be reversed to some extent. During post-treatment studies, nicotinamide can reverse an initial progression of membrane PS inversion and maintain the suppression of PS exposure over a 24 hour period [2, 13, 25, 75]. These results suggest that apoptotic injury, at least along the pathway that involves membrane PS exposure, is dynamic and reversible in nature [2, 13, 25, 75]. As a result, nicotinamide may impart an additional advantage for cell survival and function.
NICOTINAMIDE AND INTRACELLULAR SIGNAL TRANSDUCTION
Nicotinamide regulates cellular function, integrity and survival through a series of intracellular signal transduction pathways (Fig. 2). One of these pathways involves Akt, also known as protein kinase B [117, 164]. Initial studies have shown that overexpression of Akt in neurons prevents apoptosis during growth factor withdrawal [200] and that Akt is necessary to prevent cell death during growth factor absence [201]. Further work has demonstrated that Akt can be both necessary and sufficient for the survival of neurons, since expression of a dominant-negative Akt or inhibition of phosphoinositide 3 kinase (PI 3-K), necessary for Akt activation, yields apoptotic cell death during trophic factor administration [202] and precipitates cell death during oxidative stress [14, 43]. Increased Akt activity can foster cell survival during free radical exposure [98, 203], matrix detachment [204], neuronal axotomy [205], DNA damage [43, 109, 171, 206], anti-Fas antibody administration [207], oxidative stress [14, 43, 98, 208], hypoxic preconditioning [209], β-amyloid (Aβ) exposure [210], and transforming growth factor-β (TGF-β) application [211]. Activation of Akt also can prevent membrane PS exposure on injured cells and block the activation of microglia during oxidative stress [14, 43, 92]. Nicotinamide uses mechanisms that involve Akt to regulate microglial activation and proliferation [26, 29] by blocking membrane PS exposure on cells and possibly preventing the shedding of membrane PS residues that is known to occur during apoptosis [212].
Fig. 2.

Nicotinamide (NIC) modulates a variety of cellular mediators to oversee cellular metabolism, longevity, survival, and inflammatory microglial activation. Nicotinamide promotes cellular function and survival through a series of pathways that involve NAD+, cell senescence mechanisms, the serine-threonine kinase Akt and its downstream substrates of FOXO3a, Bad, and caspases. Closely to the cytoprotection by nicotinamide is the maintenance of mitochondrial membrane potential, mitochondrial energy reserves, cytochrome c (Cyto c) release, and PARP. Targeting by NIC of specific caspase pathways ultimately serves to preserve genomic integrity and prevent early apoptotic injury “tagging” for microglial disposal. NAD: β-nicotinamide adenine dinucleotide; Sir2: Silent information regulator 2; MG: microglia; Mito: mitochondria; PS: phosphatidylserine.
Akt also regulates several vital pathways that can significantly influence cell survival. One of these pathways involves Bad, a pro-apoptotic Bcl-2 family member. The activity of Bad is mediated through phosphorylation on its serine residues [15]. Phosphorylation of Bad by Akt leads to the binding of Bad with the cytosolic protein 14-3-3 to release Bcl-xL and allows Akt to block apoptosis [213]. Bcl-2 and Bcl-xL block Bax translocation to the mitochondria, maintain mitochondrial membrane potential, and prevent the release of cytochrome c from the mitochondria [12, 214]. Nicotinamide can promote the phosphorylation of Bad during oxidative stress [26]. This phosphorylation of Bad by nicotinamide can be blocked by inhibition of Akt activity, suggesting that nicotinamide phosphorylates Bad through a PI 3-K/Akt mediated pathway. In addition, Akt may promote cell survival through the inhibition of p53 transcriptional activity [215] that may be regulated by nicotinamide. Activation of p53 can promote the expression of Bax to result in apoptotic cell death [216]. Nicotinamide has been shown to either directly limit the expression of p53 [76] or prevent an NAD-dependent p53 deacetylation induced by Sir2α [217].
The Forkhead family of transcription factors represents another downstream substrate of Akt. The phosphorylation of the FOXO proteins by Akt results in their inactivation through cytoplasmic retention. The transcriptional activity of the FOXO family is closely related to the induction of apoptosis. In the nucleus, FOXO can bind to specific DNA sequences and mediate the activation of transcription that plays essential roles in the regulation of cell survival. Expression of an Akt-resistant mutant of FOXO that promotes the activity of FOXO can lead to apoptosis [218]. On the other hand, activation of Akt can lead to the inhibition of FOXO-dependent transcription and block apoptosis [219]. In particular, the Forkhead family member FOXO3a can result in apoptosis in a variety of cell types in a transcription-dependent manner following its translocation to the nucleus [219– 221]. FOXO3a can disrupt mitochondrial membrane potential and result in cytochrome c release [221].
Nicotinamide may derive its protective capacity through two separate mechanisms of post-translational modification of FOXO3a by not only maintaining inhibitory phosphorylation of FOXO3a, but also preserving the integrity of total FOXO3a and phosphorylated FOXO3a (Fig. 3). During periods of oxidative stress in the nervous system, an initial inhibitory phosphorylation of FOXO3a at the regulatory phosphorylation sites (Thr32 and Ser253) can occur [29, 112]. However, loss of phosphorylated FOXO3a expression appears to subsequently result over a 12 hour period, possibly by caspase degradation, which potentially can enhance the vulnerability of neurons to apoptotic injury during neurodegenerative disorders [29]. The loss of both FOXO3a phosphorylation and the integrity of this transcription factor may function as a significant precipitant of cellular injury. FOXO3a proteolysis occurs during cell injury yielding an amino-terminal (Nt) fragment that can become biologically active [222]. During cell injury and caspase-dependent cleavage of Akt1 [223], it is the activation of FOXO3a Nt fragments that become available and result in apoptotic cellular injury. Nicotinamide, through the phosphorylation of FOXO3a at regulatory sites that possess high affinity for Akt can prevent apoptotic cell injury [29] (Fig. 3). In addition, modulation of caspase 3 activity by nicotinamide appears to be tied to a unique regulatory mechanism that blocks the proteolytic degradation of phosphorylated FOXO3a by caspase 3. Given that FOXO3a has been shown to be a substrate for caspase 3-like proteases at the consensus sequence DELD304A [222], current work demonstrates that blockade of caspase 3 - like activity prevents the destruction of phosphorylated FOXO3a during oxidative stress [29]. In light of the dual capacity of nicotinamide to directly inhibit caspase 3 - like activity and maintain inhibitory phosphorylation of FOXO3a, these studies suggest that nicotinamide maintains a regulatory loop through the modulation of caspase 3 and the preservation of phosphorylated FOXO3a integrity.
Fig. 3.
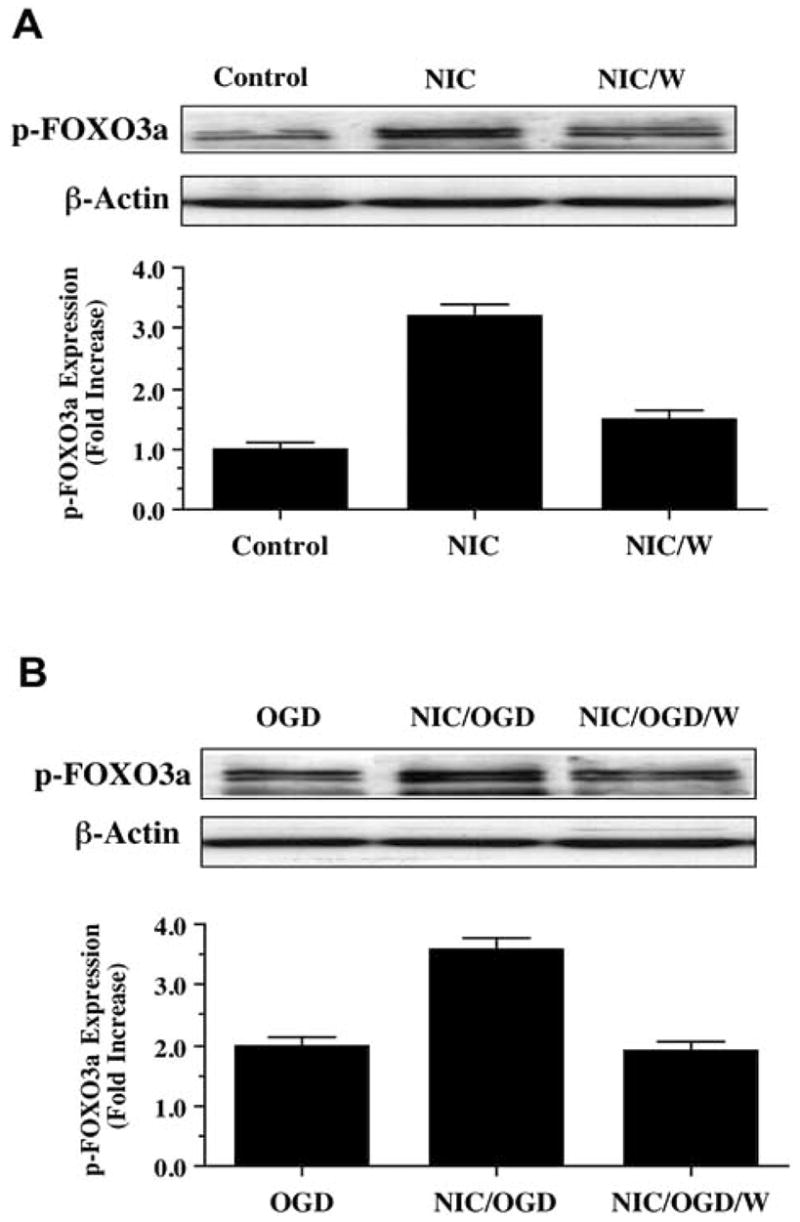
Nicotinamide (NIC) increases phosphorylation of p-FOXO3a through activation of Akt during oxidative stress. (A) Equal amounts of neuronal protein extracts (50 μg/lane) were immunoblotted at 6 hours following NIC (12.5 mM) treatment with anti - phospho-FOXO3a (p- FOXO3a) antibody. NIC increased the expression of p-FOXO3a significantly over a 6 hour period. Co-application of NIC with phosphoinositol-3 kinase inhibitor wortmannin (W, 500 μM), which inhibits phosphorylation of Akt, decreased p-FOXO3a expression during NIC application. (B) Equal amounts of neuronal protein extracts (50 μg/lane) were immunoblotted at 6 hours following oxygen glucose deprivation (OGD), NIC (12.5 mM), or NIC (12.5 mM) following a 3 hour period of OGD with anti - phospho-FOXO3a (p- FOXO3a) antibody. OGD alone increased the expression of p-FOXO3a. The expression of p-FOXO3a was further increased in neurons with NIC applied 1 hour prior to OGD. Yet, application of wortmannin (W) significantly decreased the expression of p-FOXO3a during the administration of NIC during OGD.
Nicotinamide also may foster cellular protection through modulation of caspase activity. Caspases are a family of cysteine proteases that are synthesized as inactive zymogens which are proteolytically cleaved into subunits at the onset of apoptosis [15, 30]. Caspases are composed of three domains including an N-terminal prodomain, a large subunit, and a small subunit [224]. As a result of their activation sequence, caspases are classified as either initiator caspases (also known as apical caspases) or effector caspases [225]. An initiator caspase cleaves and subsequently activates an effector caspase. The apoptotic-associated caspases include initiator caspases, such as caspase 2, 8, 9, and 10, that activate downstream effector caspases, resulting in an amplification of cascade activity. The initiator caspases consist of long N-terminal prodomains that contain caspase recruitment domains (CARDs) in caspase 2 and caspase 9 or death effector domains (DEDs) in caspase 8 and caspase 10 [226]. The effector caspases consist of caspase 3, 6, and 7 that function to directly cleave crucial cellular protein substrates to result in cell destruction.
In several cell populations, nicotinamide can prevent specific caspase activity. The caspases 1 and 3 have each been linked to the independent apoptotic pathways of genomic DNA cleavage and cellular membrane PS exposure [12, 114, 227]. The ability of nicotinamide to regulate these caspases plays an important role in cellular protection. Caspase 3 becomes a prominent mediator of genomic DNA degradation. Experimental models that use caspase 3 gene deletions or pharmacological inhibition illustrate little or no DNA fragmentation following toxic cellular insults [171, 228]. In regards to membrane PS exposure, nicotinamide appears to prevent PS externalization primarily through the inhibition of caspase 1 -like activity [26]. Yet, other caspases pathways, such as 3, 8, and 9, have been shown to be necessary to full achieve maintenance of membrane asymmetry [2, 13, 24, 25]. These caspases are also tied to the direct activation and proliferation of microglia [14, 43, 98].
Nicotinamide also prevents genomic DNA cleavage as well as cellular membrane PS exposure through caspase 8 and caspase 9 - like activities [2, 13, 24, 26, 29]. Both caspase 8 and 9 are associated with the apoptotic pathways of genomic DNA cleavage and cellular membrane PS exposure [165, 227]. Caspase 9 is activated through a process that involves the cytochrome c -Apaf-1 complex [12, 116, 229]. The precise pathways that are necessary for nicotinamide to modulate caspase pathways remain under consideration. Although some “anti-apoptotic” proteins, such as EPO [12, 113] and heat-shock protein 70 [230, 231], appear to modulate both Apaf-1 expression and cytochrome c release, protection through nicotinamide remains independent from Apaf-1 [29]. Yet, independent of Apaf-1, nicotinamide can significantly prevent cell injury by inhibiting caspase 9 - like activity directly [29]. In addition, caspase 8 serves as an upstream initiator of executioner caspases, such as caspase 3, and also leads to the mitochondrial release of cytochrome c [232, 233]. Following caspase 8 and caspase 9 activation, caspase 3 directly leads to genomic DNA degradation.
Another attractive pathway that may mediate protection by nicotinamide could involve the stress activated family of mitogen-activated protein kinases (MAPK) that includes the p38 kinases (MAPKp38) and the c-Jun N-terminal kinases (MAPKJNK). The family of MAPKs consists of the three major subgroups. The first subgroup includes the founding members of the MAPK family, ERK1 (MAPKERK1/p44) and ERK2 (MAPKERK2/p42) as well as their closest relatives. The second subgroup is termed the JNKs (MAPKJNK), since they can activate the Jun transcription factor through phosphorylating two residues near its N-terminus. The third subgroup is termed p38 MAPKs (MAPKp38), since the molecular weight of the first representative in this subgroup is found to be 38 kDa [234]. The members of both MAPKJNK and MAPKp38 pathways are also classified as stress-activated protein kinases (SAPKs) because they are activated in response to a variety of stress factors including osmotic shock, UV irradiation, inflammatory cytokines, and other stressful conditions such as oxidative stress [234].
The MAPKs are activated by phosphorylation and play a significant function during cell differentiation, growth, and death [235]. Significant activation of MAPKp38 and MAPKJNK is present in both neurons and endothelial cells during oxidative stress [2, 13, 25, 75]. In addition, MAPKJNK can promote Bax translocation through phosphorylation of 14-3-3 proteins and lead to cytochrome c release [236]. Furthermore, during cellular injury such as with cyanide-induced apoptosis, MAPKp38 can modulate Bax translocation from the cytosol to the mitochondria and result in both cytochrome c release and caspase activation [237]. Yet in a number of injury paradigms, nicotinamide does not alter the activity of either MAPKp38 or MAPKJNK [26]. These results suggest that protection by nicotinamide does not require the MAPKp38 and MAPKJNK pathways [2, 13, 25, 75].
As a caveat to the ability of nicotinamide to enhance cell survival and prevent apoptosis, under some conditions nicotinamide has been described as an agent that limits cell growth and promotes cell injury. For example, nicotinamide in the presence of transforming growth factor β-1 can block hepatic cell proliferation and lead to apoptosis with caspase 3 activation, suggesting that nicotinamide may be useful to prevent cell growth such as during hepatic fibrogenesis [125]. In addition, nicotinamide during moderate temperature hyperthermia or carbogen breathing can result in enhanced solid tumor radiosensitivity and assist with tumor load reduction [238]. It appears that the cellular environment can determine whether nicotinamide may promote or prevent apoptosis, since it has been shown that hepatic cell injury also can be reduced by nicotinamide during methotrexate administration [239].
CONCLUSIONS AND FUTURE PERSPECTIVES
Nicotinamide, a precursor for the coenzyme β-nicotinamide adenine dinucleotide (NAD+) as well as an essential nutrient, is vital for cellular function and metabolism. However, a body of new investigations has broadened the role for nicotinamide as an agent that can promote neurogenesis and cell proliferation, foster cytoprotection in multiple cell systems during injury, prevent inflammatory cell demise, and have a complex role in the cellular processes that determine cell longevity. On the flip side, recent work has identified potential cell growth limiting effects of nicotinamide during specific cellular environments that may be applicable for a narrow scope of disorders, such as to limit neoplastic growth.
Given the wide array of cellular functions regulated by nicotinamide, it becomes critical to elucidate the cellular pathways controlled by this agent. Nicotinamide can modulate cellular function through a series of cellular pathways that involve mitochondrial membrane potential, cytochrome c release, and PARP activity. In addition, nicotinamide oversees a central pathway involved in cellular proliferation, apoptotic injury, and microglial activation that is dependent upon the serine-threonine kinase Akt and its downstream substrates of Bad, FOXO3a, and caspases. Further insight into the spectrum of cellular processes modulated by nicotinamide should open the door for the future development of new therapeutic strategies for a spectrum of disorders that may involve aging, Alzheimer’s disease, diabetes, cerebral ischemia, Huntington’s disease, Parkinson’s disease, and cancer.
Acknowledgments
This research was supported by the following grants (KM): American Diabetes Association, American Heart Association (National), Bugher Foundation Award, Janssen Neuroscience Award, LEARN Foundation Award, MI Life Sciences Challenge Award, and NIH NIEHS (P30 ES06639).
References
- 1.Li F, Chong ZZ, Maiese K. Front Biosci. 2004;9:2500–2520. doi: 10.2741/1412. [DOI] [PubMed] [Google Scholar]
- 2.Maiese K, Chong ZZ. Trends Pharmacol Sci. 2003;24(5):228–32. doi: 10.1016/S0165-6147(03)00078-6. [DOI] [PubMed] [Google Scholar]
- 3.DiPalma JR, Thayer WS. Annu Rev Nutr. 1991;11:169–87. doi: 10.1146/annurev.nu.11.070191.001125. [DOI] [PubMed] [Google Scholar]
- 4.Jackson TM, Rawling JM, Roebuck BD, Kirkland JB. J Nutr. 1995;125(6):1455–61. doi: 10.1093/jn/125.6.1455. [DOI] [PubMed] [Google Scholar]
- 5.Magni G, Amici A, Emanuelli M, Orsomando G, Raffaelli N, Ruggieri S. Cell Mol Life Sci. 2004;61(1):19–34. doi: 10.1007/s00018-003-3161-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin SJ, Guarente L. Curr Opin Cell Biol. 2003;15(2):241–6. doi: 10.1016/s0955-0674(03)00006-1. [DOI] [PubMed] [Google Scholar]
- 7.Hageman GJ, Stierum RH. Mutat Res. 2001;475(1–2):45–56. doi: 10.1016/s0027-5107(01)00078-1. [DOI] [PubMed] [Google Scholar]
- 8.Sadanaga-Akiyoshi F, Yao H, Tanuma S, Nakahara T, Hong JS, Ibayashi S, Uchimura H, Fujishima M. Neurochem Res. 2003;28(8):1227–34. doi: 10.1023/a:1024236614015. [DOI] [PubMed] [Google Scholar]
- 9.Tam D, Tam M, Maynard KI. Ann N Y Acad Sci. 2005;1053:258–68. doi: 10.1196/annals.1344.023. [DOI] [PubMed] [Google Scholar]
- 10.Wang J, Zhai Q, Chen Y, Lin E, Gu W, McBurney MW, He Z. J Cell Biol. 2005;170(3):349–55. doi: 10.1083/jcb.200504028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Lisa F, Menabo R, Canton M, Barile M, Bernardi P. J Biol Chem. 2001;276(4):2571–5. doi: 10.1074/jbc.M006825200. [DOI] [PubMed] [Google Scholar]
- 12.Chong ZZ, Kang JQ, Maiese K. J Cereb Blood Flow Metab. 2003;23(3):320–30. doi: 10.1097/01.WCB.0000050061.57184.AE. [DOI] [PubMed] [Google Scholar]
- 13.Lin SH, Vincent A, Shaw T, Maynard KI, Maiese K. J Cereb Blood Flow Metab. 2000;20(9):1380–91. doi: 10.1097/00004647-200009000-00013. [DOI] [PubMed] [Google Scholar]
- 14.Kang JQ, Chong ZZ, Maiese K. Mol Pharmacol. 2003;64(3):557–69. doi: 10.1124/mol.64.3.557. [DOI] [PubMed] [Google Scholar]
- 15.Chong ZZ, Li F, Maiese K. Prog Neurobiol. 2005;75(3):207–46. doi: 10.1016/j.pneurobio.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 16.Cosi C, Marien M. Ann N Y Acad Sci. 1999;890:227–39. doi: 10.1111/j.1749-6632.1999.tb07998.x. [DOI] [PubMed] [Google Scholar]
- 17.Du L, Zhang X, Han YY, Burke NA, Kochanek PM, Watkins SC, Graham SH, Carcillo JA, Szabo C, Clark RS. J Biol Chem. 2003;278(20):18426–33. doi: 10.1074/jbc.M301295200. [DOI] [PubMed] [Google Scholar]
- 18.Smeitink JAM, van den Heuvel L, Koopman WJH, Nijtmans LGJ, Ugalde C, Willems P. Curr Neurovasc Res. 2004;1(1):29–40. doi: 10.2174/1567202043480224. [DOI] [PubMed] [Google Scholar]
- 19.Liu Y, Fiskum G, Schubert D. J Neurochem. 2002;80(5):780–7. doi: 10.1046/j.0022-3042.2002.00744.x. [DOI] [PubMed] [Google Scholar]
- 20.Turrens JF, Alexandre A, Lehninger AL. Arch Biochem Biophys. 1985;237(2):408–14. doi: 10.1016/0003-9861(85)90293-0. [DOI] [PubMed] [Google Scholar]
- 21.Wilson FH, Hariri A, Farhi A, Zhao H, Petersen KF, Toka HR, Nelson-Williams C, Raja KM, Kashgarian M, Shulman GI, Scheinman SJ, Lifton RP. Science. 2004;306(5699):1190–4. doi: 10.1126/science.1102521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bal-Price A, Brown GC. J Neurochem. 2000;75(4):1455–64. doi: 10.1046/j.1471-4159.2000.0751455.x. [DOI] [PubMed] [Google Scholar]
- 23.Klaidman L, Morales M, Kem S, Yang J, Chang ML, Adams JD., Jr Pharmacology. 2003;69(3):150–7. doi: 10.1159/000072668. [DOI] [PubMed] [Google Scholar]
- 24.Chong ZZ, Lin SH, Maiese K. J Vasc Res. 2002;39(2):131–47. doi: 10.1159/000057762. [DOI] [PubMed] [Google Scholar]
- 25.Maiese K, Lin S, Chong ZZ. Curr Med Chem Imm Endoc Metab Agents. 2001;1(3):257–267. [Google Scholar]
- 26.Chong ZZ, Lin SH, Li F, Maiese K. Curr Neurovasc Res. 2005;2(4):271–85. doi: 10.2174/156720205774322584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schinder AF, Olson EC, Spitzer NC, Montal M. J Neurosci. 1996;16(19):6125–33. doi: 10.1523/JNEUROSCI.16-19-06125.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walter DH, Haendeler J, Galle J, Zeiher AM, Dimmeler S. Circulation. 1998;98(12):1153–7. doi: 10.1161/01.cir.98.12.1153. [DOI] [PubMed] [Google Scholar]
- 29.Chong ZZ, Lin SH, Maiese K. J Cereb Blood Flow Metab. 2004;24(7):728–43. doi: 10.1097/01.WCB.0000122746.72175.0E. [DOI] [PubMed] [Google Scholar]
- 30.Chong ZZ, Li F, Maiese K. Brain Res, Brain Res, Rev. 2005;49(1):1–21. doi: 10.1016/j.brainresrev.2004.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sensi SL, Jeng JM. Curr Mol Med. 2004;4(2):87–111. doi: 10.2174/1566524043479211. [DOI] [PubMed] [Google Scholar]
- 32.Ritucci NA, Dean JB, Putnam RW. Am J Physiol. 1997;273(1 Pt 2):R433–41. doi: 10.1152/ajpregu.1997.273.1.R433. [DOI] [PubMed] [Google Scholar]
- 33.Roberts E, Jr, Chih CP. J Cereb Blood Flow Metab. 1997;17(5):560–6. doi: 10.1097/00004647-199705000-00010. [DOI] [PubMed] [Google Scholar]
- 34.Zhan RZ, Fujiwara N, Yamakura T, Taga K, Fukuda S, Endoh H, Shimoji K. Brain Res. 1997;760(1–2):179–86. doi: 10.1016/s0006-8993(97)00278-3. [DOI] [PubMed] [Google Scholar]
- 35.Vincent AM, TenBroeke M, Maiese K. J Neurobiol. 1999;40(2):171–84. doi: 10.1002/(sici)1097-4695(199908)40:2<171::aid-neu4>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 36.Vincent AM, TenBroeke M, Maiese K. Exp Neurol. 1999;155(1):79–94. doi: 10.1006/exnr.1998.6966. [DOI] [PubMed] [Google Scholar]
- 37.Ito N, Bartunek J, Spitzer KW, Lorell BH. Circulation. 1997;95(9):2303–11. doi: 10.1161/01.cir.95.9.2303. [DOI] [PubMed] [Google Scholar]
- 38.Vincent AM, Maiese K. Exp Cell Res. 1999;246(2):290–300. doi: 10.1006/excr.1998.4282. [DOI] [PubMed] [Google Scholar]
- 39.Crowley CL, Payne CM, Bernstein H, Bernstein C, Roe D. Cell Death Differ. 2000;7(3):314–26. doi: 10.1038/sj.cdd.4400658. [DOI] [PubMed] [Google Scholar]
- 40.Halestrap AP, Woodfield KY, Connern CP. J Biol Chem. 1997;272(6):3346–54. doi: 10.1074/jbc.272.6.3346. [DOI] [PubMed] [Google Scholar]
- 41.La Piana G, Marzulli D, Consalvo MI, Lofrumento NE. Arch Biochem Biophys. 2003;410(2):201–11. doi: 10.1016/s0003-9861(02)00687-2. [DOI] [PubMed] [Google Scholar]
- 42.Kennedy SG, Kandel ES, Cross TK, Hay N. Mol Cell Biol. 1999;19(8):5800–10. doi: 10.1128/mcb.19.8.5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kang JQ, Chong ZZ, Maiese K. J Neurosci Res. 2003;74(1):37–51. doi: 10.1002/jnr.10740. [DOI] [PubMed] [Google Scholar]
- 44.Southan GJ, Szabo C. Curr Med Chem. 2003;10(4):321–40. doi: 10.2174/0929867033368376. [DOI] [PubMed] [Google Scholar]
- 45.Burkle A. Bioessays. 2001;23(9):795–806. doi: 10.1002/bies.1115. [DOI] [PubMed] [Google Scholar]
- 46.Satoh MS, Lindahl T. Nature. 1992;356(6367):356–8. doi: 10.1038/356356a0. [DOI] [PubMed] [Google Scholar]
- 47.Smets LA, Loesberg C, Janssen M, Van Rooij H. Biochim Biophys Acta. 1990;1054(1):49–55. doi: 10.1016/0167-4889(90)90204-q. [DOI] [PubMed] [Google Scholar]
- 48.Saldeen J, Welsh N. Mol Cell Endocrinol. 1998;139(1–2):99–107. doi: 10.1016/s0303-7207(98)00068-9. [DOI] [PubMed] [Google Scholar]
- 49.Thies RL, Autor AP. Arch Biochem Biophys. 1991;286(2):353–63. doi: 10.1016/0003-9861(91)90051-j. [DOI] [PubMed] [Google Scholar]
- 50.Hoyt DG, Lazo JS. Biochem Pharmacol. 1993;46(10):1819–24. doi: 10.1016/0006-2952(93)90588-n. [DOI] [PubMed] [Google Scholar]
- 51.Skaper SD. Ann N Y Acad Sci. 2003;993:217–28. 287–8. doi: 10.1111/j.1749-6632.2003.tb07532.x. [DOI] [PubMed] [Google Scholar]
- 52.Klaidman LK, Yang J, Chang ML, Adams JD., Jr Curr Med Chem. 2003;10(24):2669–78. doi: 10.2174/0929867033456323. [DOI] [PubMed] [Google Scholar]
- 53.Virag L, Szabo C. Pharmacol Rev. 2002;54(3):375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- 54.Uehara N, Miki K, Tsukamoto R, Matsuoka Y, Tsubura A. Exp Eye Res. 2005 doi: 10.1016/j.exer.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 55.Kruman II, Culmsee C, Chan SL, Kruman Y, Guo Z, Penix L, Mattson MP. J Neurosci. 2000;20(18):6920–6. doi: 10.1523/JNEUROSCI.20-18-06920.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kabra DG, Thiyagarajan M, Kaul CL, Sharma SS. Brain Res Bull. 2004;62(5):425–33. doi: 10.1016/j.brainresbull.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 57.Aito H, Aalto KT, Raivio KO. Brain Res. 2004;1013(1):117–24. doi: 10.1016/j.brainres.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 58.Cole K, Perez-Polo JR. Int J Dev Neurosci. 2004;22(7):485–96. doi: 10.1016/j.ijdevneu.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 59.Takahashi K, Greenberg JH, Jackson P, Maclin K, Zhang J. J Cereb Blood Flow Metab. 1997;17(11):1137–42. doi: 10.1097/00004647-199711000-00001. [DOI] [PubMed] [Google Scholar]
- 60.Tronov VA, Konstantinov EM. Biochemistry (Mosc) 2000;65(11):1279–86. [PubMed] [Google Scholar]
- 61.Kuchmerovska T, Shymanskyy I, Donchenko G, Kuchmerovskyy M, Pakirbaieva L, Klimenko A. J Diabet Complicat. 2004;18(4):198–204. doi: 10.1016/S1056-8727(03)00039-4. [DOI] [PubMed] [Google Scholar]
- 62.Panchal M, Rholam M, Brakch N. Curr Neurovasc Res. 2004;1(4):317–323. doi: 10.2174/1567202043362117. [DOI] [PubMed] [Google Scholar]
- 63.Chong ZZ, Li FQ, Maiese K. Curr Neurovasc Res. 2005;2(1):55–72. doi: 10.2174/1567202052773508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Selkoe DJ. Physiol Rev. 2001;81(2):741–66. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 65.Birkmayer JG. Ann Clin Lab Sci. 1996;26(1):1–9. [PubMed] [Google Scholar]
- 66.Morris MC, Evans DA, Bienias JL, Scherr PA, Tangney CC, Hebert LE, Bennett DA, Wilson RS, Aggarwal N. J Neurol Neurosurg Psychiat. 2004;75(8):1093–9. doi: 10.1136/jnnp.2003.025858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Love S, Barber R, Wilcock GK. Brain. 1999;122( Pt 2):247–53. doi: 10.1093/brain/122.2.247. [DOI] [PubMed] [Google Scholar]
- 68.Adamczyk A, Czapski GA, Jesko H, Strosznajder RP. J Physiol Pharmacol. 2005;56(Suppl 2):5–13. [PubMed] [Google Scholar]
- 69.Shibata K, Shimada H, Taguchi H. Biosci Biotechnol Biochem. 1996;60(7):1204–6. doi: 10.1271/bbb.60.1204. [DOI] [PubMed] [Google Scholar]
- 70.Gaudineau C, Auclair K. Biochem Biophys Res Commun. 2004;317(3):950–6. doi: 10.1016/j.bbrc.2004.03.137. [DOI] [PubMed] [Google Scholar]
- 71.Reddy S, Salari-Lak N, Sandler S. Endocrinology. 1995;136(5):1907–12. doi: 10.1210/endo.136.5.7720637. [DOI] [PubMed] [Google Scholar]
- 72.Koppen A, Klein J, Holler T, Loffelholz K. J Pharmacol Exp Ther. 1993;266(2):720–5. [PubMed] [Google Scholar]
- 73.Williams AC, Cartwright LS, Ramsden DB. QJM. 2005;98(3):215–26. doi: 10.1093/qjmed/hci027. [DOI] [PubMed] [Google Scholar]
- 74.Porcu M, Chiarugi A. Trends Pharmacol Sci. 2005;26(2):94–103. doi: 10.1016/j.tips.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 75.Lin SH, Chong ZZ, Maiese K. J Med Food. 2001;4(1):27–38. doi: 10.1089/10966200152053686. [DOI] [PubMed] [Google Scholar]
- 76.Sonee M, Martens JR, Evers MR, Mukherjee SK. Neurotoxicology. 2003;24(3):443–8. doi: 10.1016/S0161-813X(03)00019-6. [DOI] [PubMed] [Google Scholar]
- 77.Frye RA. Biochem Biophys Res Commun. 2000;273(2):793–8. doi: 10.1006/bbrc.2000.3000. [DOI] [PubMed] [Google Scholar]
- 78.Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. Cell. 2001;107(2):149–59. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 79.Jackson MD, Schmidt MT, Oppenheimer NJ, Denu JM. J Biol Chem. 2003;278(51):50985–98. doi: 10.1074/jbc.M306552200. [DOI] [PubMed] [Google Scholar]
- 80.Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. J Biol Chem. 2002;277(47):45099–107. doi: 10.1074/jbc.M205670200. [DOI] [PubMed] [Google Scholar]
- 81.Denu JM. Trends Biochem Sci. 2005;30(9):479–83. doi: 10.1016/j.tibs.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 82.Kruszewski M, Szumiel I. DNA Repair (Amst) 2005;4(11):1306–13. doi: 10.1016/j.dnarep.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 83.Anderson RM, Bitterman KJ, Wood JG, Medvedik O, Sinclair DA. Nature. 2003;423(6936):181–5. doi: 10.1038/nature01578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chong ZZ, Li F, Balan V, Tzivion G, Maiese K. Soc Neurosci Abstr. 2005:130.15. [Google Scholar]
- 85.Gallo CM, Smith DL, Jr, Smith JS. Mol Cell Biol. 2004;24(3):1301–12. doi: 10.1128/MCB.24.3.1301-1312.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Anekonda TS, Reddy PH. J Neurochem. 2005;96(2):305–13. doi: 10.1111/j.1471-4159.2005.03492.x. [DOI] [PubMed] [Google Scholar]
- 87.Li F, Chong ZZ, Maiese K. Curr Neurovasc Res. 2005;2(4):331–40. doi: 10.2174/156720205774322557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mallat M, Marin-Teva JL, Cheret C. Curr Opin Neurobiol. 2005;15(1):101–7. doi: 10.1016/j.conb.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 89.Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, Nagata S. Science. 2004;304(5674):1147–50. doi: 10.1126/science.1094359. [DOI] [PubMed] [Google Scholar]
- 90.Hatori K, Nagai A, Heisel R, Ryu JK, Kim SU. J Neurosci Res. 2002;69(3):418–26. doi: 10.1002/jnr.10304. [DOI] [PubMed] [Google Scholar]
- 91.Lauber K, Bohn E, Krober SM, Xiao YJ, Blumenthal SG, Lindemann RK, Marini P, Wiedig C, Zobywalski A, Baksh S, Xu Y, Autenrieth IB, Schulze-Osthoff K, Belka C, Stuhler G, Wesselborg S. Cell. 2003;113(6):717–30. doi: 10.1016/s0092-8674(03)00422-7. [DOI] [PubMed] [Google Scholar]
- 92.Chong ZZ, Kang J, Li F, Maiese K. Curr Neurovasc Res. 2005;2(3):197–211. doi: 10.2174/1567202054368317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Maiese K. In: Encyclopedia of the Human Brain. 1. Ramachandran VS, editor. Elsevier Science; 2002. pp. 509–527. [Google Scholar]
- 94.Maiese K, Vincent AM. J Neurosci Res. 2000;59(4):568–80. doi: 10.1002/(SICI)1097-4547(20000215)59:4<568::AID-JNR13>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 95.Hoffmann PR, deCathelineau AM, Ogden CA, Leverrier Y, Bratton DL, Daleke DL, Ridley AJ, Fadok VA, Henson PM. J Cell Biol. 2001;155(4):649–59. doi: 10.1083/jcb.200108080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chong ZZ, Kang JQ, Maiese K. Histol Histopathol. 2003;18(1):173–89. doi: 10.14670/HH-18.173. [DOI] [PubMed] [Google Scholar]
- 97.Li F, Chong ZZ, Maiese K. Histol Histopathol. 2006;21:103–124. doi: 10.14670/hh-21.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chong ZZ, Kang JQ, Maiese K. Br J Pharmacol. 2003;138(6):1107–1118. doi: 10.1038/sj.bjp.0705161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nakano T, Ishimoto Y, Kishino J, Umeda M, Inoue K, Nagata K, Ohashi K, Mizuno K, Arita H. J Biol Chem. 1997;272(47):29411–4. doi: 10.1074/jbc.272.47.29411. [DOI] [PubMed] [Google Scholar]
- 100.Witting A, Muller P, Herrmann A, Kettenmann H, Nolte C. J Neurochem. 2000;75(3):1060–70. doi: 10.1046/j.1471-4159.2000.0751060.x. [DOI] [PubMed] [Google Scholar]
- 101.Maiese K, Chong ZZ. Restor Neurol Neurosci. 2004;22(2):87–104. [PubMed] [Google Scholar]
- 102.Sankarapandi S, Zweier JL, Mukherjee G, Quinn MT, Huso DL. Arch Biochem Biophys. 1998;353(2):312–21. doi: 10.1006/abbi.1998.0658. [DOI] [PubMed] [Google Scholar]
- 103.Mehlhorn G, Hollborn M, Schliebs R. Int J Dev Neurosci. 2000;18(4–5):423–31. doi: 10.1016/s0736-5748(00)00012-5. [DOI] [PubMed] [Google Scholar]
- 104.Benzing WC, Wujek JR, Ward EK, Shaffer D, Ashe KH, Younkin SG, Brunden KR. Neurobiol Aging. 1999;20(6):581–9. doi: 10.1016/s0197-4580(99)00065-2. [DOI] [PubMed] [Google Scholar]
- 105.Singhrao SK, Neal JW, Morgan BP, Gasque P. Exp Neurol. 1999;159(2):362–76. doi: 10.1006/exnr.1999.7170. [DOI] [PubMed] [Google Scholar]
- 106.Obal I, Jakab JS, Siklos L, Engelhardt JI. Neuroreport. 2001;12(11):2449–52. doi: 10.1097/00001756-200108080-00032. [DOI] [PubMed] [Google Scholar]
- 107.Sheng JG, Mrak RE, Griffin WS. Acta Neuropathol (Berl) 1997;94(1):1–5. doi: 10.1007/s004010050664. [DOI] [PubMed] [Google Scholar]
- 108.Wegiel J, Wang KC, Tarnawski M, Lach B. Acta Neuropathol (Berl) 2000;100(4):356–64. doi: 10.1007/s004010000199. [DOI] [PubMed] [Google Scholar]
- 109.Chong ZZ, Kang JQ, Maiese K. Exp Cell Res. 2004;296(2):196–207. doi: 10.1016/j.yexcr.2004.01.021. [DOI] [PubMed] [Google Scholar]
- 110.Liu B, Hong JS. J Pharmacol Exp Ther. 2003;304(1):1–7. doi: 10.1124/jpet.102.035048. [DOI] [PubMed] [Google Scholar]
- 111.Combs CK, Karlo JC, Kao SC, Landreth GE. J Neurosci. 2001;21(4):1179–88. doi: 10.1523/JNEUROSCI.21-04-01179.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Maiese K, Li F, Chong ZZ. Trends Pharmacol Sci. 2004;25(11):577–583. doi: 10.1016/j.tips.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 113.Maiese K, Li F, Chong ZZ. JAMA. 2005;293(1):90–5. doi: 10.1001/jama.293.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chong ZZ, Lin SH, Kang JQ, Maiese K. J Neurosci Res. 2003;71(5):659–69. doi: 10.1002/jnr.10528. [DOI] [PubMed] [Google Scholar]
- 115.Genc S, Koroglu TF, Genc K. Restor Neurol Neurosci. 2004;22:105–119. [PubMed] [Google Scholar]
- 116.Chong ZZ, Kang JQ, Maiese K. J Cereb Blood Flow Metab. 2002;22(5):503–514. doi: 10.1097/00004647-200205000-00001. [DOI] [PubMed] [Google Scholar]
- 117.Chong ZZ, Li F, Maiese K. Histol Histopathol. 2005;20(1):299–315. doi: 10.14670/hh-20.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lemmon MA, Ferguson KM, Abrams CS. FEBS Lett. 2002;513(1):71–6. doi: 10.1016/s0014-5793(01)03243-4. [DOI] [PubMed] [Google Scholar]
- 119.Reddy S, Young M, Ginn S. Histochem J. 2001;33(6):317–27. doi: 10.1023/a:1012422821187. [DOI] [PubMed] [Google Scholar]
- 120.Chen CF, Wang D, Hwang CP, Liu HW, Wei J, Lee RP, Chen HI. J Biomed Sci. 2001;8(6):446–52. doi: 10.1007/BF02256606. [DOI] [PubMed] [Google Scholar]
- 121.Moberg L, Olsson A, Berne C, Felldin M, Foss A, Kallen R, Salmela K, Tibell A, Tufveson G, Nilsson B, Korsgren O. Transplantation. 2003;76(9):1285–8. doi: 10.1097/01.TP.0000098905.86445.0F. [DOI] [PubMed] [Google Scholar]
- 122.Ungerstedt JS, Blomback M, Soderstrom T. Clin Exp Immunol. 2003;131(1):48–52. doi: 10.1046/j.1365-2249.2003.02031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Fukuzawa M, Satoh J, Muto G, Muto Y, Nishimura S, Miyaguchi S, Qiang XL, Toyota T. Immunol Lett. 1997;59(1):7–11. doi: 10.1016/s0165-2478(97)00088-6. [DOI] [PubMed] [Google Scholar]
- 124.Hiromatsu Y, Sato M, Yamada K, Nonaka K. Immunol Lett. 1992;31(1):35–9. doi: 10.1016/0165-2478(92)90007-b. [DOI] [PubMed] [Google Scholar]
- 125.Traister A, Breitman I, Bar-Lev E, Zvibel I, Harel A, Halpern Z, Oren R. Scand J Gastroenterol. 2005;40(10):1226–34. doi: 10.1080/00365520510023341. [DOI] [PubMed] [Google Scholar]
- 126.Kroger H, Hauschild A, Ohde M, Bache K, Voigt WP, Ehrlich W. Inflammation. 1999;23(2):111–5. doi: 10.1023/a:1020284810976. [DOI] [PubMed] [Google Scholar]
- 127.Soop A, Albert J, Weitzberg E, Bengtsson A, Nilsson CG, Sollevi A. Clin Exp Immunol. 2004;135(1):114–8. doi: 10.1111/j.1365-2249.2004.02315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bold JM, Gardner CR, Walker RJ. Br J Pharmacol. 1985;84(3):689–96. doi: 10.1111/j.1476-5381.1985.tb16151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lapin IP. Pharmacol Biochem Behav. 1981;14(5):589–93. doi: 10.1016/0091-3057(81)90117-9. [DOI] [PubMed] [Google Scholar]
- 130.Smith YR, Klitzman B, Ellis MN, Kull FC., Jr J Surg Res. 1989;47(5):465–9. doi: 10.1016/0022-4804(89)90103-0. [DOI] [PubMed] [Google Scholar]
- 131.Sakai Y, Jiang J, Kojima N, Kinoshita T, Miyajima A. Cell Transplant. 2002;11(5):435–41. [PubMed] [Google Scholar]
- 132.Vaca P, Berna G, Martin F, Soria B. Transplant Proc. 2003;35(5):2021–3. doi: 10.1016/s0041-1345(03)00735-8. [DOI] [PubMed] [Google Scholar]
- 133.Minami K, Okuno M, Miyawaki K, Okumachi A, Ishizaki K, Oyama K, Kawaguchi M, Ishizuka N, Iwanaga T, Seino S. Proc Natl Acad Sci USA. 2005;102(42):15116–21. doi: 10.1073/pnas.0507567102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Guruprasad KP, Vasudev V, Anilkumar MN, Chethan SA. Mutagenesis. 2002;17(1):1–8. doi: 10.1093/mutage/17.1.1. [DOI] [PubMed] [Google Scholar]
- 135.Inden M, Kim D, Gu Y, Kitamura Y, Kondo J, Tsuchiya D, Taniguchi T, Shimohama S, Akaike A, Sumi S, Inoue K. J Pharmacol Sci. 2004;96(1):53–64. doi: 10.1254/jphs.fpj04010x. [DOI] [PubMed] [Google Scholar]
- 136.Miura M, Kameda Y. J Comp Neurol. 2005;492(3):334–48. doi: 10.1002/cne.20731. [DOI] [PubMed] [Google Scholar]
- 137.Shen CC, Huang HM, Ou HC, Chen HL, Chen WC, Jeng KC. J Biomed Sci. 2004;11(4):472–81. doi: 10.1007/BF02256096. [DOI] [PubMed] [Google Scholar]
- 138.Kiuchi K, Kondo M, Ueno S, Moriguchi K, Yoshizawa K, Miyake Y, Matsumura M, Tsubura A. Curr Eye Res. 2003;26(6):355–62. doi: 10.1076/ceyr.26.5.355.15435. [DOI] [PubMed] [Google Scholar]
- 139.Kiuchi K, Yoshizawa K, Shikata N, Matsumura M, Tsubura A. Exp Eye Res. 2002;74(3):383–92. doi: 10.1006/exer.2001.1127. [DOI] [PubMed] [Google Scholar]
- 140.Reber F, Geffarth R, Kasper M, Reichenbach A, Schleicher ED, Siegner A, Funk RH. Graefes Arch Clin Exp Ophthalmol. 2003;241(3):213–25. doi: 10.1007/s00417-002-0528-1. [DOI] [PubMed] [Google Scholar]
- 141.Wallis RA, Panizzon KL, Girard JM. Brain Res. 1996;710(1–2):169–77. doi: 10.1016/0006-8993(95)01278-8. [DOI] [PubMed] [Google Scholar]
- 142.Yang J, Klaidman L, Chang M, Kem S, Sugawara T, Chan P, Adams J. Pharmacol Biochem Behav. 2002;73(4):901–910. doi: 10.1016/s0091-3057(02)00939-5. [DOI] [PubMed] [Google Scholar]
- 143.Gupta S, Kaul CL, Sharma SS. Neurol Res. 2004;26(1):103–7. doi: 10.1179/016164104773026624. [DOI] [PubMed] [Google Scholar]
- 144.Sakakibara Y, Mitha AP, Ayoub IA, Ogilvy CS, Maynard KI. Brain Res. 2002;931(1):68–73. doi: 10.1016/s0006-8993(02)02263-1. [DOI] [PubMed] [Google Scholar]
- 145.Brewer KL, Hardin JS. Acad Emerg Med. 2004;11(2):125–30. doi: 10.1111/j.1553-2712.2004.tb01421.x. [DOI] [PubMed] [Google Scholar]
- 146.Isbir CS, Ak K, Kurtkaya O, Zeybek U, Akgun S, Scheitauer BW, Sav A, Cobanoglu A. Eur J Cardiothorac Surg. 2003;23(6):1028–33. doi: 10.1016/s1010-7940(03)00110-6. [DOI] [PubMed] [Google Scholar]
- 147.Macleod MR, O’Collins T, Howells DW, Donnan GA. Stroke. 2004;35(5):1203–8. doi: 10.1161/01.STR.0000125719.25853.20. [DOI] [PubMed] [Google Scholar]
- 148.Zingarelli B, Salzman AL, Szabo C. Shock. 1996;5(4):258–64. doi: 10.1097/00024382-199604000-00005. [DOI] [PubMed] [Google Scholar]
- 149.Giulumian AD, Meszaros LG, Fuchs LC. J Cardiovasc Pharmacol. 2000;36(6):758–63. doi: 10.1097/00005344-200012000-00011. [DOI] [PubMed] [Google Scholar]
- 150.Braun RD, Lanzen JL, Turnage JA, Rosner G, Dewhirst MW. Radiat Res. 2001;155(5):724–33. doi: 10.1667/0033-7587(2001)155[0724:eotibc]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 151.Autor AP, Bonham AC, Thies RL. J Toxicol Environ Health. 1984;13(2–3):387–95. doi: 10.1080/15287398409530505. [DOI] [PubMed] [Google Scholar]
- 152.Bowes J, Piper J, Thiemermann C. Br J Pharmacol. 1998;124(8):1760–6. doi: 10.1038/sj.bjp.0702009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Cox MJ, Sood HS, Hunt MJ, Chandler D, Henegar JR, Aru GM, Tyagi SC. Am J Physiol Heart Circ Physiol. 2002;282(4):H1197–205. doi: 10.1152/ajpheart.00483.2001. [DOI] [PubMed] [Google Scholar]
- 154.Reddy S, Bibby NJ, Wu D, Swinney C, Barrow G, Elliott RB. Diabetes Res Clin Pract. 1995;29(2):83–92. doi: 10.1016/0168-8227(95)01109-9. [DOI] [PubMed] [Google Scholar]
- 155.Hu Y, Wang Y, Wang L, Zhang H, Zhao B, Zhang A, Li Y. Chin Med J (Engl) 1996;109(11):819–22. [PubMed] [Google Scholar]
- 156.Liu HK, Green BD, Flatt PR, McClenaghan NH, McCluskey JT. Endocr Res. 2004;30(1):61–8. doi: 10.1081/erc-120028485. [DOI] [PubMed] [Google Scholar]
- 157.Olmos PR, Hodgson MI, Maiz A, Manrique M, De Valdes MD, Foncea R, Acosta AM, Emmerich MV, Velasco S, Muniz OP, Oyarzun CA, Claro JC, Bastias MJ, Tora LA. Diabet Res Clin Pract. :2005. doi: 10.1016/j.diabres.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 158.Crino A, Schiaffini R, Ciampalini P, Suraci MC, Manfrini S, Visalli N, Matteoli MC, Patera P, Buzzetti R, Guglielmi C, Spera S, Costanza F, Fioriti E, Pitocco D, Pozzilli P. J Pediatr Endocrinol Metab. 2005;18(8):749–54. doi: 10.1515/jpem.2005.18.8.749. [DOI] [PubMed] [Google Scholar]
- 159.Eto N, Miyata Y, Ohno H, Yamashita T. Nephrol Dial Transplant. 2005;20(7):1378–84. doi: 10.1093/ndt/gfh781. [DOI] [PubMed] [Google Scholar]
- 160.Doonan F, Cotter TG. Curr Neurovasc Res. 2004;1(1):41–53. doi: 10.2174/1567202043480215. [DOI] [PubMed] [Google Scholar]
- 161.Koyama R, Ikegaya Y. Curr Neurovasc Res. 2004;1(1):3–10. doi: 10.2174/1567202043480242. [DOI] [PubMed] [Google Scholar]
- 162.Ferretti P. Curr Neurovasc Res. 2004;1(3):215–229. doi: 10.2174/1567202043362397. [DOI] [PubMed] [Google Scholar]
- 163.Li F, Chong ZZ, Maiese K. Neurosignals. 2004;13(6):265–89. doi: 10.1159/000081963. [DOI] [PubMed] [Google Scholar]
- 164.Chong ZZ, Kang JQ, Maiese K. Antioxid Redox Signal. 2004;6(2):277–87. doi: 10.1089/152308604322899341. [DOI] [PubMed] [Google Scholar]
- 165.Maiese K. Histol Histopathol. 2001;16(2):633–44. doi: 10.14670/HH-16.633. [DOI] [PubMed] [Google Scholar]
- 166.Cai Z, Manalo DJ, Wei G, Rodriguez ER, Fox-Talbot K, Lu H, Zweier JL, Semenza GL. Circulation. 2003;108(1):79–85. doi: 10.1161/01.CIR.0000078635.89229.8A. [DOI] [PubMed] [Google Scholar]
- 167.Bose J, Gruber AD, Helming L, Schiebe S, Wegener I, Hafner M, Beales M, Kontgen F, Lengeling A. J Biol. 2004;3(4):15. doi: 10.1186/jbiol10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Mari C, Karabiyikoglu M, Goris ML, Tait JF, Yenari MA, Blankenberg FG. Eur J Nucl Med Mol Imaging. 2004;31(5):733–9. doi: 10.1007/s00259-004-1473-5. [DOI] [PubMed] [Google Scholar]
- 169.Hong JR, Lin GH, Lin CJ, Wang WP, Lee CC, Lin TL, Wu JL. Development. 2004;131(21):5417–27. doi: 10.1242/dev.01409. [DOI] [PubMed] [Google Scholar]
- 170.Lin SH, Maiese K. J Cereb Blood Flow Metab. 2001;21(3):262–75. doi: 10.1097/00004647-200103000-00010. [DOI] [PubMed] [Google Scholar]
- 171.Chong ZZ, Kang JQ, Maiese K. Circulation. 2002;106(23):2973–9. doi: 10.1161/01.cir.0000039103.58920.1f. [DOI] [PubMed] [Google Scholar]
- 172.Bombeli T, Karsan A, Tait JF, Harlan JM. Blood. 1997;89(7):2429–42. [PubMed] [Google Scholar]
- 173.Vincent AM, Maiese K. J Histochem Cytochem. 1999;47(5):661–72. doi: 10.1177/002215549904700508. [DOI] [PubMed] [Google Scholar]
- 174.Blankenberg FG. Curr Pharm Des. 2004;10(13):1457–67. doi: 10.2174/1381612043384790. [DOI] [PubMed] [Google Scholar]
- 175.Blankenberg F, Mari C, Strauss HW. Q J Nucl Med. 2003;47(4):337–48. [PubMed] [Google Scholar]
- 176.Dombroski D, Balasubramanian K, Schroit AJ. J Autoimmun. 2000;14(3):221–9. doi: 10.1006/jaut.2000.0365. [DOI] [PubMed] [Google Scholar]
- 177.Jessel R, Haertel S, Socaciu C, Tykhonova S, Diehl HA. J Cell Mol Med. 2002;6(1):82–92. doi: 10.1111/j.1582-4934.2002.tb00313.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Torriglia A, Chaudun E, Courtois Y, Counis MF. Biochimie. 1997;79(7):435–8. doi: 10.1016/s0300-9084(97)86153-6. [DOI] [PubMed] [Google Scholar]
- 179.Sun XM, Cohen GM. J Biol Chem. 1994;269(21):14857–60. [PubMed] [Google Scholar]
- 180.Weber JT. Curr Neurovasc Res. 2004;1(2):151–171. doi: 10.2174/1567202043480134. [DOI] [PubMed] [Google Scholar]
- 181.Madaio MP, Fabbi M, Tiso M, Daga A, Puccetti A. Eur J Immunol. 1996;26(12):3035–41. doi: 10.1002/eji.1830261232. [DOI] [PubMed] [Google Scholar]
- 182.Torriglia A, Chaudun E, Chany-Fournier F, Jeanny JC, Courtois Y, Counis MF. J Biol Chem. 1995;270(48):28579–85. doi: 10.1074/jbc.270.48.28579. [DOI] [PubMed] [Google Scholar]
- 183.Montague JW, Hughes F, Jr, Cidlowski JA. J Biol Chem. 1997;272(10):6677–84. doi: 10.1074/jbc.272.10.6677. [DOI] [PubMed] [Google Scholar]
- 184.Pandey S, Walker PR, Sikorska M. Biochemistry. 1997;36(4):711–20. doi: 10.1021/bi962387h. [DOI] [PubMed] [Google Scholar]
- 185.Rao VL. Neurochem Int. 2002;41(2–3):161–70. doi: 10.1016/s0197-0186(02)00038-4. [DOI] [PubMed] [Google Scholar]
- 186.Goldshmit Y, Erlich S, Pinkas-Kramarski R. J Biol Chem. 2001;276(49):46379–85. doi: 10.1074/jbc.M105637200. [DOI] [PubMed] [Google Scholar]
- 187.Pugazhenthi S, Nesterova A, Jambal P, Audesirk G, Kern M, Cabell L, Eves E, Rosner MR, Boxer LM, Reusch JE. J Neurochem. 2003;84(5):982–96. doi: 10.1046/j.1471-4159.2003.01606.x. [DOI] [PubMed] [Google Scholar]
- 188.Sanchez-Alcazar JA, Ault JG, Khodjakov A, Schneider E. Cell Death Differ. 2000;7(11):1090–100. doi: 10.1038/sj.cdd.4400740. [DOI] [PubMed] [Google Scholar]
- 189.Kiprianova I, Freiman TM, Desiderato S, Schwab S, Galmbacher R, Gillardon F, Spranger M. J Neurosci Res. 1999;56(1):21–7. doi: 10.1002/(SICI)1097-4547(19990401)56:1<21::AID-JNR3>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 190.Maiese K, Boccone L. J Cereb Blood Flow Metab. 1995;15(3):440–9. doi: 10.1038/jcbfm.1995.55. [DOI] [PubMed] [Google Scholar]
- 191.Maiese K, Boniece I, DeMeo D, Wagner JA. J Neurosci. 1993;13(7):3034–40. doi: 10.1523/JNEUROSCI.13-07-03034.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Culmsee C, Junker V, Wolz P, Semkova I, Krieglstein J. Eur J Immunol. 1998;342(2–3):193–201. doi: 10.1016/s0014-2999(97)01499-4. [DOI] [PubMed] [Google Scholar]
- 193.Maiese K, Vincent AM. Cell Mol Neurobiol. 2000;20(3):383–400. doi: 10.1023/A:1007070311203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Maiese K, TenBroeke M, Kue I. J Neurochem. 1997;68(2):710–4. doi: 10.1046/j.1471-4159.1997.68020710.x. [DOI] [PubMed] [Google Scholar]
- 195.Maiese K, Vincent A, Lin SH, Shaw T. J Neurosci Res. 2000;62(2):257–272. doi: 10.1002/1097-4547(20001015)62:2<257::AID-JNR10>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 196.Vincent AM, Mohammad Y, Ahmad I, Greenberg R, Maiese K. J Neurosci Res. 1997;50(4):549–64. doi: 10.1002/(SICI)1097-4547(19971115)50:4<549::AID-JNR6>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 197.Chong ZZ, Maiese K. Histol Histopathol. 2004;19(2):495–504. doi: 10.14670/hh-19.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Fontaine V, Mohand-Said S, Hanoteau N, Fuchs C, Pfizenmaier K, Eisel U. J Neurosci. 2002;22(7):RC216. doi: 10.1523/JNEUROSCI.22-07-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Leyton L, Quest AF. Biol Res. 2004;37(1):29–43. doi: 10.4067/s0716-97602004000100004. [DOI] [PubMed] [Google Scholar]
- 200.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Cell. 1997;91(2):231–41. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 201.Philpott KL, McCarthy MJ, Klippel A, Rubin LL. J Cell Biol. 1997;139(3):809–15. doi: 10.1083/jcb.139.3.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Crowder RJ, Freeman RS. J Neurosci. 1998;18(8):2933–43. doi: 10.1523/JNEUROSCI.18-08-02933.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Matsuzaki H, Tamatani M, Mitsuda N, Namikawa K, Kiyama H, Miyake S, Tohyama M. J Neurochem. 1999;73(5):2037–46. [PubMed] [Google Scholar]
- 204.Rytomaa M, Lehmann K, Downward J. Oncogene. 2000;19(39):4461–8. doi: 10.1038/sj.onc.1203805. [DOI] [PubMed] [Google Scholar]
- 205.Namikawa K, Honma M, Abe K, Takeda M, Mansur K, Obata T, Miwa A, Okado H, Kiyama H. J Neurosci. 2000;20(8):2875–86. doi: 10.1523/JNEUROSCI.20-08-02875.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Henry MK, Lynch JT, Eapen AK, Quelle FW. Blood. 2001;98(3):834–41. doi: 10.1182/blood.v98.3.834. [DOI] [PubMed] [Google Scholar]
- 207.Suhara T, Mano T, Oliveira BE, Walsh K. Circ Res. 2001;89(1):13–9. doi: 10.1161/hh1301.092506. [DOI] [PubMed] [Google Scholar]
- 208.Yamaguchi H, Wang HG. Oncogene. 2001;20(53):7779–86. doi: 10.1038/sj.onc.1204984. [DOI] [PubMed] [Google Scholar]
- 209.Wick A, Wick W, Waltenberger J, Weller M, Dichgans J, Schulz JB. J Neurosci. 2002;22(15):6401–7. doi: 10.1523/JNEUROSCI.22-15-06401.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Martin D, Salinas M, Lopez-Valdaliso R, Serrano E, Recuero M, Cuadrado A. J Neurochem. 2001;78(5):1000–8. doi: 10.1046/j.1471-4159.2001.00472.x. [DOI] [PubMed] [Google Scholar]
- 211.Conery AR, Cao Y, Thompson EA, Townsend CM, Jr, Ko TC, Luo K. Nat Cell Biol. 2004;6(4):366–72. doi: 10.1038/ncb1117. [DOI] [PubMed] [Google Scholar]
- 212.Simak J, Holada K, Vostal JG. BMC Cell Biol. 2002;3(1):11. doi: 10.1186/1471-2121-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Li Y, Tennekoon GI, Birnbaum M, Marchionni MA, Rutkowski JL. Mol Cell Neurosci. 2001;17(4):761–7. doi: 10.1006/mcne.2000.0967. [DOI] [PubMed] [Google Scholar]
- 214.Putcha GV, Deshmukh M, Johnson EM., Jr J Neurosci. 1999;19(17):7476–85. doi: 10.1523/JNEUROSCI.19-17-07476.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Yamaguchi A, Tamatani M, Matsuzaki H, Namikawa K, Kiyama H, Vitek MP, Mitsuda N, Tohyama M. J Biol Chem. 2001;276:5256–5264. doi: 10.1074/jbc.M008552200. [DOI] [PubMed] [Google Scholar]
- 216.Miyashita T, Reed JC. Cell. 1995;80(2):293–9. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 217.Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Cell. 2001;107(2):137–48. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 218.Tang ED, Nunez G, Barr FG, Guan KL. J Biol Chem. 1999;274(24):16741–6. doi: 10.1074/jbc.274.24.16741. [DOI] [PubMed] [Google Scholar]
- 219.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Cell. 1999;96(6):857–68. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 220.Gilley J, Coffer PJ, Ham J. J Cell Biol. 2003;162(4):613–22. doi: 10.1083/jcb.200303026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Dijkers PF, Birkenkamp KU, Lam EW, Thomas NS, Lammers JW, Koenderman L, Coffer PJ. J Cell Biol. 2002;156(3):531–42. doi: 10.1083/jcb.200108084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Charvet C, Alberti I, Luciano F, Jacquel A, Bernard A, Auberger P, Deckert M. Oncogene. 2003;22(29):4557–68. doi: 10.1038/sj.onc.1206778. [DOI] [PubMed] [Google Scholar]
- 223.Widmann C, Gibson S, Johnson GL. J Biol Chem. 1998;273(12):7141–7. doi: 10.1074/jbc.273.12.7141. [DOI] [PubMed] [Google Scholar]
- 224.Earnshaw WC, Martins LM, Kaufmann SH. Annu Rev Biochem. 1999;68:383–424. doi: 10.1146/annurev.biochem.68.1.383. [DOI] [PubMed] [Google Scholar]
- 225.Shi Y. Cell. 2004;117(7):855–8. doi: 10.1016/j.cell.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 226.Hofmann K, Bucher P, Tschopp J. Trends Biochem Sci. 1997;22(5):155–6. doi: 10.1016/s0968-0004(97)01043-8. [DOI] [PubMed] [Google Scholar]
- 227.Takahashi H, Nakamura S, Asano K, Kinouchi M, Ishida-Yamamoto A, Iizuka H. Exp Cell Res. 1999;249(2):291–8. doi: 10.1006/excr.1999.4476. [DOI] [PubMed] [Google Scholar]
- 228.Keramaris E, Stefanis L, MacLaurin J, Harada N, Takaku K, Ishikawa T, Taketo MM, Robertson GS, Nicholson DW, Slack RS, Park DS. Mol Cell Neurosci. 2000;15(4):368–79. doi: 10.1006/mcne.2000.0838. [DOI] [PubMed] [Google Scholar]
- 229.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cell. 1997;91(4):479–89. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 230.Latchman DS. Curr Neurovasc Res. 2004;1(1):21–27. doi: 10.2174/1567202043480206. [DOI] [PubMed] [Google Scholar]
- 231.Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM, Green DR. Nat Cell Biol. 2000;2(8):469–75. doi: 10.1038/35019501. [DOI] [PubMed] [Google Scholar]
- 232.Engels IH, Stepczynska A, Stroh C, Lauber K, Berg C, Schwenzer R, Wajant H, Janicke RU, Porter AG, Belka C, Gregor M, Schulze-Osthoff K, Wesselborg S. Oncogene. 2000;19(40):4563–73. doi: 10.1038/sj.onc.1203824. [DOI] [PubMed] [Google Scholar]
- 233.Stegh AH, Barnhart BC, Volkland J, Algeciras-Schimnich A, Ke N, Reed JC, Peter ME. J Biol Chem. 2002;277(6):4351–60. doi: 10.1074/jbc.M108947200. [DOI] [PubMed] [Google Scholar]
- 234.Haddad JJ. Prog Neurobiol. 2004;73(5):359–77. doi: 10.1016/j.pneurobio.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 235.Chong ZZ, Lin SH, Kang JQ, Maiese K. Cell Mol Neurobiol. 2003;23(4–5):561–78. doi: 10.1023/A:1025158314016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Tsuruta F, Sunayama J, Mori Y, Hattori S, Shimizu S, Tsujimoto Y, Yoshioka K, Masuyama N, Gotoh Y. EMBO J. 2004 doi: 10.1038/sj.emboj.7600194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Shou Y, Li L, Prabhakaran K, Borowitz JL, Isom GE. Toxicol Sci. 2003;75(1):99–107. doi: 10.1093/toxsci/kfg157. [DOI] [PubMed] [Google Scholar]
- 238.Griffin RJ, Ogawa A, Williams BW, Song CW. Immunol Invest. 2005;34(3):343–59. doi: 10.1081/imm-200066270. [DOI] [PubMed] [Google Scholar]
- 239.Kroger H, Hauschild A, Ohde M, Bache K, Voigt WP, Thefeldt W, Kruger D. Gen Pharmacol. 1999;33(2):203–6. doi: 10.1016/s0306-3623(98)00232-8. [DOI] [PubMed] [Google Scholar]