


Abstract
The Trypanosoma brucei exoribonuclease, TbDSS-1, has been implicated in multiple aspects of mitochondrial RNA metabolism. Here, we investigate the role of TbDSS-1 in RNA processing and surveillance by analyzing 12S rRNA processing intermediates in TbDSS-1 RNAi cells. RNA fragments corresponding to leader sequence upstream of 12S rRNA accumulate upon TbDSS-1 depletion. The 5′ extremity of 12S rRNA is generated by endonucleolytic cleavage, and TbDSS-1 degrades resulting upstream maturation by-products. RNAs with 5′ ends at position −141 and 3′ ends adjacent to the mature 5′ end of 12S rRNA are common and invariably possess oligo(U) tails. 12S rRNAs with mature 3′ ends and unprocessed 5′ ends also accumulate in TbDSS-1 depleted cells, suggesting that these RNAs represent dead-end products normally destined for decay by TbDSS-1 in an RNA surveillance pathway. Together, these data indicate dual roles for TbDSS-1 in degradation of 12S rRNA maturation by-products and as part of a mitochondrial RNA surveillance pathway that eliminates stalled 12S processing intermediates. We further provide evidence that TbDSS-1 degrades RNAs originating upstream of the first gene on the minor strand of the mitochondrial maxicircle suggesting that TbDSS-1 also removes non-functional RNAs generated from other regions of the mitochondrial genome.
INTRODUCTION
The degradation of RNA is a key process involved in controlling RNA abundance and, thus, gene expression (1,2). RNA decay is also required for the 5′ and 3′ trimming of precursor transcripts to produce mature RNAs, for the removal of maturation by-products, and for the elimination of aberrant or improperly processed RNAs that inevitably form during RNA synthesis and maturation (3). In eukaryotes, a complex set of mechanisms has evolved to check the quality of mRNA and to distinguish between those RNAs that are or will become mature and RNAs that are non-functional and need to be degraded. Two distinct mRNA surveillance pathways, nonsense-mediated decay and non-stop decay, facilitate the detection and destruction of mRNAs with premature or absent termination codons, respectively (4,5). In addition, a nuclear poly(A) polymerase complex is involved in a polyadenylation-mediated RNA surveillance mechanism that degrades unmodified initiator 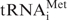 , rRNA precursors and snoRNP precursors (6,7). Similar quality control mechanisms also apparently exist in mitochondria. In yeast, recent data indicate that the mitochondrial degradosome complex composed of DSS-1 exoribonuclease and SUV3 RNA helicase plays a central role in RNA surveillance, degrading aberrant and unprocessed RNAs. Yeast strains lacking degradosome components strongly accumulate mitochondrial mRNA and rRNA precursors untrimmed at their 5′ and 3′ termini and display variations in the steady-state levels of mature mRNAs (8,9). These cells also exhibit massive accumulation of mitochondrial introns, indicating a role for DSS-1 in decay of these maturation by-products (8). In Arabidopsis mitochondria, polynucleotide phosphorylase (PNPase) degrades rRNA and tRNA maturation by-products and RNA transcripts that are expressed to high levels from regions lacking known genes (10). Thus, mitochondria of divergent species have evolved different mechanisms for the removal of aberrant or improperly processed RNAs and maturation by-products. A recent study in yeast showed that growth defects resulting from mutations in the mitochondrial degradosome can be rescued simply by mutations that reduce the rate of mitochondrial transcription (11). This observation underscores the vital importance of pathways dedicated to removing non-functional RNAs.
, rRNA precursors and snoRNP precursors (6,7). Similar quality control mechanisms also apparently exist in mitochondria. In yeast, recent data indicate that the mitochondrial degradosome complex composed of DSS-1 exoribonuclease and SUV3 RNA helicase plays a central role in RNA surveillance, degrading aberrant and unprocessed RNAs. Yeast strains lacking degradosome components strongly accumulate mitochondrial mRNA and rRNA precursors untrimmed at their 5′ and 3′ termini and display variations in the steady-state levels of mature mRNAs (8,9). These cells also exhibit massive accumulation of mitochondrial introns, indicating a role for DSS-1 in decay of these maturation by-products (8). In Arabidopsis mitochondria, polynucleotide phosphorylase (PNPase) degrades rRNA and tRNA maturation by-products and RNA transcripts that are expressed to high levels from regions lacking known genes (10). Thus, mitochondria of divergent species have evolved different mechanisms for the removal of aberrant or improperly processed RNAs and maturation by-products. A recent study in yeast showed that growth defects resulting from mutations in the mitochondrial degradosome can be rescued simply by mutations that reduce the rate of mitochondrial transcription (11). This observation underscores the vital importance of pathways dedicated to removing non-functional RNAs.
The nature of gene expression in the mitochondria of Trypanosoma brucei necessitates a key role for RNA degradation in both the control of gene expression and in the elimination of non-functional RNAs. Transcription is polycistronic (12–14) and, thus, both endonuclease cleavage and exonuclease trimming are presumably required to form mature RNAs from polycistronic precursors. Many of the adjacent genes overlap extensively, such that it is impossible to produce two mature monocistronic RNAs from the same precursor molecule (13,14). This overlapping gene arrangement indicates that a substantial volume of non-functional RNA will be generated during pre-mRNA maturation. Further, a majority of mitochondrial RNAs in T. brucei require an extensive editing process involving uridine insertion and deletion to form translatable mRNAs (15,16). Improperly edited RNAs are abundant in the steady-state RNA pool, and it is unknown whether these RNAs are intermediates destined to become properly edited or aberrantly processed RNAs that need to be removed (17,18). The maturation of mRNAs, rRNAs and guide RNAs (gRNAs) also requires proper 3′ end modification through addition of non-encoded tails (19,20). Therefore, the potential exists for the generation of a large volume of non-functional RNA that needs to be removed from the system. This suggests that an RNA surveillance system must exist for the rapid identification and degradation of improperly processed RNAs, and that numerous by-products of RNA maturation must also be efficiently recognized and degraded.
We previously identified an exoribonuclease in T. brucei mitochondria, that we termed TbDSS-1 (21). This protein is an RNR exoribonuclease family member and a homolog of Saccharomyces cerevisiae degradosome component, DSS-1. Targeted disruption of TbDSS-1 using RNA interference (RNAi) demonstrated that the protein is essential for growth in procyclic form T. brucei (21). TbDSS-1 depletion results in aberrant levels of several mitochondrial RNA species, including never edited, unedited and edited mRNAs as well as gRNAs (21). The diversity of phenotypes associated with TbDSS-1 down-regulation suggests that this protein participates in multiple aspects of mitochondrial RNA metabolism. Similar to yeast, T. brucei apparently lacks a homolog of the PNPase that functions in RNA surveillance in Arabidopsis mitochondria (Kao,C.-Y. and Read,L., unpublished data) (1). This strongly suggests that TbDSS-1 could have a critical role in RNA maturation, RNA surveillance and the removal of maturation by-products in the mitochondria of trypanosomes. In this article, we investigate the potential involvement of TbDSS-1 in these aspects of mitochondrial RNA metabolism using the 12S/9S rRNA locus as a model system. We chose to focus our analysis on processing and decay of the transcript containing the 12S and 9S rRNAs because it is one of the simplest and most well-studied primary transcripts in T. brucei mitochondria. T. brucei mitochondrial rRNAs do not undergo RNA editing and the 12S and 9S genes do not overlap, simplifying analysis of precursor RNA processing. Additionally, previous studies have shown that 12S and 9S rRNAs are initially transcribed as a precursor with a 5′ flanking region that is rapidly processed and degraded (22). Here, we find that multiple RNA fragments corresponding to the leader sequence upstream of the 5′ extremity of 12S rRNA accumulate in TbDSS-1 depleted cells. RNA maturation by-products whose 3′ ends are immediately adjacent to the mature 5′ end of 12S rRNA are a prominent class of RNAs that accumulate in TbDSS-1 depleted cells, and, unexpectedly, sequence analysis revealed that the majority of these RNAs possess oligo(U) tails. These data indicate that the 5′ extremity of 12S rRNA is generated by endonucleolytic cleavage and that TbDSS-1 is responsible for the degradation of upstream 12S rRNA-processing intermediates. We also observe that 12S rRNAs with properly processed 3′ ends and unprocessed 5′ ends accumulate in TbDSS-1 depleted cells, suggesting that these RNAs represent non-functional processing intermediates that are normally removed by an RNA surveillance pathway involving TbDSS-1. We further demonstrate that TbDSS-1 is dispensable for proper 3′ processing of 12S rRNA. In addition, analysis of RNAs that accumulate upon TbDSS-1 down-regulation suggests a pathway for 12S rRNA maturation. Finally, northern blot data suggest that TbDSS-1 also functions in decay of RNAs originating from the region upstream of the first gene on the minor strand of the mitochondrial genome. Overall, our results indicate a role for TbDSS-1 in RNA surveillance and degradation of 12S rRNA maturation by-products, and suggest that the enzyme plays a wider role in decay of similar products from other regions in the mitochondrial genome.
MATERIALS AND METHODS
Oligonucleotides used in this study
The oligonucleotides used in this study are listed as follows with restriction sites underlined. P1 (5′ GTAAATTCAAATTTAAACTCTCATGTCCCAAA 3′), P2 (5′ TTGGATTACAATTTAAATTTTTAAAAGC 3′), P3 (5′ TAAATTCAAGTTCAAACCTCCAAATCCC 3′), P4 (5′ TTTAAGTTATCAATAAAAAAC 3′), P5 (5′ TTTTCTAATTAA ATAAAATACACATAATAAAC 3′), P6 (5′ GCTTTTAAAAATTTAAAATTTGTAATCCAA 3′), 12S-1(5′ GCTTGTTAACCTGCTCGAAC 3′), 12S-2 (5′ CGGGATCCATTAAAAATAAATGTGTTTCATCGTC 3′), 12S-3 (5′ CATTATTATAATATTCTTCTTAA TTG G 3′), 12S-4 (5′ TATATTTATTTGGTACATATAGAAC 3′) and 12S–9S3 (5′ ATTATTGAAAAAAGAAAGAAGAATA 3′).
All numbers referring to primers given here are as in M94286, accession number for T. brucei kinetoplast maxicircle genes. The reverse primers P3, P1, P5, P2, 12S-3, 12S-4 and 12S-1 correspond to nucleotides 373–399, 1013–1044, 1302–1333, 1334–1363, 1370–1396, 1626–1650 and 2331–2350, respectively. The forward primers P4, P6, 12S-2 and 12S–9S3 correspond to nucleotides 1046–1066, 1334–1363, 2351–2376 and 2508–2532, respectively. Sense primer RPS12-5′-T7 and antisense primer RPS12UE were used to generate a 221 nt PCR template for synthesizing the riboprobe for the flanking region 5′ to the minor strand mRNAs, as previously described (23). All primers and their respective locations are shown in appropriate figures below.
Trypanosome growth and RNA analysis
Procyclic form T. brucei with a construct for the induction of RNAi of TbDSS-1 integrated into the rDNA spacer, were grown in SDM-79 supplemented with 15% fetal bovine serum in the presence of G418 (15 μg/ml), hygromycin (50 μg/ml) and phleomycin (2.5 μg/ml). Generation and initial characterization of these cells were previously published (21). For induction of double-stranded RNA (dsRNA), cells were cultured in the presence of 1 μg/ml tetracycline. Total RNA was purified from 1.3 × 109 to 4.8 × 109 cells (Purescript RNA isolation kit; Gentra Systems). Two separate inductions (including monitoring of growth and RNA isolation) were performed, and most analyses were repeated with RNA from both inductions.
Northern hybridization analysis
For northern hybridization analysis, 10–30 μg of total RNA was electrophoresed on 1.5% formaldehyde–agarose gels and transferred to nylon membrane. Blots were probed with the indicated oligonucleotide probes (P1, P2 and P3) labeled with [γ-32P] ATP and polynucleotide kinase (Invitrogen) for detection of 12S rRNA 5′ flanking region and hybridization was performed at 42°C (P2 and P3) or 50°C (P1), followed by five washes in 6× SSPE/1% sarkosyl at room temperature and one wash in 1× SSPE/1% SDS at 42°C or 50°C. The membrane was stripped after each hybridization and reprobed. Riboprobes for the P1–P3 region and the flanking region 5′ to the minor strand mRNAs were generated by in vitro transcription with incorporation of [α-32P] UTP, and hybridization was performed as previously described (24). Membranes were analyzed either by autoradiography or by phosphoimager analysis on a Bio-Rad Personal FX Phosphoimager using Quantity One software. Most analyses were repeated with RNA from two separate inductions.
PCR methods and cloning
Circular RT-PCR (cRT-PCR) was used to determine both the 5′ and 3′ extremities of 12S rRNA and its flanking regions (25). Five micrograms of total RNA isolated from TbDSS-1 RNAi cells was incubated with 40 U of T4 RNA ligase (New England Biolabs) in the supplied buffer supplemented with 2 U of RNase OUT (Invitrogen) and 1.5 U DNase I (Invitrogen) in a total volume of 25 μl. After phenol–chloroform extraction and ethanol precipitation, cDNAs were synthesized using Superscript III RNaseH– reverse transcriptase (Invitrogen) and designated reverse primer as indicated in the Results section. The region containing the junction of the 5′ and 3′ extremities was then amplified by RT-PCR using forward and reverse primers as indicated. PCR amplification was performed as follows: 1–2 successive rounds of 35 cycles at 94°C for 1 min, 40–56°C for 30°s, 72°C for 1 min. Annealing temperatures varied depending upon primer pair: 40°C (P3/P4), 52°C (P5/P6), 56°C (P2/12S-2) and 54°C (12S-2/12S-3). All cRT-PCR reactions were cloned using the TOPO cloning kit (Invitrogen) and multiple clones were randomly sequenced. All reactions were performed 2 to 3 times, and the majority of analyses were repeated on two RNA preparations from separate RNAi inductions.
RESULTS
RNAs containing 12S 5′ flanking region sequence accumulate in TbDSS-1 depleted cells
To determine whether TbDSS-1 plays a role in precursor transcript processing and/or RNA surveillance in the mitochondria of T. brucei, we began by analyzing RNAs generated from the primary transcript containing 12S and 9S rRNAs in TbDSS-1 RNAi cells (Figure 1A). This transcript is one of the simplest and most well-studied primary transcripts in T. brucei mitochondria. The 12S and 9S genes do not overlap, and in organello labeling experiments previously showed that 12S and 9S ribosomal RNAs are initially transcribed as a precursor with a ≥1.2 kb 5′ flanking region that is rapidly processed and degraded (22). Thus, the promoter is located in the variable region of the maxicircle at least 1.2 kb upstream of the 5′ end of the 12S rRNA (22). To test whether TbDSS-1 is involved in the maturation and/or turnover of rRNA precursor transcripts, we utilized our previously constructed TbDSS-1 RNAi cell line (21). Procyclic form T. brucei harboring an integrated tetracycline-inducible expression system for the induction of TbDSS-1 RNAi was grown in the presence or absence of tetracycline (21). Cell growth and down-regulation of TbDSS-1 mRNA was monitored for each RNA preparation used and was consistently similar to what was previously published (21). Cells grew normally for the first 4 days of induction but growth ceased by Day 6 after tetracycline addition. TbDSS-1 mRNA levels were reduced by an average of 50% and 30% of uninduced levels by Days 2 and 4, respectively, and mRNA was undetectable by Day 6.
Figure 1.

RNAs containing 12S rRNA 5′ flanking sequence accumulate in TbDSS-1 RNAi cells. (A) Location of primers P1–P3 on a schematic of 12S/9S rRNA genes and upstream flanking region. Arrows indicate the antisense orientation of the primers. Sizes of the different regions of the 12S rRNA locus are indicated in nts. (B–C) Northern hybridization analysis of RNAs containing 12S rRNA flanking sequence. Total RNA from uninduced (−tet) and induced (+tet) cells on Days 2, 4 and 6 after tetracycline addition was separated on a 1.5% agarose/formaldehyde gel, transferred to a nylon membrane and hybridized with 5′ [32P]-labeled P1, P3 or P2 oligo probe or internally radiolabeled P1/P3 riboprobe. Arrows indicate those transcripts that accumulate upon TbDSS-1 depletion, which have been labeled I–VI. Tubulin RNA levels (bottom) were monitored as a loading control.
To analyze changes in the metabolism of rRNA precursor transcripts upon TbDSS-1 down-regulation, we used northern hybridization analysis with antisense primers P1, P2 and P3, which are located various distances upstream of the mature 5′ end of 12S rRNA within the flanking region (Figure 1A). Northern blots probed with primer P1, which is located 320 nt upstream of the 5′ end of 12S rRNA, revealed an increase in RNAs ranging in size from ∼400 to 1600 nt in TbDSS-1 cells on Days 4 and 6 following tetracycline induction of RNAi (Figure 1B, left). Specifically, four RNAs, labeled I–IV, were detected only upon TbDSS-1 depleted on Days 4 and 6. To further map the regions contained in these accumulating RNAs, we used two additional probes, one upstream and one downstream of P1. Using primer P3, which is located 644 nt farther upstream within the flanking region, we observed three discrete RNAs of ∼650, 1300 and 1600 nt whose abundance was increased in TbDSS-1 depleted cells (Figure 1B, middle). The two larger RNAs, labeled I and II, appear to be the same size as the larger products I and II detected by primer P1 suggesting these RNAs span both P1 and P3 primer locations within the 12S flanking region. The smaller RNA of ∼650 nt does not correspond to any of the RNAs detected in the P1 blot, suggesting the 3′ of this RNA is upstream of the P1 primer location. To further clarify the sizes of the accumulating RNAs, we also performed the analysis with a more sensitive riboprobe that encompasses the area spanning from primer P1 to P3. Using the riboprobe P1/P3, we detected three discrete bands of ∼1300, 1600 and 1900 nt (Figure 1B, right). All three RNAs were strongly up-regulated in TbDSS-1 depleted cells compared to parallel uninduced cells. The two smaller bands at 1300 and 1600 nt apparently correspond to RNAs I and II detected in the P1 and P3 blots. Thus, the three different probes spanning the P1–P3 region identified a slightly different pattern of accumulating RNAs, presumably due to differences in the sequences and affinities of the probes. However, RNAs of 1300 and 1600 nt were consistently identified with all three probes. We next used primer P2, which is located immediately upstream of 12S rRNA gene, as a probe, and observed a broad band of just over 200 nt that accumulated very strongly upon TbDSS-1 depletion (Figure 1C). The decrease in the level of this RNA on Day 6 as compared to Day 4 after tetracycline induction may suggest compensation by another mechanism in the RNAi cells, since the tubulin control blot indicates equal loading of RNA in these samples. The location of the primers used in this analysis suggests that the major smaller RNA detected with primer P2 encompasses the region immediately upstream of the 5′ end of mature 12S rRNA since it was not observed with primers P1 and P3. Together, the northern hybridization data demonstrate that multiple RNA species containing various portions of the 12S rRNA 5′ flanking region accumulate in cells in which TbDSS-1 has been down-regulated, suggesting that TbDSS-1 is involved in the degradation of these RNA species. Additionally, because multiple RNAs of discrete lengths were observed, it suggests that endonucleolytic cleavage may be involved in processing the 5′ flanking region of rRNA precursors.
To more precisely identify the RNA species that accumulate in TbDSS-1 RNAi cells, we mapped their 5′ and 3′ extremities. To this end, we used the cRT-PCR technique in which RNAs are circularized by T4 RNA ligase prior to cDNA synthesis, and PCR is used to amplify the region encompassing ligated 5′ and 3′ ends (25). We first performed cRT-PCR experiments using primer P1 for cDNA synthesis and primers P3 and P4 for PCR amplification (Figure 2A). Primer P4 corresponds to a sense primer located immediately downstream of primer P1. Following two successive rounds of PCR amplification, we observed a smear of products from ∼400 to 1100 nt containing three distinct bands of ∼400, 600 and 800 nt (Figure 2B). These products were not observed in uninduced cells, from which only a single band of 300 nt was amplified. Cloning and sequencing of the 300 nt product from uninduced cells yielded only sequences that were apparent artifacts of the PCR reaction due to mis-priming. To define the sizes and 5′ and 3′ boundaries of the RNAs that accumulate in induced TbDSS-1 RNAi cells, we randomly cloned and sequenced over 20 cDNA products from both the Day 4, and Day 6 samples. However, we were unable to identify sequences corresponding to the sizes of the major accumulating cDNAs observed by gel electrophoresis, with all of the sequenced cDNAs being less than 400 bp. These cDNAs isolated from induced cells corresponded exclusively to 12S 5′ flanking region sequence and possessed 5′ ends between −1008 and −1188 and 3′ ends between −165 and −255 relative to the mature 12S rRNA 5′ end. One-third of these RNAs possessed short non-encoded 3′ oligo(U) or AU tails. Similar RNAs were not detected in randomly cloned and sequenced cDNAs from uninduced cells. While the PCR amplification and cloning steps certainly favor the detection of shorter compared with longer transcripts, the cRT-PCR results shown in Figure 2B are consistent with the northern blot analysis showing that fragments of the 12S rRNA 5′ flanking region are stabilized in TbDSS-1 RNAi cells. Thus, we conclude TbDSS-1 is responsible for degrading 12S rRNA precursors corresponding to the leader region upstream of the mature 12S 5′ end.
Figure 2.

Characterization of RNAs containing 12S 5′ flanking sequence in TbDSS-1 RNAi cells. (A) Location of primers P1, P3 and P4 used to map RNA 5′ and 3′ extremities by cRT-PCR. Arrows indicate the orientations of the primers. Vertical dotted lines indicate the distance in nt from the 12S rRNA 5′ end. (B) Negative image of an ethidium bromide-stained gel of cRT-PCR products amplified from P1 primed cDNA using primer P3 in combination with primer P4. RNA was isolated from cells uninduced (−tet) and induced with tetracycline (+tet) on Days 4 and 6 (D4, D6) following tetracycline addition. M, DNA marker consisting of bands with 100 bp increments. The positions of the 400, 600 and 800 bp markers is indicated on the left. The presence or absence of reverse transcriptase during the cDNA synthesis step is indicated by +RT or −RT, respectively.
The 5′ end of 12S rRNA is generated by an endonucleolytic cleavage event in which upstream products are oligouridylated and subsequently degraded by TbDSS-1
We next wanted to more closely analyze the abundant products that were detected by northern hybridization analysis of TbDSS-1 depleted cells using primer P2 (Figure 1C). For this analysis, we performed cRT-PCR using primer P2 for cDNA synthesis and primers P5 and P6 for PCR amplification (Figure 3A). Primer P5 is antisense, located immediately upstream of primer P2, while primer P6 is the sense sequence of P2 (Figure 3A). After two successive PCR amplifications, we observed a smear containing discrete bands at ∼150 bp (I), 300 bp (II) and 450 bp (III) in TbDSS-1 RNAi cells induced with tetracycline (Figure 3B). We also observed a band of ∼250 nt that was equally abundant in samples from both uninduced and induced cells. To characterize the RNAs that are stabilized in TbDSS-1 depleted cells, we randomly cloned and sequenced 15 cDNAs amplified from these cells. Sequence analysis revealed that, with one exception, the 3′ extremities of the clones from Groups I to III mapped at or within 1 nt downstream of the reported 5′ end of mature 12S rRNA as determined by Sloof et al. (26) (Figure 3C). We detected one clone with a 3′ extremity extending 27 nt into the 12S rRNA sequence. Thus, the primary difference between Group I and III RNAs is their 5′ extremity and the distance to which they extend into the 12S 5′ flanking region (Figure 3C). Surprisingly, all of the clones in Group I possessed non-encoded 3′ extensions, whereas such extensions were completely absent in cDNAs from Groups II and III. Eight of the clones from Group I possessed 3′ oligo(U) tails ranging from 3 to 5 nt, and two additional clones possessed AU tails. Sequence analysis from the cells not induced with tetracycline revealed that the major product at 250 bp also had a 3′ extremity mapping within 1 nt of the reported mature 5′ of 12S rRNA and a 5′ extremity ∼250 nt upstream of the mature 5′ end of 12S rRNA. The striking abundance, in induced samples, of clones with a 5′ extremity at −141 relative to the mature 12S 5′ end suggests that this site is a favored endonuclease cleavage site within the 12S rRNA 5′ flanking sequence. The location of the 3′ ends of accumulating 12S rRNA maturation by-products suggests that a second endonucleolytic cleavage occurs to create the 12S 5′ flanking region fragments that are stabilized upon TbDSS-1 depletion. Specifically, a precise cleavage occurs within a 2 nt region that defines the 5′ end of the mature 12S rRNA. It is of interest that a majority (10 out of 14) of the accumulating products whose 3′ ends are adjacent to the mature 5′ end of 12S rRNA possess oligo(U) tails. Thus, just as 3′ cleavage of T. brucei 12S and 9S rRNAs is linked to uridylation of the 3′ ends of mature rRNAs, so is cleavage at the 12S 5′ end linked to uridylation of the upstream cleavage product. This finding suggests that both 5′ and 3′ ends of 12S rRNA may be generated by the same machinery. Notably, all of the clones that possess a non-encoded 3′ extension have 5′ ends mapping to position −141, suggesting a functional link between cleavage at −141 and 3′ uridylation of the resulting RNA (see Discussion section). Based on their accumulation in TbDSS-1 depleted cells, small uridylated RNAs corresponding to the region immediately upstream of the 12S rRNA 5′ end are apparently major substrates for TbDSS-1.
Figure 3.

Characterization of RNAs containing flanking sequence immediately adjacent to the 12S 5′ end in TbDSS-1 RNAi cells. (A) Location of primers P2, P5 and P6 used to map RNA 5′ and 3′ extremities by cRT-PCR. Arrows indicate the orientations of the primers. (B) Negative image of an ethidium bromide-stained gel of cRT-PCR products amplified from P2 primed cDNA using primer P5 in combination with primer P6. M, DNA marker consisting of bands with 100 bp increments. The positions of the 200 and 400 bp markers is indicated on the left. Arrows indicate those major transcripts that accumulate upon tetracycline induction, which have been labeled I–III. Other labels as in Figure 2. (C) Schematic of experimentally determined 5′ and 3′ extremities from induced (+tet; Day 4 and Day 6) clones. The numbering of the 3′ and 5′ ends refers to the nts upstream of the 5′ end of 12S rRNA. Non-encoded U and A residues are boxed in gray. The numbers on the right indicate the frequency with which individual clones were isolated. The first clone in Group II has a 3′ extremity that extends 27 nt into 12S rRNA gene.
TbDSS-1 does not play a role in processing the 3′ end of 12S rRNA
We next wanted to determine whether the 3′ end of 12S rRNA is generated by an endonucleolytic cleavage event or involves 3′ to 5′ trimming, potentially by TbDSS-1. Northern blot analysis from our previous studies did not show a difference in the abundance of mature 12S rRNA between uninduced and induced TbDSS-1 RNAi cells (21). However, the technique used in those studies was not sensitive enough to detect the size difference of 3′ extended 12S rRNA transcripts that would arise if the 18 nt 12S–9S intergenic region had not been removed (Figure 1A). To determine whether TbDSS-1 functions in 12S rRNA generation by 3′ trimming, we performed cRT-PCR experiments using primers located within the 12S rRNA gene. The locations of primer 12S-1, used to initiate cDNA synthesis, as well as the location of primers 12S-2 and 12S-3, used for PCR amplification, are shown in Figure 4A. If TbDSS-1 is involved in 3′ trimming, we would expect to observe 3′ extended clones using this method. A discrete band of 250 bp was detected in both uninduced and induced cells at similar levels (Figure 4B). Cloning and sequencing of PCR products revealed that they encode 12S rRNAs correctly processed at their 5′ extremities, mapping within the 2 nt region that corresponds precisely to the 3′ extremities of the 12S 5′ flanking region maturation by-products identified in the previous experiment (compare Figure 4C 5′ ends with Figure 3C 3′ ends). The correlation of the 5′ and 3′ ends of PCR products derived by these two strategies provides further support for the role of endonucleolytic cleavage in the generation of 12S rRNA 5′ ends. Regarding the 3′ ends of the clones obtained using the strategy in Figure 4A, 11 out of 12 cDNAs from the uninduced samples and 9 out of 11 from the induced samples had mature 12S 3′ extremities and possessed oligo(U) tails ranging from 6 to 18 U residues, similar to what has been previously reported (19). These results show that there is no change in the abundance of mature 12S rRNA upon TbDSS-1 depletion, which is consistent with our previous northern blot results (21). In addition to mature 12S rRNA, we detected a small number of 3′-truncated 12S transcripts with 3′ extremities mapping 32–88 nt upstream of the mature 12S rRNA 3′ end (Figure 4C). These truncated products did not possess oligo(U) tails. The 3′-truncated 12S transcripts most likely represent degradation intermediates that exist in low abundance in these cells. The results shown here suggest that TbDSS-1 does not play a role in degrading these 3′ truncated 12S rRNAs since there was no significant increase of these RNAs in TbDSS-1 depleted cells. Because the cRT-PCR analyses presented in Figure 4 did not reveal any accumulation of 12S transcripts with extended 3′ extremities in TbDSS-1 RNAi cells, we conclude that TbDSS-1 does not play a direct role in processing the 3′ of 12S rRNA.
Figure 4.

TbDSS-1 is not required for correct 12S rRNA 3′ end processing. (A) Location of primers 12S-1, 12S-2 and 12S-3 used to map the 5′ and 3′ extremities of 12S rRNA in TbDSS-1 RNAi cells by cRT-PCR. Arrows indicate the orientations of the primers. (B) Negative image of an ethidium bromide-stained gel of cRT-PCR products amplified from 12S-1 primed cDNA using primer 12S-2 in combination with 12S-3. RNA was isolated from uninduced cells (−tet) and cells induced with tetracycline for 6 days (+tet). M, DNA marker consisting of bands with 100 bp increments. The positions of the 100 and 200 bp makers are indicated on the left. Other labels as in Figure 2. (C) Schematic of experimentally determined 5′ and 3′ extremities. Solid arrowheads indicate RNAs from induced cells; open arrowheads indicate RNAs from uninduced cells. The number of clones obtained at a given site is indicated above or below each arrowhead. The lengths of the oligo(U) tails of those clones with a mature 3′ extremity are indicated.
5′ extended 12S rRNAs accumulate in TbDSS-1 depleted cells
The results in Figure 4 demonstrate that TbDSS-1 is not required for maturation of the 12S rRNA 3′ end by exonucleolytic trimming. However, it remains possible that 12S rRNAs that are unprocessed at their 5′ and/or 3′ ends accumulate upon TbDSS-1 down-regulation, and that these classes of unprocessed RNAs are not detectable in the presence of overwhelming amounts of mature 12S rRNA using the PCR strategy presented in Figure 4. To specifically detect 5′ or 3′ unprocessed 12S rRNA, we utilized primer combinations consisting of an internal 12S rRNA primer and a primer corresponding to 5′ or 3′ flanking sequence. We first screened for 5′ unprocessed 12S rRNAs by cRT-PCR using 12S-1 primed cDNAs. The resulting cDNAs were then subjected to PCR amplification using a forward primer, 12S-2, located within the 3′ end of the 12S gene in combination with a reverse primer, P2, located within the 12S 5′ flanking region (Figure 5A, top). A major band of ∼350–400 nt accumulated in TbDSS-1 RNAi cells on Days 4 and 6 post-induction (Figure 5B). Cloning and random sequencing of cDNAs from induced cells revealed that the 5′ extremities of these cDNAs mapped within the 12S 5′ flanking region, in a range between residues 136 and 462 upstream of the mature 12S 5′ end (data not shown). The 5′ ends of 11 out of the 16 induced clones mapped at or near −141 upstream of the 12S 5′ end. Of these, two clones mapped to −136, two mapped to −140, and 7 mapped to −141. The −141 site is same 5′ end that was observed for all 10 induced clones in Figure 3C, again suggesting that −141 (and potentially the surrounding region to −136) is a preferred nuclease cleavage site. Sequence analysis of the 3′ ends revealed that the same 11 cDNAs had mature 3′ extremities corresponding to the reported 12S 3′ end and possessed oligo(U) tails ranging from 6 to 19 U residues (Figure 5C) (19). The five remaining cDNAs from the induced cells had 5′ ends extending from 223 to 462 nt upstream of the mature 12S 5′ end and were truncated within the 12S rRNA, 90 to 144 bp upstream of the mature 12S rRNA 3′ end. Clones that were truncated within the mature 12S rRNA sequence did not contain oligo(U) tails, similar to the truncated clones presented in Figure 4C. Thus, RNAs consisting of mature, oligouridylated 12S rRNA with attached 5′ flanking sequence are stabilized upon TbDSS-1 depletion, demonstrating that TbDSS-1 functions in the degradation of these processing intermediates. These results suggest that 12S rRNAs properly processed at their 3′ ends but unprocessed at their 5′ ends are specific targets for an RNA surveillance pathway involving TbDSS-1.
Figure 5.

Characterization of 5′ extended 12S rRNA precursors in TbDSS-1 RNAi cells. (A) Location of primers P2, 12S-1 and 12S-2 (top) and 12S-4, 12S-1 and 12S–9S3 (bottom) used to map the 5′ and 3′ extremities of unprocessed 12S rRNA in TbDSS-1 RNAi cells by cRT-PCR. Arrows indicate the orientations of the primers. (B) Negative image of an ethidium bromide-stained agarose gel of cRT-PCR products amplified from 12S-1 primed cDNA using primer P2 in combination with 12S-2. M, DNA marker consisting of bands with 100 bp increments. The position of the 100 and 400 bp markers are indicated on the left. (C) Schematic of experimentally determined 3′ extremities of 12S rRNA sequences in induced, solid arrowhead, samples on Day 4 after tetracycline addition. The sequences of primer 12S-2 and the 5′ end of 9S rRNA are underlined and double underlined, respectively. The 12S–9S intergenic region is shown in bold. The number of clones obtained with the respective 3′ end is indicated above each arrowhead. An asterisk indicates the positions of oligo(U) tails of 6–19 nt.
To determine whether TbDSS-1 functions in degradation of 3′ unprocessed 12S rRNAs, we performed cRT-PCR using 12S-1 primed cDNAs and two different primer pairs in which one primer was specific for mature 12S sequence and the other selected for the 12S–9S intergenic region (Figure 5A, bottom). Despite up to three rounds of PCR amplification with different primer pairs, no PCR products were detected in either the uninduced or induced samples. Thus, we conclude that 3′ extended 12S rRNAs are present in extremely low abundance. The absence of any accumulation of 3′ unprocessed 12S rRNA in TbDSS-1 depleted cells using this PCR strategy suggests that TbDSS-1 does not function in degrading this class of RNAs. Moreover, the paucity of 3′ unprocessed 12S rRNAs in the steady-state RNA population indicates that processing of the 3′ end of 12S rRNA is a very rapid event following transcription of the precursor RNA.
RNAs 5′ to the minor strand mRNAs accumulate in TbDSS-1 depleted cells
Since we observed the accumulation of 12S 5′ flanking region RNAs in TbDSS-1 RNAi cells, we decided to expand our analysis and determine if RNAs 5′ to the minor strand of the mitochondrial maxicircle genome accumulate upon TbDSS-1 depletion. This region of the genome is not as well characterized as the 12S/9S primary transcript and the location of the upstream promoter is unknown. We designed a riboprobe upstream of the ND3 gene 5′ to the minor strand mRNAs (Figure 6A). Northern blot analysis of TbDSS-1 RNAi cells revealed a major product of ∼1750 nt that was present at the same level in the uninduced and induced cells (data not shown, shorter exposure). Upon longer exposure however RNAs of ∼1400, 400 and 200 nt were detected only in cells depleted for TbDSS-1 (Figure 6B). These RNAs were 4- to 6-fold more abundant upon TbDSS-1 depletion after compensating for ethidium bromide-stained ribosomal RNAs. Therefore, various RNAs containing regions 5′ to the minor strand mRNAs accumulate in TbDSS-1 RNAi cells. This is similar to what was observed for 12S 5′ flanking region RNAs (i.e. the region just upstream of the coding sequence of the major strand), which are also targets for TbDSS-1 degradation. These results suggest that TbDSS-1 is involved in removing maturation by-products that originate from multiple regions of maxicircle genome.
Figure 6.

Characterization of RNA 5′ to the minor strand mRNAs in TbDSS-1 RNAi cells. (A) Schematic of a portion of the T. brucei maxicircle showing major and minor strand genes above and below the line respectively. Black line represents location of probe. (B) Northern hybridization analysis of RNAs containing regions 5′ to the minor strand mRNAs. Thirty micrograms of RNA isolated from uninduced (−tet) and induced (+tet) cells on Day 6 after tetracycline addition was subjected to northern blot analysis with a internally radiolabeled riboprobe. Ethidium bromide staining of ribosomal RNA bands is shown as a loading control.
DISCUSSION
This study shows that the mitochondrial exoribonuclease, TbDSS-1, plays a role in the degradation of 12S rRNA processing intermediates and maturation by-products. Abundant RNAs derived from the 12S rRNA locus accumulated upon TbDSS-1 depletion. These include fragments of the flanking region upstream of the mature 5′ extremity of 12S rRNA (Figures 1–3), 5′ flanking sequence plus truncated 12S rRNA sequence, as well as full-length 12S rRNAs with extended 5′ ends (Figure 5). A large percentage of the RNAs that we sequenced from TbDSS-1 depleted cells possess 5′ termini at or near −141 relative to the 12S 5′ end (Figure 3, RNAs I), and these RNAs invariably have non-encoded uridines at their 3′ ends. The accumulation of discrete populations of RNAs from the 12S 5′ flanking region suggests that degradation of this RNA involves the action of both an endonuclease that acts on preferential target regions and an exoribonuclease. Based on our results, TbDSS-1 comprises at least one of the exoribonucleases that functions in this pathway. Sequence data further suggest that the 5′ extremity of 12S rRNA is generated by endonucleolytic cleavage and that TbDSS-1 is responsible for the degradation of the resulting upstream maturation by-products. We also show that TbDSS-1 is dispensable for proper 3′ processing of 12S rRNA, and that the abundance of mature 12S rRNA does not change in TbDSS-1 RNAi cells consistent with our previous results (21). Together, these data demonstrate that one function of TbDSS-1 is the elimination of non-functional by-products of RNA processing in T. brucei mitochondria. In addition, our results implicate TbDSS-1 as part of an RNA surveillance pathway that removes apparently stalled processing intermediates in the 12S rRNA maturation pathway. Northern blot data also suggest that TbDSS-1 functions similarly with respect to maturation by-products derived from the region upstream of the first gene on the minor strand of the mitochondrial genome (Figure 6).
In some regards, the phenotype of TbDSS-1 depleted cells is reminiscent of yeast DSS-1 knockouts (9). Yeast mitochondria lacking DSS-1 massively accumulate excised group 1 introns, similar to the greatly increased abundance of 5′ flanking sequences excised from 12S rRNA precursors in T. brucei (Figure 1). In addition, yeast cells depleted of DSS-1 demonstrate an accumulation of unprocessed mitochondrial mRNAs and rRNAs, while in some cases corresponding mature RNAs of the proper size are abundant (8). In the yeast studies, it was concluded that the accumulation of precursor RNAs was not due to impairment of RNA processing, but rather due to the lack of precursor RNA degradation (8). A similar situation exists in trypanosome mitochondria, where we observe no change in the abundance of mature 12S rRNA upon TbDSS-1 depletion, but TbDSS-1 down-regulation leads to the accumulation of 12S processing intermediates that are improperly processed at their 5′ ends (Figure 5). Accumulation of these processing intermediates in TbDSS-1 depleted cells suggests they are dead-end products that are normally destined for decay by TbDSS-1. Since 3′ unprocessed 12S rRNAs do not accumulate in TbDSS-1 RNAi cells, our data suggest that some aspect of the 5′ flanking sequence is recognized by TbDSS-1 or an associated protein as part of an RNA surveillance pathway that detects stalled processing intermediates.
During our sequence analysis, we unexpectedly discovered that a large percentage of processing by-products whose 3′ ends are adjacent to the mature 5′ end of 12S rRNA possess oligo(U) tails (Figure 3). Previous studies, as well as data reported here, also demonstrate that both mature 12S and 9S rRNAs in T. brucei mitochondria are oligouridylated at their 3′ ends, although the mechanism of 3′ end formation is unknown (19). Our data suggest that the same machinery, composed of at least an endonuclease and a terminal uridylyltransferase (TUTase), may cleave both the 5′ and 3′ ends of 12S rRNA and deposit an oligo(U) tail on the upstream cleavage fragment. T. brucei mitochondria contain multiple TUTases that could account for the addition of oligo(U) tails to rRNA maturation by-products observed in this study (27,28). This includes RET1, which adds Us to gRNA 3′ ends, RET2, which is a component of the editosome, and additional TUTase homologs whose functions and subcellular localizations are unknown. Preliminary data from RET1 depleted cells revealed no significant decrease in the length or presence of the oligo(U) tails on the 12S flanking region maturation by-products (Mattiacio,J.L. and Read,L.K., unpublished data), suggesting either that RET1 does not uridylate the TbDSS-1 substrates identified here, or that mitochondrial TUTases are functionally redundant in this case.
The sequence characteristics of accumulating 12S rRNA processing intermediates and by-products in TbDSS-1 knockdown cells provide insight into the rRNA processing pathway. One striking aspect of the major accumulating RNAs is that the primary class of RNAs that undergo 3′ oligouridylation are those whose 5′ ends are at or near −141 nt relative to the 12S 5′ end. This correlation holds true regardless of whether 3′ ends immediately abut the 5′ end of 12S rRNA or map to the mature 12S rRNA 3′ end. Those RNAs whose 5′ ends are located upstream of −141 nt generally did not possess oligo(U) tails at their 3′ ends, whether their 3′ ends immediately abutted the 5′ end of 12S rRNA or were truncated within 12S rRNA gene. We did observe a few clones with whose 3′ ends mapped upstream of −141 that possessed small oligo(U) or AU tails (see discussion of Figure 3 in Results section). These data are consistent with the recent report from Madej et al. (29), suggesting that all RNA transcripts encoded by a kinetoplast genome are prone to some degree of oligouridylation. However, this modest degree of overall uridylation contrasts with the absolute correlation between oligouridylation of RNAs whose 5′ ends are at or near −141. These observations indicate that there is some specificity to the oligouridylation of 12S rRNA processing intermediates and by-products. Moreover, endonucleolytic cleavage at or near −141 was strictly correlated with cleavage at the mature 12S 5′ or 3′ end. RNAs whose 3′ ends were within the 12S coding region invariably had 5′ ends upstream of −141. These results suggest a functional link between endonucleolytic cleavage at −141, the 12S 5′ end and the 12S 3′ end. Taken together, our data are consistent with a model for a major pathway of 12S rRNA processing (Figure 7) that involves the following steps: (i) initial cleavage of the 12S precursor transcript at −141 nt within the 5′ flanking region, followed by recruitment of the processing machinery, (ii) cleavage and uridylation at 12S rRNA 3′ end and finally and (iii) cleavage and uridylation at 12S rRNA 5′ end (Figure 7A, Step 3, right). Complete processing results in mature 12S rRNA with an oligo(U) tail and 12S 5′ flanking region with an oligo(U) tail (Figure 7A, Step 3, right). The 5′ flanking fragment is then degraded by TbDSS-1. If, during this process, the 12S rRNA processing machinery prematurely dissociates after Step 2, the resulting product represents a dead-end processing intermediate that needs to be removed from the system by TbDSS-1 (Figure 7A, Step 3, left). The above scenario does not rule out alternate processing pathways. For example, while cleavage at −141 is common, it is not an absolute requirement for cleavage at the 12S 5′ end. RNAs with 5′ ends upstream of −141 and 3′ ends abutting the 12S 5′ end or extending into 12S rRNA also accumulated upon TbDSS-1 depletion (Figure 7B). These RNAs may result from an alternate processing pathway, or may represent incorrectly processed RNAs that are subject to TbDSS-1-mediated decay.
Figure 7.

Model for 12S rRNA processing and the role of TbDSS-1 in T. brucei mitochondria (see text in Discussion section for details).
In summary, we have identified a role for TbDSS-1 exoribonuclease in degrading products resulting from 12S rRNA maturation. This includes both the removal of maturation by-products and degradation of stalled processing intermediates in what appears to be a type of RNA surveillance pathway. The studies reported here, in combination with our previous analyses of mRNA levels and gRNA levels in TbDSS-1 depleted cells (17), indicate that this enzyme plays multiple roles in RNA degradation in T. brucei mitochondria. TbDSS-1 likely has additional functions in processing and surveillance of pre-mRNAs, where it may interface with other RNA processing components involved in editing and cleavage/polyadenylation. Indeed, our previous studies demonstrated that TbDSS-1 is associated with a multicomponent complex (17), and we are currently exploring the interaction of TbDSS-1 with other RNA processing factors. These studies will allow us to further explore the functions of TbDSS-1 in processing and surveillance of pre-mRNAs and gRNAs, and may provide insight into the coordination of multiple RNA processing events in trypanosome mitochondria.
ACKNOWLEDGEMENTS
We thank Melissa Ajunwa for technical assistance. We are also grateful to Dr Christopher Ryan for his very thorough critical reading of the manuscript. This work was supported by NIH grant RO1 AI47329 to L.K.R. J.L.M. was supported in part by NIH training grant #T32 AI07614. Funding to pay the Open Access publication charges for this article was provided by NIH grant R56 AI47329 to L.K.R.
Conflict of interest statement. None declared.
REFERENCES
- 1.Gagliardi D, Stepien PP, Temperley RJ, Lightowlers RN, Chrzanowska-Lightowlers ZM. Messenger RNA stability in mitochondria: different means to an end. Trends Genet. 2004;20:260–267. doi: 10.1016/j.tig.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 2.van Hoof A, Parker R. Messenger RNA degradation: beginning at the end. Curr. Biol. 2002;12:R285–R287. doi: 10.1016/s0960-9822(02)00802-3. [DOI] [PubMed] [Google Scholar]
- 3.Vasudevan S, Peltz SW. Nuclear mRNA surveillance. Curr. Opin. Cell Biol. 2003;15:332–337. doi: 10.1016/s0955-0674(03)00051-6. [DOI] [PubMed] [Google Scholar]
- 4.Wagner E, Lykke-Andersen J. mRNA surveillance: the perfect persist. J. Cell Sci. 2002;115:3033–3038. doi: 10.1242/jcs.115.15.3033. [DOI] [PubMed] [Google Scholar]
- 5.Frischmeyer PA, van Hoof A, O’Donnell K, Guerrerio AL, Parker R, Dietz HC. An mRNA surveillance mechanism that eliminates transcripts lacking termination codons. Science. 2002;295:2258–2261. doi: 10.1126/science.1067338. [DOI] [PubMed] [Google Scholar]
- 6.Vanacova S, Wolf J, Martin G, Blank D, Dettwiler S, Friedlein A, Langen H, Keith G, Keller W. A new yeast poly(A) polymerase complex involved in RNA quality control. PLoS Biol. 2005;3:e189. doi: 10.1371/journal.pbio.0030189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.LaCava J, Houseley J, Saveanu C, Petfalski E, Thompson E, Jacquier A, Tollervey D. RNA degradation by the exosome is promoted by a nuclear polyadenylation complex. Cell. 2005;121:713–724. doi: 10.1016/j.cell.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 8.Dziembowski A, Piwowarski J, Hoser R, Minczuk M, Dmochowska A, Siep M, van der Spek H, Grivell L, Stepien PP. The yeast mitochondrial degradosome. Its composition, interplay between RNA helicase and RNase activities and the role in mitochondrial RNA metabolism. J. Biol. Chem. 2003;278:1603–1611. doi: 10.1074/jbc.M208287200. [DOI] [PubMed] [Google Scholar]
- 9.Dziembowski A, Malewicz M, Minczuk M, Golik P, Dmochowska A, Stepien PP. The yeast nuclear gene DSS1, which codes for a putative RNase II, is necessary for the function of the mitochondrial degradosome in processing and turnover of RNA. Mol. Gen. Genet. 1998;260:108–114. doi: 10.1007/s004380050876. [DOI] [PubMed] [Google Scholar]
- 10.Holec S, Lange H, Kuhn K, Alioua M, Borner T, Gagliardi D. Relaxed transcription in Arabidopsis mitochondria is counterbalanced by RNA stability control mediated by polyadenylation and polynucleotide phosphorylase. Mol. Cell. Biol. 2006;26:2869–2876. doi: 10.1128/MCB.26.7.2869-2876.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rogowska AT, Puchta O, Czarnecka AM, Kaniak A, Stepien PP, Golik P. Balance between transcription and RNA degradation is vital for Saccharomyces cerevisiae mitochondria: reduced transcription rescues the phenotype of deficient RNA degradation. Mol. Biol. Cell. 2006;17:1184–1193. doi: 10.1091/mbc.E05-08-0796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grams J, McManus MT, Hajduk SL. Processing of polycistronic guide RNAs is associated with RNA editing complexes in Trypanosoma brucei. Embo. J. 2000;19:5525–5532. doi: 10.1093/emboj/19.20.5525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koslowsky DJ, Yahampath G. Mitochondrial mRNA 3′ cleavage/polyadenylation and RNA editing in Trypanosoma brucei are independent events. Mol. Biochem. Parasitol. 1997;90:81–94. doi: 10.1016/s0166-6851(97)00133-3. [DOI] [PubMed] [Google Scholar]
- 14.Read LK, Myler PJ, Stuart K. Extensive editing of both processed and preprocessed maxicircle CR6 transcripts in Trypanosoma brucei. J. Biol. Chem. 1992;267:1123–1128. [PubMed] [Google Scholar]
- 15.Simpson L, Aphasizhev R, Gao G, Kang X. Mitochondrial proteins and complexes in Leishmania and Trypanosoma involved in U-insertion/deletion RNA editing. RNA. 2004;10:159–170. doi: 10.1261/rna.5170704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stuart K, Panigrahi AK. RNA editing: complexity and complications. Mol. Microbiol. 2002;45:591–596. doi: 10.1046/j.1365-2958.2002.03028.x. [DOI] [PubMed] [Google Scholar]
- 17.Decker CJ, Sollner-Webb B. RNA editing involves indiscriminate U changes throughout precisely defined editing domains. Cell. 1990;61:1001–1011. doi: 10.1016/0092-8674(90)90065-m. [DOI] [PubMed] [Google Scholar]
- 18.Koslowsky DJ, Bhat GJ, Read LK, Stuart K. Cycles of progressive realignment of gRNA with mRNA in RNA editing. Cell. 1991;67:537–546. doi: 10.1016/0092-8674(91)90528-7. [DOI] [PubMed] [Google Scholar]
- 19.Adler BK, Harris ME, Bertrand KI, Hajduk SL. Modification of Trypanosoma brucei mitochondrial rRNA by posttranscriptional 3′ polyuridine tail formation. Mol. Cell. Biol. 1991;11:5878–5884. doi: 10.1128/mcb.11.12.5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blum B, Simpson L. Guide RNAs in kinetoplastid mitochondria have a nonencoded 3′ oligo(U) tail involved in recognition of the preedited region. Cell. 1990;62:391–397. doi: 10.1016/0092-8674(90)90375-o. [DOI] [PubMed] [Google Scholar]
- 21.Penschow JL, Sleve DA, Ryan CM, Read LK. TbDSS-1, an essential Trypanosoma brucei exoribonuclease homolog that has pleiotropic effects on mitochondrial RNA metabolism. Eukaryot. Cell. 2004;3:1206–1216. doi: 10.1128/EC.3.5.1206-1216.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Michelotti EF, Harris ME, Adler B, Torri AF, Hajduk SL. Trypanosoma brucei mitochondrial ribosomal RNA synthesis, processing and developmentally regulated expression. Mol. Biochem. Parasitol. 1992;54:31–41. doi: 10.1016/0166-6851(92)90092-x. [DOI] [PubMed] [Google Scholar]
- 23.Kao CY, Read LK. Opposing effects of polyadenylation on the stability of edited and unedited mitochondrial RNAs in Trypanosoma brucei. Mol. Cell. Biol. 2005;25:1634–1644. doi: 10.1128/MCB.25.5.1634-1644.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Corell RA, Myler P, Stuart K. Trypanosoma brucei mitochondrial CR4 gene encodes an extensively edited mRNA with completely edited sequence only in bloodstream forms. Mol. Biochem. Parasitol. 1994;64:65–74. doi: 10.1016/0166-6851(94)90135-x. [DOI] [PubMed] [Google Scholar]
- 25.Kuhn J, Binder S. RT-PCR analysis of 5′ to 3′-end-ligated mRNAs identifies the extremities of cox2 transcripts in pea mitochondria. Nucleic Acids Res. 2002;30:439–446. doi: 10.1093/nar/30.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sloof P, Van den Burg J, Voogd A, Benne R, Agostinelli M, Borst P, Gutell R, Noller H. Further characterization of the extremely small mitochondrial ribosomal RNAs from trypanosomes: a detailed comparison of the 9S and 12S RNAs from Crithidia fasciculata and Trypanosoma brucei with rRNAs from other organisms. Nucleic Acids Res. 1985;13:4171–4190. doi: 10.1093/nar/13.11.4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aphasizhev R. RNA uridylyltransferases. Cell. Mol. Life Sci. 2005;62:2194–2203. doi: 10.1007/s00018-005-5198-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aphasizhev R, Aphasizheva I, Simpson L. Multiple terminal uridylyltransferases of trypanosomes. FEBS Lett. 2004;572:15–18. doi: 10.1016/j.febslet.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 29.Madej MJ, Alfonzo JD, Huttenhofer A. Small ncRNA transcriptome analysis from kinetoplast mitochondria of Leishmania tarentolae. Nucleic Acids Res. 2007;35:1544–1554. doi: 10.1093/nar/gkm004. [DOI] [PMC free article] [PubMed] [Google Scholar]