

Abstract
We present a helical unwinding assay for reversibly binding DNA ligands that uses closed circular DNA, topoisomerase I (Topo I), and two-dimensional agarose gel electrophoresis. Serially diluted Topo I relaxation reactions at constant DNA/ligand ratio are performed, and the resulting apparent unwinding of the closed circular DNA is used to calculate both ligand unwinding angle (φ) and intrinsic association constant (Ka). Mathematical treatment of apparent unwinding is formally analogous to that of apparent extinction coefficient data for optical binding titrations. Extrapolation to infinite DNA concentration yields the true unwinding angle of a given ligand and its association constant under Topo I relaxation conditions. Thus this assay delivers simultaneous structural and thermodynamic information describing the ligand–DNA complex. The utility of this assay has been demonstrated by using calichearubicin B (CRB), a synthetic hybrid molecule containing the anthraquinone chromophore of (DA) and the carbohydrate domain of calicheamicin γ1I. The unwinding angle for CRB calculated by this method is −5.3 ± 0.5°. Its Ka value is 0.20 × 106 M−1. For comparison, the unwinding angles of ethidium bromide and DA have been independently calculated, and the results are in agreement with canonical values for these compounds. Although a stronger binder to selected sites, CRB is a less potent unwinder than its parent compound DA. The assay requires only small amounts of ligand and offers an attractive option for analysis of DNA binding by synthetic and natural compounds.
Keywords: DNA unwinding, drug binding constants, daunorubicin, calicheamicin, ethidium bromide
By now there is little doubt that intercalation is a phenomenon of widespread biological importance. The intercalation model (1) describes a lengthening of the DNA helix and a concomitant unwinding of the phosphodiester backbone to accommodate a molecule of intercalator. As this unwinding varies with intercalator, the unwinding angle (φ) is a valuable biophysical parameter when evaluating the ability of a compound to interact with DNA. It represents the number of degrees by which the DNA is unwound about its helical axis per ligand molecule bound.
To date, most experimental approaches for measuring φ have exploited the special conformational characteristics of closed circular DNA (ccDNA). As isolated from prokaryotes and viruses, ccDNA is negatively supercoiled, that is to say, its writhe (Wr), linking difference (ΔLk), and superhelical density (σ, the average number of superhelical turns per 10 bp) are negative.§ Upon binding by intercalator, ccDNA is unwound so that ΔLk and σ each become less negative (Fig. 1B). For a certain critical amount of bound intercalator, all superhelicity disappears and the ccDNA exists as a relaxed circle in which the helical axis lies flat in a plane. For this example, ΔLk, Wr, and σ equal zero (Fig. 1C). Further titration with intercalator past this topological equivalence point continues to unwind the helix, introducing supercoils in an opposite sense so that both ΔLk and σ become positive (Fig. 1D).
Figure 1.

Schematic Topo I relaxation reactions of ccDNA at no, low ([I]L), medium ([I]M), and high ([I]H) ligand binding. a, Topo I relaxation; b, ligand removal by organic extraction. Ligand molecules are represented as nodules along the helical axis of the ccDNA shown. Reaction A lacks ligand, so the ccDNA product is fully relaxed with ΔLk = 0. In reaction B, enough ligand is bound to titrate out 2 superhelical turns (sht) so that the ccDNA now contains −3 sht. After a and b, these 2 titrated sht are manifested in a final ΔLk of −2. Reaction C depicts just enough bound ligand to fully titrate all 5 sht, so product and starting ccDNAs are indistinguishable. Finally, D shows high-ligand binding to +2 sht beyond the equivalence point of C. In this example, the ccDNA product bears this additional superhelicity in its ΔLk of −7.
Early hydrodynamic methods of studying σ and φ were based on changes in the sedimentation velocity (4–8), buoyant density (9–11), and viscosity (8) of ccDNA when bound by intercalator. The discovery of the enzyme topoisomerase I (Topo I) (12, 13) permitted other experimental approaches to both φ and σ, which were extended to studies of complexes between DNA and such topology-perturbing proteins as EcoRI (14), Xenopus transcription factor A (15), RNA polymerase (16–18), and the lac repressor (19). Topo I facilitates the thermodynamic equilibration between supercoiled and relaxed forms of ccDNA by creating a transient single-stranded nick at which superhelical strain can be relieved. Methods for the determination of φ and σ that have used Topo I and intercalating ligand are based on the processes shown in Fig. 1.
When ccDNA that has been intercalated to some extent is acted on by Topo I, a fully relaxed species is generated whose twist (Tw) and Wr reflect the presence of bound intercalator. In this fully relaxed species, Wr = ΔLk = 0. When this intercalator is removed, the sudden change in Tw is partially offset by a compensatory Wr so that Lk, an invariant topological property intrinsic to any intact molecule of ccDNA, remains unchanged. This change in Wr manifests itself conformationally as a reappearance of superhelicity, the magnitude of which is determined by the original amount of intercalator bound at the time of enzymatic religation. Quantitating this superhelicity in some way enables a calculation of φ or σ. In reality, the resulting ccDNA is not homogeneous and actually exists as a family of topoisomers differing by integral increments of ΔLk. Thermal conformational fluctuations at the time of ccDNA religation give rise to a Boltzman distribution of topoisomers around an average ΔLk (20–23). This central ΔLk (ΔLkc) is taken as the average most abundant topoisomer in the Boltzman population and is used as a comparative value between populations arising from Topo I reaction on ccDNA with various intercalator concentrations.
Probably the simplest method to quantitate such topoisomeric reaction products is resolution by agarose gel electrophoresis. In this method, the speed of topoisomer migration through a gel is directly proportional to degree of superhelicity. Electrophoretic band counting methods (20, 21, 24) differ from earlier procedures in that they eliminate the requirement for costly equipment such as the ultracentrifuge and are in general easier to perform. However, because band counting methods resolve topoisomeric reaction products in only one dimension, they suffer from severely limited resolution at the topological extremes of either fully relaxed or highly supercoiled ccDNA.
Unwinding as a Viable Measure of Extent of Ligand Binding.
The present method to calculate φ relies on resolving topoisomeric reaction products in two electrophoretic dimensions (25, 26) and analyzing the degrees of unwinding in a way that is analogous to the classic treatment of optical binding data (27). The incorporation of a second gel dimension, run perpendicular to the first, allows the clear resolution of topoisomers, which otherwise comigrate in just one dimension.
Mathematical analysis of reaction products uses the Scatchard binding isotherm adjusted for nearest neighbor exclusion by bound ligand (28, 29), presented in closed form by McGhee and Von Hippel (30):
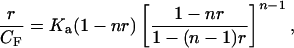 |
1 |
where CF is the free ligand concentration, r is the ratio of bound ligand to base pairs, Ka is the intrinsic association constant under the chosen conditions, and n is the number of sites (normally base pairs) covered by one molecule of bound ligand. Eq. 1 simplifies to linear form at low binding when r ≪ 1/n:
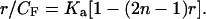 |
2 |
Traditionally, r has been calculated optically via the measured apparent ligand extinction coefficient difference between free and bound states, Δɛap. With increasing DNA concentration, Δɛap approaches Δɛ, and the fraction of bound ligand is Δɛap/Δɛ. With CB = (Δɛap/Δɛ)CT,
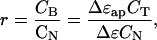 |
3 |
where CB, CT, and CN are, respectively, the concentrations of bound ligand, total ligand, and total DNA. With increasing dilution CF ≈ CT, so substitution of Eq. 3 into Eq. 2 and expansion to first order in (Δɛ − Δɛap) yields the linear equation:
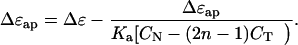 |
4 |
A plot of Δɛap vs. Δɛap/[CN − (2n − 1)CT] intercepts the ordinate at Δɛ as CN approaches infinity. The slope of the extrapolation yields −1/Ka directly.
It is Eq. 4 that has been brought to bear upon the problem of unwinding angle determination. For this task, Δɛap and Δɛ are replaced by, respectively, φap and φ, the apparent unwinding elicited by incomplete binding of ligand where r ≪ 1/n and the unwinding angle of the ligand itself at extrapolated infinite CN. Hence, Eq. 4 can be rewritten to describe the binding equilibrium of ligand to DNA by monitoring apparent unwinding:
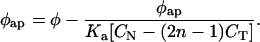 |
5 |
The resulting line’s interception with the ordinate yields φ and the negative inverse of its slope gives Ka under the temperature and ionic conditions of the Topo I relaxation reaction.
Topo I relaxation reactions containing identical total amounts of DNA and ligand are run in a series of ascending volumes. As total reaction volume increases, dissociation of the ligand–DNA complex is promoted by the parallel decrease of both DNA and ligand concentrations. The bp/ligand ratio remains unchanged (and high) throughout, ensuring the low r required by the mathematical approximations above. Reactions are treated and resolved as described below to determine ΔLkc, the Boltzman center of mass of integrated topoisomer band intensity representing the average most populous topoisomer (Fig. 2). For a given distribution of topoisomeric reaction products, φap is determined as follows:
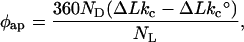 |
6 |
where ΔLkc and ΔLk°c are the Boltzman centers of topoisomer band intensity of, respectively, a reaction with ligand and a control reaction containing no ligand (this difference is ΔΔLkc). ND and NL represent, respectively, the actual numbers of ccDNA and ligand molecules in a particular reaction. The ligand is presumed to be randomly distributed throughout all the ccDNA molecules present in the reaction mixture. The factor of 360 represents 360° per ΔLk in the ccDNA, so φap carries the expected units of degrees. With φap, Eq. 5 can be used to obtain φ and Ka. The required estimation of n is discussed below.
Figure 2.

Example calculation of φap. Photos of EB-stained gels are shown above with their corresponding Boltzman distributions below. The results shown are from reactions containing (Left) (•) and lacking (Right) (○) the unwinding ligand DAUN, and values for ΔLk as well as electrophoretic dimensions are indicated on the gels. ΔLkc is −0.951 for the ligand-containing reaction, and the maximum of the ligand-free control reaction, ΔLk°c, is +1.49. This implies a ΔΔLkc of −2.44. With NL = 1.46 × 1014 ligand per reaction and ND = 1.17 × 1012 ccDNA per reaction, Eq. 6 gives φap = −7.03°. This becomes one point in a binding isotherm as shown in Fig. 3.
Intercalation Directed by a Selective Minor Groove Binder: Calichearubicin B (CRB).
In the intercalation of DNA by the antineoplastic agent daunorubicin (DA), the daunosamine sugar is indispensible (Fig. 4). Although only the aromatic rings of the DA anthraquinone are directly intercalated, it is the daunosamine sugar that rests in the minor groove to electrostatically stabilize the drug–DNA complex (31, 32). Indeed, in the absence of this sugar, daunorubicinone does not exhibit any appreciable intercalative propensity at physiologically relevant ionic strength and pH (data not shown). Yet for all its stability, the DA–DNA interaction demonstrates only minimal sequence specificity (33, 34).
Figure 4.

Structures of DA, MGOS, and CRB. Note that the connectivity in CRB is through the β-anomer of B.
In contrast, the enediyne antibiotic calicheamicin γ1I (CLM) exhibits marked sequence specificity in its cleavage of double-stranded DNA (36, 37). In this reaction, it is the extended pentacyclic carbohydrate domain of CLM that recognizes and directs aglycone cleavage to polypyrimidine–polypurine (Py⋅Pu) tracts in DNA (38, 39). This sequence selectivity has been proposed to result from a number of factors, including specific van der Waals contacts between atoms on the CLM carbohydrate and the DNA (40), preorganization of the CLM carbohydrate into an extended rigid structure (41, 42), and a greater inherent propensity for distortion of local DNA topology at preferred sequences, which promotes binding by CLM (43, 44). Of these, the latter two have emerged as the most critical for sequence selective binding by CLM. The specificity observed seems to be determined more by general DNA shape (minor groove width and depth, increased helical repeat, and bending) than by distinct contacts between CLM and DNA. With their deepened and narrowed minor groove and their slightly increased helical repeat, Py⋅Pu tracts present CLM with an environment spatially amenable to binding by the extended carbohydrate (45, 46).
With these considerations in mind, we wondered whether a synthetic union of the CLM carbohydrate with the DA anthraquinone might result in a molecule in which the binding specificity of the former is conferred to the intercalation of the latter. Such a molecule is CRB, shown in Fig. 4. The two domains of CRB are joined by an ethoxyethyl spacer, judged by computer modeling studies as sufficient to accommodate both anthraquinone intercalation and sensing of minor groove environment by the carbohydrate. Preliminary studies of the binding interaction between CRB and DNA indicated that the molecule binds preferentially to Py⋅Pu tracts (47), suggesting that the known sequence selectivity of the carbohydrate domain is in fact conferred to the hybrid molecule. Furthermore, the absorption profile obtained upon binding CRB to bulk DNA correlated essentially exactly in intensity attenuation and shift of λmax to that of natural DA, suggesting that the CRB chromophore is fully intercalated (47).
These findings spurred the attempt to quantify this binding behavior by the present helical unwinding assay. However, the mathematical treatment of this binding had to reflect the fact that CRB does not bind to bulk DNA in the nonspecific manner shown by other compounds such as DA and ethidium bromide (EB). For binding by CRB, the number of potential binding sites was calculated from the known sequence of the ccDNA used in the helical unwinding assay. In the CRB studies, CN is therefore the concentration of potential binding sites rather than of base pairs.
MATERIALS AND METHODS
Materials.
Purified pUC19 DNA was isolated from transformed JM101 Escherichia coli by the Qiagen protocol. Assay by one-dimensional (1D) agarose gel electrophoresis confirmed that the DNA was more than 98% supercoiled ccDNA. Calf thymus Topo I and ultrapure agarose were purchased from GIBCO/BRL. EB and DA were purchased from Aldrich Chemicals and used without further purification. Microcon centrifugal concentrators were purchased from Amicon. All water used in reactions, gel casting, and running buffers was double-distilled.
Reactions.
Before each reaction series, ligand concentration as determined by absorbance by using the following molar extinction coefficients: ɛ480 = 5,600 M−1⋅cm−1 for EB and ɛ480 = 11,500 M−1⋅cm−1 for DA and CRB. DNA was prepared from stock solutions as a single premixture containing appropriate buffer for the smallest-volume reaction. This premixture was then distributed equally into the reaction tubes. Ample excess premixture was prepared so that the base-pair concentration could be determined exactly by absorbance. For this, a molar extinction coefficient of ɛ260 = 13,200 M(bp)−1⋅cm−1 was used.
Reactions containing identical amounts of ccDNA and ligand were run in a series of volumes spanning two orders of magnitude (20 μl to 2 ml) in intervals of factors of two. DNA concentration was typically between 175 and 275 μM (bp) in the smallest-volume reaction. Ligand concentration depended on the potency of ligand unwinding (see Discussion) but was typically between 5 and 10 μM in the smallest-volume reaction. Reactions of higher volume were supplemented with appropriate additional buffer stock to ensure constant ionic strength in all reactions.
Unwinding reactions were run in 50 mM Tris⋅HCl, pH 7.5/50 mM KCl/10 mM MgCl2/0.5 mM DTT/100 μM EDTA/BSA (30 μg/ml). Complete relaxation of the ccDNA was achieved using 10–11 units of Topo I per μg of ccDNA in each reaction and was ensured by reacting an additional control sample containing ccDNA but no ligand. After reaction for 4 h at 37°C, samples were extracted with an equal volume of 1:1 PhOH/CHCl3 buffered to pH 7.5 with 50 mM Tris⋅HCl to remove protein and ligand. The phases were partitioned and the aqueous phase from each sample was concentrated to approximately 10 μl by centrifugation. Small (about 0.5 μl) aliquots were removed for preliminary analysis by 1% 1D agarose gel electrophoresis. Control reactions lacking unwinding ligand were identically reacted, extracted, and concentrated as those containing ligand.
Nonuniform binding of EB during gel staining might be expected to introduce artifacts in observed band intensity that could skew the calculated value of ΔLkc. Both the superhelical state of the ccDNA and the absolute amount of ccDNA in a given band might be expected to cause such nonuniformity in EB binding. Two sets of control experiments were performed to investigate these possibilities. In one, identical amounts of ccDNA intercalated by various amounts of EB were completely relaxed and treated as described above so that the product ccDNAs spanned a range of superhelicities. These products were resolved by 1D agarose gel electrophoresis and visualized by EB staining. For the range of superhelicities relevant for the present method, the observed intensity (quantitated as described below) showed no systematic dependence on ΔLk (data not shown). In another control, various absolute amounts of ccDNA homogeneous in ΔLk were resolved by 1D agarose gel electrophoresis and visualized by EB staining. Quantitation of total topoisomer band intensity (see below) showed a linear dependence of intensity on the amount of ccDNA up to 1.2 μg of ccDNA, after which point the intensity saturated (data not shown). Because the present method involves no two-dimensional topoisomer band containing more than 0.6 μg of ccDNA, all quantitations of band intensity are well within the linear range required for reliable quantitation.
Electrophoresis.
Two-dimensional gel casting units (each 17 × 17 cm) were constructed according to established protocols (25). Gels were cast with 250 ml of 1% agarose in 80 mM TBE (80 mM Tris borate/1 mM EDTA) to a final gel thickness of approximately 8 mm. A single loading well with a diameter of 1 mm was made in a corner of each gel. The first dimension was electrophoresed 20 h in 1 liter of 80 mM TBE running buffer at 1.9 V/cm. After the first dimension, the gel was rotated 90° in its casting tray, chloroquine was added in a small volume to a final concentration of 1.1 μg/ml and the gel was soaked with light agitation for 1 h. Electrophoresis in the second dimension then proceeded at 1.9 V/cm for an additional 18 h.
Visualization and Analysis.
After electrophoresis, gels were soaked in EB (1 μg/ml) for 2 h, destained in water for 1 h, and photographed on a UV transilluminator with Polaroid 667 film. The photographs were scanned with a Hewlett–Packard Scan Jet 3P and the photonegatives of individual topoisomer bands were quantitated by using nih image software. Resulting datasets were analyzed by using kaleidagraph software, with an appropriate baseline being subtracted from each plot prior to integration of total topoisomer band intensity. The integrated intensities were then plotted to give a Boltzman distribution from which ΔLkc was calculated by fitting to I = IM exp[−w(ΔLk − ΔLkc)], where I and IM are, respectively, integrated band intensity and maximum band intensity at the peak of the topoisomer distribution and w is the width of the distribution. ΔLkc was then used to obtain φap via Eq. 6 (see above). Fig. 2 shows an example of topoisomeric reaction products from reactions with and without ligand, and the corresponding Boltzman distributions of topoisomer band intensity.
RESULTS
EB and DA as Comparative Unwinders.
To demonstrate the accuracy of the method and provide comparative values for CRB, helical unwinding assays were performed on the known intercalators DA and EB. The results of these reactions are displayed graphically in Fig. 3. The values of n used were 2 for EB and 4 for DA (see Discussion).
Figure 3.

Binding isotherms for CRB (○, n = 1), DA (□, n = 4), and EB (•, n = 2). Unwinding data are plotted according to Eq. 5. Interception of the ordinate at extrapolated infinite CN yields φ directly, and the negative inverse of the slope yields Ka. An additional hypothetical isotherm for DA (▪, n = 6) has been included to show that the intercept value of φ does not depend on the value of n used.
Fig. 3 shows that the line for EB intercepts the ordinate at −28.7 ± 2.9° and the line for DA intercepts at −12.1 ± 1.2°, both values in good agreement with canonical values for the unwinding angles (Table 1). The value for ΔLk°c, the Gaussian center of topoisomeric mass for a control reaction lacking any unwinding ligand, was found to be +1.5° for the ccDNA used. This value (discussed below) is an average of 5 ligand-free controls and is used as ΔLk°c in all calculations of ΔΔLkc.
Table 1.
Summary of unwinding and binding parameters for CRB, DA, and EB
| Ligand | φ | φc | Ka, ×106 M−1 |
|---|---|---|---|
| CRB | −5.30° | — | 0.39 |
| DA | −12.1° | −12° | 0.14 |
| EB | −28.7° | −26° | 0.02 |
φ is the unwinding angle calculated by the present method. φc is the canonical unwinding angle for a given ligand, obtained as the intercept of a respective isotherm with the ordinate in Fig. 3. Ka is the intrinsic association constant calculated as the negative inverse of a respective isotherm’s slope in Fig. 3.
Unwinding by CRB.
Methidiumpropyl EDTA·Fe(II) footprinting data for CRB and the methyl glycoside of the CLM oligosaccharide (MGOS) on a SphI–SgrAI restriction fragment of pBR322 showed cleavage enhancement at identical sites for CRB and MGOS, each corresponding to Py⋅Pu sites of 4 bp. The observed sites d(CTTC)⋅d(AGGA) and d(TTTC)⋅ d(GAAA) extend the list of other polypyrimidine sites observed for MGOS and CLM in the literature, suggesting that it is indeed more the general steric environment of the minor groove in such motifs that facilitates carbohydrate binding rather than distinct ligand–DNA contacts of certain unique sequences. With this in mind, 380 potential binding sites were calculated in pUC19 (the ccDNA used in the experiments) by taking all possible 4-bp combinations of cytidine and thymidine (or guanosine and adenosine for the complementary strand). Here CN is taken as the concentration of such sites in an unwinding reaction and n = 1. Fig. 3 shows the graphical representation of these data, giving an unwinding angle of −5.3 ± 0.5°. The association constants for CRB, DA, and EB obtained by taking the negative inverse of the slopes in Fig. 3 are summarized in Table 1.
DISCUSSION
Comparing the Binding of CRB and DA.
The higher Ka of CRB relative to its parent intercalator DA would seem to indicate an enhanced thermodynamic complex stabilization gained by replacing the daunosamine sugar with the tethered CLM carbohydrate. However, the attenuated φ of CRB (−5.3 ± 0.5°) relative to DA (−12.1 ± 1.2°) suggests that other factors are also at play.
One interpretation is that the unwinding effect of the chromophore in CRB is partially canceled by a positive winding by the bound carbohydrate at its minor groove site. Such partial cancellation would diminish apparent CRB unwinding relative to DA. In fact, the carbohydrate domain has been reported to bend DNA in NMR solution studies (41, 46). Although Topo I experiments using MGOS alone indicate a positive ΔΔLk (data not shown), this effect is only observable with levels of MGOS in excess of actual binding sites (saturating conditions such as these invalidate the equations of the assay; see below), and even then, the effect is only slight (ΔΔLk ≈ +0.75). It is therefore implausible that the much lower levels of this weak winder in CRB would be capable of compensating the relatively potent unwinding effect of the anthraquinone by more than half. The observation of a slightly positive ΔΔLk for MGOS confirms earlier hypotheses that methidiumpropyl/EDTA Fe(II) cleavage enhancement at MGOS-bound sites derives from local topological perturbation of DNA (39, 47). However, the present assay is incapable of distinguishing ΔTw from ΔWr in the global ΔLk actually observed. It thus remains unknown whether this slight winding effect by MGOS is because of twisting, writhing, or a combination of both.
Mathematical analysis of the CRB data identical to that used for DA, using Eq. 5 with CN = [bp]TOT and n = 4 indicates a 3-fold higher total binding affinity of DA relative to CRB. This treatment ignores any inherent sequence preference of CRB and serves only to compare the apparent association constants of the two drugs for DNA of average sequence. Because the apparent affinity analyzed this way is smaller for CRB than for DA, we conclude that the sequence selectivity of the CLM carbohydrate serves to restrict the binding of CRB from sites otherwise available to the parent intercalator DA.
The selectivity of CRB in its binding of DNA is supported by methidiumpropyl/EDTA·Fe(II) footprinting studies in which patterns of cleavage enhancement for CRB and MGOS correlate exactly. Adjusting CN to reflect the consensus Py⋅Pu motif required for CRB binding and setting n = 1 accounts for CRB binding selectivity in Eq. 5 and raises the KA value for CRB above that for DA. However, the Ka values in Table 1, which are based on this reasoning, imply only an approximately 3-fold improvement of CRB affinity for sites of appropriate sequence over the affinity of DA for bulk or average-sequence DNA. Thus the carbohydrate side chain provides only a modest advantage over the daunosamine sugar in contributing to affinity for the target sequences. The major effect of exchanging the carbohydrate portion is to prevent binding of CRB to the bulk sequences to which DA can readily bind. This result is not entirely expected, given the high DNA binding affinity of MGOS that can be inferred from its ability to inhibit DNA cutting by CLM at MGOS concentrations only an order of magnitude higher than CLM (39).
Although the synthetic union of the DA chromophore and the CLM carbohydrate has resulted in a molecule combining minor groove sequence recognition with intercalative unwinding, this has been at the cost of unwinding propensity. The data support a model in which the chromophore- and carbohydrate-binding domains in the DNA twist toward each other by +6 to 7° so that simultaneous binding by each drug domain is possible. In this model, the −12° unwinding angle of the DA chromophore is partially canceled in its contribution to the global parameter ΔLk so that the net φ observed is only −5.3°. Such a scenario encompasses earlier spectrophotometric evidence supporting full intercalation by the CRB chromophore by comparison to that of DA (47), as well as the finding that MGOS alone does not engender appreciable positive twist in the DNA at its binding site (see above). These findings suggest that the ethoxyethyl linker in CRB is of a slightly nonoptimum length to allow simultaneous bifunctional drug–DNA interactions without a deformation of the DNA helix in the form of positive twisting. This is not unreasonable given the unavoidable constraint to segments of integral bond length in the synthesis of the spacer separating the two domains of CRB. This postulated imperfect steric match may be the reason why the carbohydrate side chain does not contribute more strongly than it does to the affinity of CRB for its target sequences.
The Mutual Dependence of φ and σ.
A difficulty with past methods for determination of φ involves the mutual dependence of φ and σ. Traditionally, determination of one of these two parameters required prior knowledge of the other, precluding any a priori calculation of either. In addition, it has become common practice in most of the past methods to calibrate the experimental φ of an unknown ligand to that of EB, itself accepted to be about −26 ± 2° (9). Hence any calculated unwinding angle was necessarily a relative one. Although initial electrophoretic band counting was in fact capable of providing an absolute measure of φ, the method still presupposed prior knowledge of σ for the ccDNA used in the reactions and a measure of the maximum bound mole fraction of ligand per base pair (v).
In contrast, the present assay for the simultaneous determinination of φ and Ka requires no prior knowledge of either σ or v, using instead sufficiently high CN/CT ratios so that extrapolation to infinite CN is possible. At this infinity point, the ligand is assumed to be essentially completely bound to the DNA, and φap = φ. The prerequisites for the present method are (i) that the Kd of the ligand–DNA complex be sufficiently low to warrant the assumption of near complete ligand binding at high CN, (ii) knowledge of the size (in base pairs) of the ccDNA used in the relaxation reactions, and (iii) an estimate of the number of potential binding sites along a molecule of the ccDNA used.
Choice of n.
Consideration of Eq. 5 reveals the need for an estimate of n, the number of potential binding sites covered by one molecule of bound ligand. For intercalating ligands such as DA and EB, which show only minimal sequence selectivity in their binding to bulk DNA, each base pair in the ccDNA may be seen as making an equal contribution to the number of total viable binding sites. Under these conditions, potential binding sites overlap one another and the neighbor exclusion model (28–30) applies, with n greater than unity [notice that substitution of n = 1 into Eq. 1 regenerates the original Scatchard binding isotherm (27) in which independent potential binding sites do not overlap]. If the serially diluted reactions are run at a sufficiently high DNA/ligand ratio, then CT ≪ CN and the choice of n in Eq. 5 has little effect on the resulting plot. For less potent unwinders, it may be necessary to run reactions of higher ligand concentration to generate a quantifiable ΔΔLkc. In these situations, the CT/CN quotient rises and the choice of n becomes more important. However even in this case, n affects only the slope of the line in Eq. 5 (the source of Ka) and has no bearing upon the line’s intercept with the ordinate, from which the unwinding angle is determined (Fig. 3). Given a reasonable estimate of the number of potential binding sites along the ccDNA, this method is, therefore, capable of generating a good measure of φ regardless of the chosen n. For exceedingly weak unwinders, a detectable ΔΔLkc is sometimes possible only by addition of excess ligand so that all potential binding sites remain saturated even at low CN. At this point, Eq. 5 becomes invalid. The limit of the method’s applicability is thus reached as the CT/CN ratio approaches unity.
The choices of n = 2 and n = 4 for the respective ligands EB and DA are based on literature values for these compounds (48, 49). Because of DA’s importance as a comparative model for anthracycline binding in the present study of CRB, its interaction with bulk DNA was carefully monitored in fluorescence quenching titrations under Topo I reaction buffer conditions. Analysis of fluorescence emission intensity by Eq. 1 corroborated the value of n = 4 used for DA in the unwinding calculation (data not shown). The value of n = 1 for the present CRB binding calculation reflects the fact that the number of base pairs in a potential CRB binding site was preset to 4 in the calculation of these sites on the ccDNA. Because CN in this case represents the concentration of sites and one molecule of CRB is assumed to cover one site, n is set to unity. The mathematical treatment of CRB unwinding data is thus consistent with a classical Scatchard interpretation in which independent potential binding sites do not overlap. This is in fact almost entirely the case with the potential CRB binding sites in the ccDNA used.
Regarding ΔLk°c and ΔLkc.
When relaxing ccDNA with Topo I in the absence of ligand, theory predicts that the average most populous topoisomer should center on the fully relaxed species for which ΔLk°c = 0 (Fig. 1A). Yet the value for ΔLk°c used in the above calculations is not zero, but +1.5. This shift of +1.5 superhelical turns away from the expected zero results from the fact that the enzymatic relaxation reaction is carried out at higher ionic strength than the subsequent two-dimensional agarose gel analysis. However, this has no effect on the calculation of ΔΔLkc, because both ligand-free and ligand-containing samples are reacted and treated identically. Because superhelicity of ccDNA depends on ionic strength, it is of paramount importance to ensure identical salt conditions for all samples during reaction. To this end, supplementing samples of higher volume with appropriate additional reaction buffer is essential. It is then merely a matter of convention which topoisomer one chooses to use as a reference to compare control and ligand-containing samples. For this, the Boltzman center of topoisomer band intensity is the most obvious and most accurately calculable point of reference for the determination of ΔΔLkc.
In summary, the present method is unique among related assays for its efficiency in describing ligand–DNA interactions both structurally and thermodynamically with a single experiment. The assay’s ease of performance and its requirement for only extremely small amounts of ligand make it ideal for the analysis of synthetic and natural compounds intended as potential DNA-interacting drugs. Disadvantages include the restriction to solution conditions suitable for Topo I activity and the requirement that the ligand produce a change in ΔLk so that its binding can be detected.
Acknowledgments
This work was supported by the National Institutes of Health (Grants 5-T32-AI 07404-05, CA 28824, and HL 28549).
ABBREVIATIONS
- ccDNA
closed circular DNA
- Topo I
topoisomerase I
- CRB
calichearubicin B
- DA
daunorubicin
- CLM
calicheamicin γ1I
- EB
ethidium bromide
- Wr
writhe
- ΔLk
linking difference
- Tw
twist
- 1D
one dimensional
- MGOS
methyl glycoside of the CLM oligosaccharide
Footnotes
A commentary on this article begins on page 4092.
Lk = Tw + Wr, where Tw is the twist about the helical axis and Wr is the writhe of the helical axis through three-dimensional space. Whereas Tw and Wr are each geometric properties that vary with environmental conditions such as temperature and ionic strength, Lk is a topological property that is invariant in the absence of single-stranded nicks. ΔLk is defined as the difference in superhelical turns between a fully relaxed species for which Wr = 0 and species j of nonzero Wr. Although ΔLk can assume a fractional value, Lk is integral by definition. Excellent treatments of ccDNA topology exist in the literature (2, 3).
References
- 1.Lerman L S. J Mol Biol. 1961;3:18–30. doi: 10.1016/s0022-2836(61)80004-1. [DOI] [PubMed] [Google Scholar]
- 2.Bates A D, Maxwell A. In: DNA Topology. Rickwood D, Male D, editors. Oxford: Oxford Univ. Press; 1993. pp. 17–61. [Google Scholar]
- 3.Cozzarelli N R, Boles T C, White J H. In: DNA Topology and Its Biological Effects. Cozzarelli N R, Wang W C, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1990. pp. 139–184. [Google Scholar]
- 4.Wang J C. J Mol Biol. 1969;43:263–272. doi: 10.1016/0022-2836(69)90266-6. [DOI] [PubMed] [Google Scholar]
- 5.Bauer W, Vinograd J. J Mol Biol. 1968;33:141–171. doi: 10.1016/0022-2836(68)90286-6. [DOI] [PubMed] [Google Scholar]
- 6.Waring M. J Mol Biol. 1970;54:247–279. doi: 10.1016/0022-2836(70)90429-8. [DOI] [PubMed] [Google Scholar]
- 7.Crawford L V, Waring M J. J Mol Biol. 1967;25:23–30. doi: 10.1016/0022-2836(67)90276-8. [DOI] [PubMed] [Google Scholar]
- 8.Révet B M J, Schmir M, Vinograd J. Nat New Biol. 1971;229:10–13. doi: 10.1038/newbio229010a0. [DOI] [PubMed] [Google Scholar]
- 9.Wang J C. J Mol Biol. 1974;89:783–801. doi: 10.1016/0022-2836(74)90053-9. [DOI] [PubMed] [Google Scholar]
- 10.Gray H B, Jr, Upholt W B, Vinograd J. J Mol Biol. 1971;62:1–19. doi: 10.1016/0022-2836(71)90127-6. [DOI] [PubMed] [Google Scholar]
- 11.Radloff R, Bauer W, Vinograd J. Proc Natl Acad Sci USA. 1967;57:1514–1521. doi: 10.1073/pnas.57.5.1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang J C, Liu L F. In: Molecular Genetics. Taylor J H, editor. New York: Academic; 1979. , Part III, pp. 65–88. [Google Scholar]
- 13.Wang J C. In: Nucleic Acid Research. Mizobuchi K, Watanabe I, Watson J D, editors. New York: Academic; 1983. pp. 549–566. [Google Scholar]
- 14.Kim R, Modrich P, Kim S-H. Nucleic Acids Res. 1984;12:7285–7292. doi: 10.1093/nar/12.19.7285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hanas J S, Bogenhagen D F, Wu C-W. Nucleic Acids Res. 1984;12:1265–1276. doi: 10.1093/nar/12.2.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beard P, Hughes M, Nyfeler K, Hoey M. Eur J Biochem. 1984;143:39–45. doi: 10.1111/j.1432-1033.1984.tb08336.x. [DOI] [PubMed] [Google Scholar]
- 17.Amouyal M, Buc H. J Mol Biol. 1987;195:795–808. doi: 10.1016/0022-2836(87)90485-2. [DOI] [PubMed] [Google Scholar]
- 18.Saucier J-M, Wang J C. Nat New Biol. 1972;239:167–170. doi: 10.1038/newbio239167a0. [DOI] [PubMed] [Google Scholar]
- 19.Kim R, Kim S-H. Cold Spring Harbor Symp Quant Biol. 1982;47:451–454. doi: 10.1101/sqb.1983.047.01.053. [DOI] [PubMed] [Google Scholar]
- 20.Keller W. Proc Natl Acad Sci USA. 1975;72:4876–4880. doi: 10.1073/pnas.72.12.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shure M, Vinograd J. Cell. 1976;8:215–226. doi: 10.1016/0092-8674(76)90005-2. [DOI] [PubMed] [Google Scholar]
- 22.Depew R E, Wang J C. Proc Natl Acad Sci USA. 1975;72:4275–4279. doi: 10.1073/pnas.72.11.4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pulleyblank D E, Shure M, Tang D, Vinograd J, Vosberg H-P. Proc Natl Acad Sci USA. 1975;72:4280–4284. doi: 10.1073/pnas.72.11.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dougherty G. Biosci Rep. 1983;3:453–460. doi: 10.1007/BF01121956. [DOI] [PubMed] [Google Scholar]
- 25.Bowater R, Aboul-Ela F, Lilley D M J. Methods Enzymol. 1992;212:105–120. doi: 10.1016/0076-6879(92)12007-d. [DOI] [PubMed] [Google Scholar]
- 26.Lee C-H, Mizusawa H, Kakefuda T. Proc Natl Acad Sci USA. 1981;78:2838–2842. doi: 10.1073/pnas.78.5.2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scatchard G. Ann NY Acad Sci. 1949;51:660–672. [Google Scholar]
- 28.Crothers D M. Biopolymers. 1968;6:575–584. doi: 10.1002/bip.1968.360060411. [DOI] [PubMed] [Google Scholar]
- 29.Müller W, Crothers D M. J Mol Biol. 1968;35:251–290. doi: 10.1016/s0022-2836(68)80024-5. [DOI] [PubMed] [Google Scholar]
- 30.McGhee J D, Von Hippel P H. J Mol Biol. 1974;86:469–489. doi: 10.1016/0022-2836(74)90031-x. [DOI] [PubMed] [Google Scholar]
- 31.Moore M H, Hunter W N, d’Estaintot B L, Kennard O. J Mol Biol. 1989;206:693–705. doi: 10.1016/0022-2836(89)90577-9. [DOI] [PubMed] [Google Scholar]
- 32.Frederick C A, Williams L D, Ughetto G, van der Marel G A, van Boom J H, Rich A, Wang A H-J. Biochemistry. 1990;29:2538–2549. [PubMed] [Google Scholar]
- 33.Chaires J B, Fox K R, Herrera J E, Britt M, Waring M J. Biochemistry. 1987;26:8227–8236. doi: 10.1021/bi00399a031. [DOI] [PubMed] [Google Scholar]
- 34.Roche C J, Thomson J A, Crothers D M. Biochemistry. 1994;33:926–935. doi: 10.1021/bi00170a011. [DOI] [PubMed] [Google Scholar]
- 35.Nicolaou K C, Dai W-M. Angew Chem Int Ed Engl. 1991;30:1387–1416. [Google Scholar]
- 36.Zein N, Sinha A M, McGahren W J, Ellestad G A. Science. 1988;240:1198–1201. doi: 10.1126/science.3240341. [DOI] [PubMed] [Google Scholar]
- 37.Zein N, Poncin M, Nilakantan R, Ellestad G A. Science. 1989;244:697–699. doi: 10.1126/science.2717946. [DOI] [PubMed] [Google Scholar]
- 38.Drak J, Iwasawa N, Danishefsky S, Crothers D M. Proc Natl Acad Sci USA. 1991;88:7464–7468. doi: 10.1073/pnas.88.17.7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aiyar J, Danishefsky S J, Crothers D M. J Am Chem Soc. 1992;114:7552–7554. [Google Scholar]
- 40.Hawley R C, Kiessling L L, Schreiber S L. Proc Natl Acad Sci USA. 1989;86:1105–1109. doi: 10.1073/pnas.86.4.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walker S, Murnick J, Kahne D. J Am Chem Soc. 1993;115:7954–7961. [Google Scholar]
- 42.Walker S, Valentine K, Kahne D. J Am Chem Soc. 1990;112:6428–6429. [Google Scholar]
- 43.Uesugi M, Sugiura Y. Biochemistry. 1993;32:4622–4627. doi: 10.1021/bi00068a020. [DOI] [PubMed] [Google Scholar]
- 44.Kuduvalli P N, Townsend C A, Tullius T D. Biochemistry. 1995;34:3899–3906. doi: 10.1021/bi00012a005. [DOI] [PubMed] [Google Scholar]
- 45.Mah A, Price M A, Townsend C, Tullius T D. Tetrahedron. 1994;50:1361–1378. [Google Scholar]
- 46.Ikemoto N, Kumar R A, Ling T, Ellestad G A, Danishefsky S J, Patel D J. Proc Natl Acad Sci USA. 1995;92:10506–10510. doi: 10.1073/pnas.92.23.10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Depew K M, Zeman S M, Boyer S H, Denhart D J, Ikemoto N, Danishefsky S J, Crothers D M. Angew Chem Int Ed Engl. 1996;35:2797–2801. [Google Scholar]
- 48.Wu H-W, Dattagupta N, Hogan M, Crothers D M. Biochemistry. 1980;19:626–634. doi: 10.1021/bi00545a004. [DOI] [PubMed] [Google Scholar]
- 49.Chaires J B, Dattagupta N, Crothers D M. Biochemistry. 1983;22:284–292. doi: 10.1021/bi00271a009. [DOI] [PubMed] [Google Scholar]