

Abstract
Batten disease (neuronal ceroid lipofuscinoses, NCLs) are a group of inherited childhood diseases that result in severe brain atrophy, blindness and seizures, leading to premature death. To date eight different genes have been identified, each associated with a different form. Linkage analysis indicated a CLN5 form in a colony of affected New Zealand Borderdale sheep. Sequencing studies established the disease-causing mutation to be a substitution at a consensus splice site (c.571+G>A), leading to the excision of exon 3 and a truncated putative protein. A molecular diagnostic test has been developed based on the excision of exon 3. Sequence alignments support the gene product being a soluble lysosomal protein. Western blotting of isolated storage bodies indicates the specific storage of subunit c of mitochondrial ATP synthase. This flock is being expanded as a large animal model for mechanistic studies and trial therapies.
Keywords: CLN5, NCL, Batten disease, mRNA splicing, animal model, Borderdale sheep, lysosomal storage disease
Introduction
The neuronal ceroid lipofuscinoses (NCLs, Batten disease) are a group of fatal inherited neurodegenerative diseases affecting an estimated 1 in 12,500 live births world-wide (Rider and Rider, 1999). They are characterised by severe brain atrophy and the accumulation of fluorescent lysosome derived organelles (storage bodies) in neurons and most other cells throughout the body. Retinal degeneration is also a common feature. Affected children start life normally but then develop progressive visual failure, mental and motor deterioration. They sleep poorly, suffer nightmares, hallucinations, fits and seizures which are difficult to control, and die between infancy and early adulthood. Adult onset cases have also been reported. Presently there are no effective therapies.
A number of different mutations in at least eight genes underlie the group (see www.ucl.ac.uk/ncl), and more have been suggested. The clinical features, characteristic pathologies and ultrastructure of each form have been well described along with the genetics and biochemical characteristics (Goebel et al., 1999; Haltia, 2003; Haltia, 2006). Better understanding of the genetics has led to improved diagnosis and an increase in the number of cases reported.
The NCLs are lysosomal storage diseases in which protein is stored in lysosome derived organelles. Mutations in lysosomal enzymes are responsible for some forms. Mutations i) in palmitoyl protein thioesterase 1 (PPT1) cause the infantile CLN1 form (Vesa et al., 1995), ii) in cathepsin D cause a congenital form (Siintola et al., 2006; Tyynelä et al., 2000), iii) in tripeptidyl peptidase 1 (TPP1) cause the classical late infantile CLN2 form (Sleat et al., 1997) and iv) in a soluble lysosomal protein of unknown function cause the variant late infantile CLN5 form (Bessa et al., 2006; Holmberg et al., 2004; Holmberg et al., 2000; Pineda-Trujillo et al., 2005; Savukoski et al., 1998). Another group of proteins, all of unknown function, reside in the lysosomal membrane or membranes of pre-lysosomal compartments, and are associated with i) the juvenile CLN3 form (Cao et al., 2006; Ezaki et al., 2003; Fossale et al., 2004), ii) the Northern epilepsy CLN8 form (Lonka et al., 2004), iii) the CLN6 variant late infantile form (Heine et al., 2004; Mole et al., 2004) and iv) the CLN7 Turkish variant (Siintola et al., 2007).
The specific storage of subunit c of mitochondrial ATP synthase in most NCLs was unequivocally established by direct protein sequencing (Chen et al., 2004; Fearnley et al., 1990; Palmer et al., 1989; Palmer et al., 1992; Tyynelä et al., 1997) and inferred from immunohistochemical studies. Sphingolipid activator proteins A and D are stored in the CLN1 infantile form (Tyynelä et al., 1993). Traditionally it was thought that neuropathology and gliosis were consequences of this storage but a recent study in the CLN6 form in sheep revealed that activation began prenatally, long before significant storage or neuron loss (Kay et al., 2006; Oswald et al., 2005). Indications from mouse models suggest that this may be general to the NCLs (Cooper, 2003; Pontikis et al., 2004) and other lysosomal storage diseases (Jeyakumar et al., 2003; Ohmi et al., 2003; Wada et al., 2000). Glial activation has also been reported in other neurodegenerative conditions (Hunot and Hirsch, 2003; Minagar et al., 2002; Neumann, 2001; Stoll and Jander, 1999).
Despite these advances in genetics and biochemistry, understanding of the pathobiology of the NCLs is still limited, yet understanding of the inter-connections between the gene lesions, subunit c storage and neurodegeneration is central to determining the options for therapy. Studies in humans are very restricted and progress depends on studying genetically defined animal models. Several mouse models are very useful but in general do not show the severe cortical atrophy, profound neuronal loss and retinal degeneration characteristic of the sheep and human diseases. Sheep have gyrencephalic human-like brains and are a convenient size for experimentation. Being production domestic animals they are used to human handling and straightforward and economic to maintain.
Recently an NCL was reported in New Zealand Borderdale sheep (Jolly et al., 2002). We report here that this disease is caused by a nucleotide substitution at a consensus splice site in the CLN5 gene (c.571+1G>A) and excision of exon 3, and the establishment of a flock for research particularly relevant to the human CLN5 late infantile form and to soluble enzyme forms of NCLs in general.
Materials and methods
Animals
Animals were mated in single pairs or by artificial insemination and maintained under standard New Zealand pastoral conditions on university research farms with adjacent animal hospital facilities. All procedures accorded to NIH guidelines and the New Zealand Animal Welfare Act (1999). Affected lambs were diagnosed at 2–3 months of age by histopathology of needle brain biopsies (Dickson et al., 1989) and/or analysis of mRNA extracted from blood (see below).
Leucocyte isolation and enzyme assays
Blood was collected from the jugular vein of affected and normal animals into heparinized tubes and mixed by inversion with 2.5 volumes of ice-cold lysis solution (150 mM NH4Cl, 10 mM KCl, 0.1 mM Na2EDTA). Leucocytes were pelleted by centrifugation, 3000 rpm, 10 min, 4°C and washed 2× in 10 ml of TKME buffer, pH 7.4 (50 mM Tris, 25 mM KCl, 5 mM MgCl2, 1 mM EDTA), 4°C, by resuspension and repelleting. The pellets were freeze dried, sent to Rotterdam, The Netherlands, and enzyme assays for PPT1, TPP1 and cathepsin D performed as previously described (van Diggelen et al., 2001).
Genomic DNA and BAC (bacterial artificial chromosome) clones
Sheep genomic DNA was extracted from heparinized venous blood by NaCl fractionation (Montgomery and Sise, 1990), from Whatman FTA cards (Whatman, Brentford, Middlesex, UK), or from EDTA blood samples using the QIAamp DNA mini extraction kit (Qiagen, Hilden, Germany). Four BAC clones (126L2, 16J4, 223D24 and 433I11) were kindly provided by Dr. Sue Galloway (AgResearch Limited, Dunedin, New Zealand) after screening of the ovine BAC library CHORI-243 (BACPAC Resources, Children's Hospital Oakland Research Institute, Oakland, CA, USA) with the bovine CLN5 mRNA sequence (GenBank accession no. NM_001046299) (Houweling et al., 2006). DNA was extracted from the BAC clones using a Qiagen plasmid midi prep kit.
Linkage analysis
Microsatellite markers were genotyped on 6% polyacrylamide gels in a LICOR 4200 analyzer (LICOR Biosciences, NE, USA) after PCR amplification using M13 tailed primers and a modified “touchdown” PCR cycle protocol (Oetting et al., 1995). This consisted of 10 μl PCRs with a template of 50 ng of genomic DNA in 1× QBuffer (Qiagen), 2.5 mM MgCl2, 200 mM dNTP, 0.08 pmol of each primer, 0.4 pmol of IRD700 (Licor) and 1 unit of Hotstar Taq polymerase (Qiagen). The cycles used were: 95°C for 15 min; 5 cycles of 95°C for 45 sec, 68°C annealing for 1 min 30 sec decreasing by 2°C/cycle and 72°C for 1 min; 4 cycles of 95°C for 45 sec, 58°C for 1 min decreasing by 2°C/cycle and 72°C for 1 min and finally 25 cycles of 95°C for 45 sec, 50°C for 1 min and 72°C for 1 min, followed by 72°C for 5 min.
Digital gel images of each microsatellite were transferred to RFLP scan software (BD Biosciences Bioimaging/Scanalytics, Rockville, MD, USA) and individual genotypes determined. The program CRI-MAP (Green, 1992) was used to determine linkage between the NCL phenotype and selected markers. The CLN6 SNP genotype was determined as previously described (Tammen et al., 2006).
Sequencing
PCR products were separated on agarose gels, excised and purified using the Perfectprep Gel Cleanup kit (Eppendorf, Hamburg, Germany) before sequencing using the PCR primers. Direct BAC sequencing was performed on 1.5 μg of purified BAC DNA per reaction and using only one primer.
Sequence reactions were performed using BigDye terminator v3.1 Cycle sequencing kits and the 5X BigDye v1.1/3.1 sequencing buffer (Applied Biosystems, Foster City, CA, USA) after pre-sequencing clean up of excess dye terminator with CleanSEQ dye-terminator removal (Agencout Bioscience Corporation, Beverly, MA, USA). Samples were run on an ABI PRISM 3100-Avant Genetic Analyser (Applied Biosystems).
Sequence compilation and analysis
The ovine sequence produced (GenBank accession no. NM_001082595) was analysed and aligned to CLN5 mRNA and protein sequences published on NCBI using Genedoc (Nicholas et al., 1997). Alignments were made to cattle (NM_001046299), human (NM_006493), mouse (NM_001033242) and dog (NM_001011556) sequences (Fig. 1).
Fig. 1. Alignment of human, mouse, dog, cattle and sheep CLN5 inferred polypeptides (N=normal and A=affected).

Black or grey backgrounds indicate amino acids conserved in all or several species. The premature terminal affected ovine sequence [Sheep_(A)] is highlighted by ******.
mRNA analysis and genotyping
Total RNA was isolated from whole blood using a total RNA blood purification kit (Invitrogen, Carlsbad, CA, USA) and fractions of each sample converted to cDNA using SuperScript III RNase H− reverse transcriptase (Invitrogen) and oligo d(T)15 as the primer. CLN5 cDNA was amplified using primers E2F2 and E4R3 under the following conditions: 95°C for 15 min, 35 cycles at 95°C for 30 sec, 55°C for 30 sec and 72°C for 1 min, followed by 72°C for 5 min. The products were separated on 1.5% agarose gels to identify genotypes (Fig. 3B), and confirmed by sequencing the 528 bp product generated by PCR using genomic DNA as the template and E3F1 and I3R285 as primers.
Fig. 3.
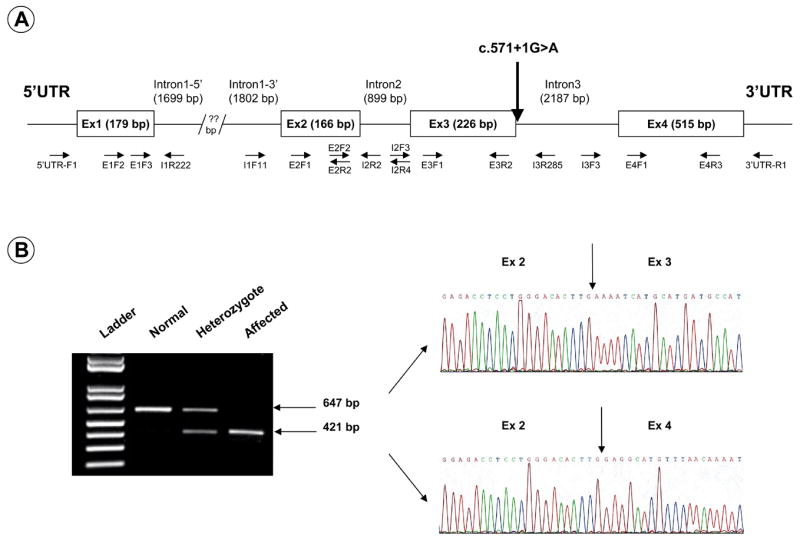
(A) Map of ovine CLN5. Note the position of the mutation observed in the Borderdale sheep (c.571+1G>A) as well as the position of the primers used in the sequencing of the gene. (B) CLN5 splice variant. (Left panel) Reverse transcription-PCR amplification of CLN5 between exons 2 and 4 yield normal transcripts retaining exon 3 on an agarose gel (647 bp) or affected transcripts lacking exon 3 (421 bp). Heterozygote transcripts contain both bands. (Right panel) Sequencing of the two bands confirmed the exact lack of exon 3 in the smaller band. Arrows indicate the boundaries of each exon.
Isolation and characterisation of storage bodies
Storage bodies were isolated from affected liver and brain, after previously described methods (Palmer et al., 1988). Liver, 10 g, was vigorously homogenised in 100 ml of deionised water at 4°C, sonicated 1 min and filtered through glass wool. The pellet after centrifugation (40 min, 1,400 gmax, 4°C) was washed 2× by resuspension in deionised water and centrifugation (15 min, 48,000 gmax, 4°C) and the soft buff overlay of the darker firmer storage body pellet was washed off each time. Brain grey matter, 5 g in 50 ml deionised water, was homogenised, sonicated and filtered as above, made up to a density of 1.18 by addition of solid CsCl, centrifuged 1 h at 48,000 gmax, the floated myelin and supernatant removed and the pelleted storage bodies washed as above.
Storage bodies were dissolved in 2% lithium dodecyl sulphate (LDS) and proteins separated by electrophoresis on 7×10 cm 15% 29:1 acrylamide:bisacrylamide gels (Laemmli, 1970) using 1% LDS as the detergent, 3.5 h, 100V. Gels were silver stained as previously described (Fearnley et al., 1990) or used for Western blotting. Proteins were transferred to nitrocellulose membranes (Amersham Biosciences, Little Chalfont, UK), 1 h, 200 mA which were blocked in 5% non-fat dry milk in Tris buffered saline (TBS), pH 8, overnight, 4°C, and incubated with antibodies against subunit c of mitochondrial ATP synthase (Palmer et al., 1995), 1/2000, 4 h, room temperature. The membranes were washed 4× with TBS containing 0.05% Tween 20, incubated with anti–rabbit IgG peroxydase conjugate (Sigma, St Louis, MO, USA), 1/10,000, 1 h, room temperature and the immune complexes revealed using chemiluminescent detection reagents (Pierce Biotechnology, Rockford, IL, USA).
Electron microscopy
Samples for electron microscopy were fixed in 2% glutaraldehyde in phosphate buffered saline, 36 h, 4°C, post-fixed in osmium tetroxide and thin sections stained with lead citrate and uranyl acetate for transmission electron microscopy.
Results
Establishment of the Borderdale flock
A colony of affected Borderdale sheep has been established following the diagnosis of NCL among 650 Borderdale ewes randomly mated each year to rams from the same stud (Jolly et al., 2002). One to three affected lambs were first noticed each year by the way they lagged behind when the flock was moved. Clinical signs noted were difficulty in moving through gateways with the rest of the flock, apparent blindness, low head carriage, a tendency to walk in circles and to move towards humans rather than away from them. The affected ewes were easily caught and appeared to be deaf. These clinical diagnoses were later confirmed by histological, immunohistological and electron microscopy studies on samples taken at post mortem, which indicated the possibility of a novel ovine NCL (Jolly et al., 2002).
Three affected ewes identified in 2002 were superovulated and artificially inseminated with semen from unaffected Coopworth rams, yielding 24 lambs after embryo transfer to surrogate ewes. The following year a similarly affected ewe was superovulated and inseminated with obligate heterozygous ram lamb semen from the cross above and two resultant ram lambs were diagnosed as affected on histology of brain biopsy samples. Subsequently the flock has been expanded by crossing obligate heterozygous ewes with affected rams, augmented by superovulation, artificial insemination and embryo transfer (Kay et al., 1999).
Location of the genetic defect
Defects in CLN1, 2 and cathepsin D were eliminated by assays of activities of the relevant enzymes in leucocytes from affected and control animals (Table 1). The activities of palmitoyl protein thioesterase 1, the enzyme coded by CLN1, were not significantly different between affected and control animals. The activities of tripeptidyl peptidase 1 (CLN2) and cathepsin D in the affected sheep were about twofold those in controls, indicating that catalytically active enzyme is coded for by both genes in affected sheep and suggesting that transcription may be up-regulated.
Table 1.
Activities of lysosomal enzymes in leucocytes from affected and control sheep
| PPT1a | TPP1 a | cathepsin D a | |
|---|---|---|---|
| Affected (n=3) | 66.03 ± 20.6b | 29.73 ± 6.7c | 602.3 ± 223.2d |
| Control (n=25) | 50.58 ± 12.9b | 13.80 ± 3.1c | 308.2 ± 115d |
Mean leucocyte activities of PPT1, palmitoyl protein thioesterase (CLN1); TPP1, tripeptidyl peptidase 1 (CLN2) and cathepsin D; in nmol of substrate/mg of protein/hour ± S.D. for n animals.
Not statistically different, p> .05.
Statistically different, p< .001.
Linkage analysis indicated a lesion in the region likely to contain ovine CLN5. A total of 87 sheep; 23 Coopworth sheep of unknown status, 15 affected sheep and 49 heterozygous sheep were genotyped for linkage analysis. No linkage was found to the single nucleotide polymorphism, c.822G>A (Table 2), within ovine CLN6 linked to this form of NCL in affected Merino and South Hampshire sheep (Tammen et al., 2006), thus the disease-causing mutation is not in CLN6 or elsewhere in this region of ovine chromosome (OAR) 7. In addition, crossing of affected Borderdales with CLN6 affected South Hampshires resulted in phenotypically normal lambs (n=3).
Table 2.
Two-point linkage analysis between NCL and markers on OAR7 and OAR10
| Ovine CLN6 SNP c.822G>A | BMS975 | ILSTS056 | OARH441 | BMS585 | |
|---|---|---|---|---|---|
| LODa score | 0.00 | 1.82 | 2.32 | 1.19 | 0.91 |
| Recombination fraction (⊖) | 0.50 | 0.08 | 0.07 | 0.15 | 0.16 |
| Chromosome | OAR7 | OAR10 | OAR10 | OAR10 | OAR10 |
Log of disequilibrium
CLN5 is located on human chromosome 13, bovine chromosome (BTA) 12 and predicted to be located on OAR 10 (Houweling et al., 2006). Comparative mapping using Oxford grid (http://oxgrid.angis.org.au/) for BTA12 and OAR10 and the virtual sheep genome map of OAR10 (www.livestockgenomics.csiro.au/sheep/mapcreator/) refined the predicted location of ovine CLN5 to the close vicinity of microsatellite markers BMS975 and ILSTS056 on OAR10. Linkage analysis (Table 2) linked this NCL in Borderdale sheep to four microsatellite markers in this region: ILSTS056 (Kemp et al., 1995), OARHH41 (Henry et al., 1993), BMS585 and BMS975 (Stone et al., 1995). Marker order calculated using CRI-MAP (Table 3) and the NCL data set is congruent with the existing sheep map and supported the predicted location of ovine CLN5. Consequently the sequence of ovine CLN5 was determined in control and affected animals.
Table 3.
CRI-MAP prediction of marker location
| Marker order and location | Distance from NCL (cM) |
|---|---|
| AGLA226 | 15.3 |
| OARH441 | 17.7 |
| BMS585 | 6.1 |
| ILSTS056 | 3.0 |
| BMS975 | 6.8 |
Sequencing of ovine CLN5 and identification of the disease-causing mutation
A variety of strategies were required to determine the ovine CLN5 sequence and identify the disease-causing mutation. Most of the sequence was determined exploiting the high degree of homology between ovine and bovine CLN5 (Houweling et al., 2006), by using primers designed from the bovine sequence (Fig. 3A and Table 4). This strategy provided the sequences of exons 2, 3 and 4. Introns 2 and 3 were determined by PCR amplification on genomic DNA using primers in exons 2, 3 and 4. The 3′ end of exon 1 was provided using a primer designed from the bovine sequence in exon 1 and an intronic primer designed from the bovine sequence towards the 3′ end of intron 1, but the 5′ end of exon 1 could not be determined this way. The sequence of this GC-rich region (84.4% in exon 1) was determined by direct sequencing of BAC DNA using an intron 1 reverse primer. This also provided 515 nucleotides of 5′ UTR and the sequence was confirmed on genomic DNA using forward primers based on that sequence.
Table 4.
CLN5 primer sequence information
| Name | Sequence | Location (bp)* |
|---|---|---|
| 5′UTR-F1 | 5′-AGT TCT GCG CAT GCT CGG CTC G-3′ | 5′UTR |
| E1F2 | 5′-CGC ACT GCT CTG GCT GGC GG-3′ | exon1 (96-115) |
| E1F3 | 5′-GTG GCC GGT GCC CTA CAA-3′ | exon1 (162-179) |
| I1R222 | 5′-ACA GTC ACT TTG GAA CTG GCT CAA CG-3′ | intron1 |
| I1F11 | 5′-TAT AGA CTG TGT GCT CAG TC-3′ | intron1 |
| E2F1 | 5′-GAT CCT TAT TGT CAA GCT A-3′ | exon2 (205-223) |
| E2F2 | 5′-GAC CTC CTG GGA CAC TTG-3′ | exon2 (328-345) |
| E2R2 | 5′-AAG TGT CCC AGG AGG TC-3′ | exon2 (328-344) |
| I2R4 | 5′-TTG GTG TTC CTC TTC AGT TCA-3′ | intron2 |
| I2R2 | 5′-CAG GCC CTG GGC ATG AAG TGA C-3′ | intron2 |
| I2F3 | 5′-ATG AGG GCA GGG AGT TAG AG-3′ | intron2 |
| E3F1 | 5′-ATG CAT GAT GCC ATT GGA TT-3′ | exon3 (352-371) |
| E3R2 | 5′-CTA GAA CTA ACG TCC CAT TTT CCT-3′ | exon3 (533-556) |
| I3R285 | 5'-CAG AGA ACA AAC TAG TGG TCA CC-3′ | intron3 |
| I3F3 | 5′-TCA GAG GTA TCT TGT GCC TT-3′ | intron3 |
| E4F1 | 5′-GCA TGT TTA ACA AAA TGG CAA-3′ | exon4 (575-595) |
| E4R3 | 5′-GAA CTC TCT GTG TAT AAC CAC-3′ | exon4 (949-969) |
| 3′UTR-R1 | 5′-ACT GAT GTG CTG GTA GAA AGA A-3′ | 3′UTR |
Primer position based on the ovine CLN5 sequence submitted to Genbank (accession number NM_001082595)
The ovine CLN5 gene contains 4 exons and encodes a predicted 361 amino acid protein (Fig. 1). Affected and normal cDNA sequences were identical but a number of polymorphic variations were found in the introns. Among these a G to A substitution of the first nucleotide in intron 3, positioned between coding cDNA nucleotides 571 and 572 (c.571+1G>A, Fig. 3A) segregates with the disease and was identified as the disease-causing mutation (Fig. 2). This mutation disrupts the normal splicing consensus sequence at the 5′ end of the intron 3 (donor site), normally GU (Black, 2003). In the affected Borderdales this anticipated donor site has become AU, not expected to be recognised by the splicing machinery, resulting in the excision of exon 3.
Fig. 2. Electropherograms of a normal (top panel), an affected (middle panel) and a heterozygote Borderdale sheep (bottom panel).
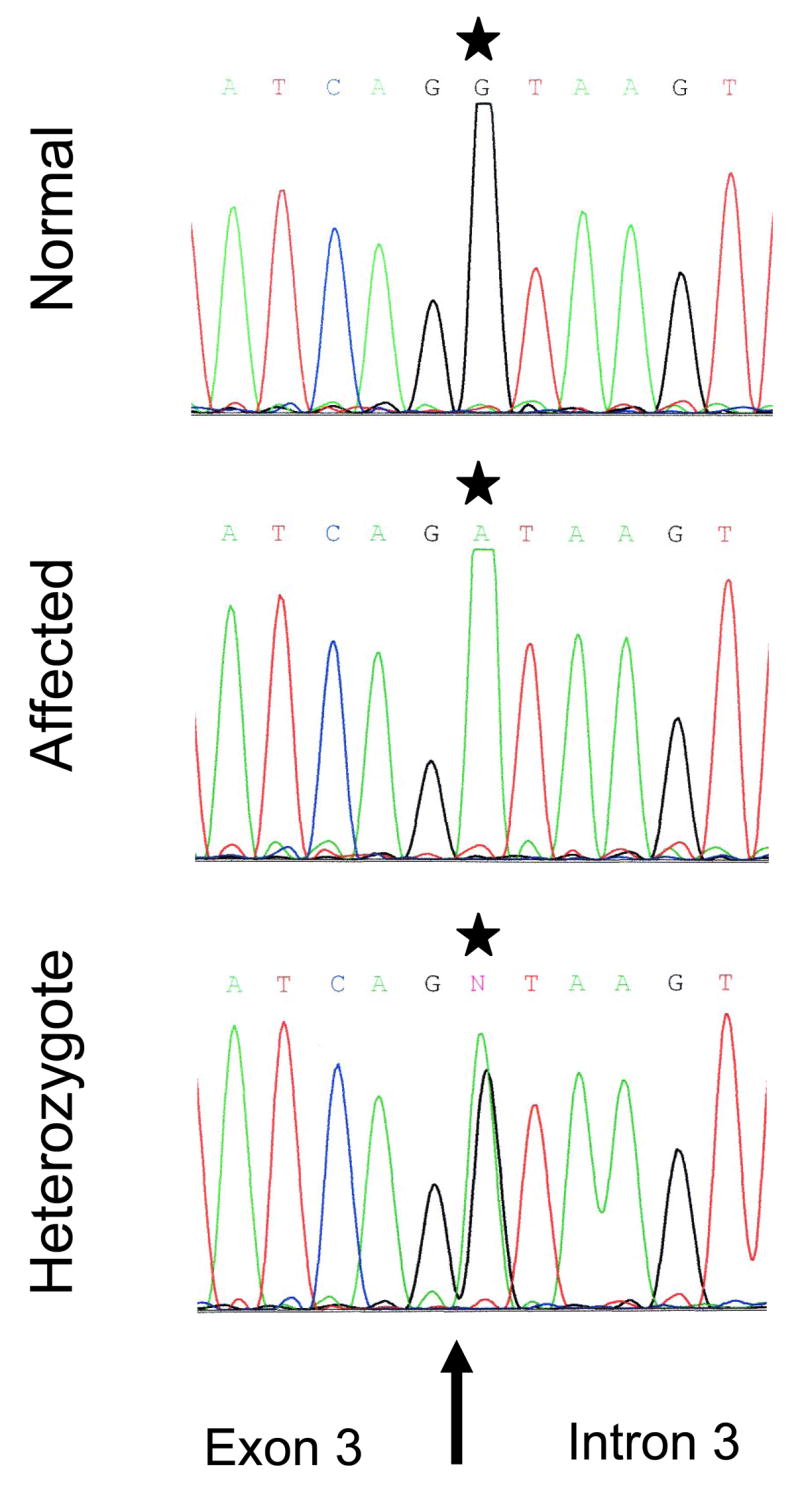
The affected sheep chromatograph shows the substitution c.571+1G>A (*). Note the presence of both alleles in the heterozygote animal. The arrow indicates the exon 3/intron 3 boundary.
This was confirmed by studies of mRNA isolated from affected, normal and heterozygous sheep (Fig. 3B). Reverse transcriptase-PCR using primers located in exons 2 and 4 showed that exon 3 is entirely missing in cDNA synthesised from total RNA from brain, skeletal muscle and leucocytes of affected animals, diagnosed by clinical signs or at biopsy, whereas it is present in cDNA samples from all control animals. Normal animals give bands of 647 bp, affected animals bands of 421 bp and every heterozygote sample tested to date (n=58) yields both the full length and truncated transcripts. The disease association of this mutation has been tested on over 114 animals with complete reliability and it is now used as a rapid and simple diagnostic test to determine the genetic status of lambs shortly after birth.
Storage body structure, isolation and composition
Storage bodies were isolated from brain and liver of affected sheep by well established methods which yielded clean preparations of their contents, as judged by electron microscopy (Fig. 4). The isolates retained the morphology of storage bodies in situ, shown here as membranous whorls in cytoplasmic organelles adjacent to the endoplasmic reticulum in a pancreatic cell (Fig. 4A). These were similar to those observed in brain storage bodies (Fig. 4B) while the membranes appeared to be more condensed in isolated liver storage bodies (Fig. 4C).
Fig 4. Electron micrographs.
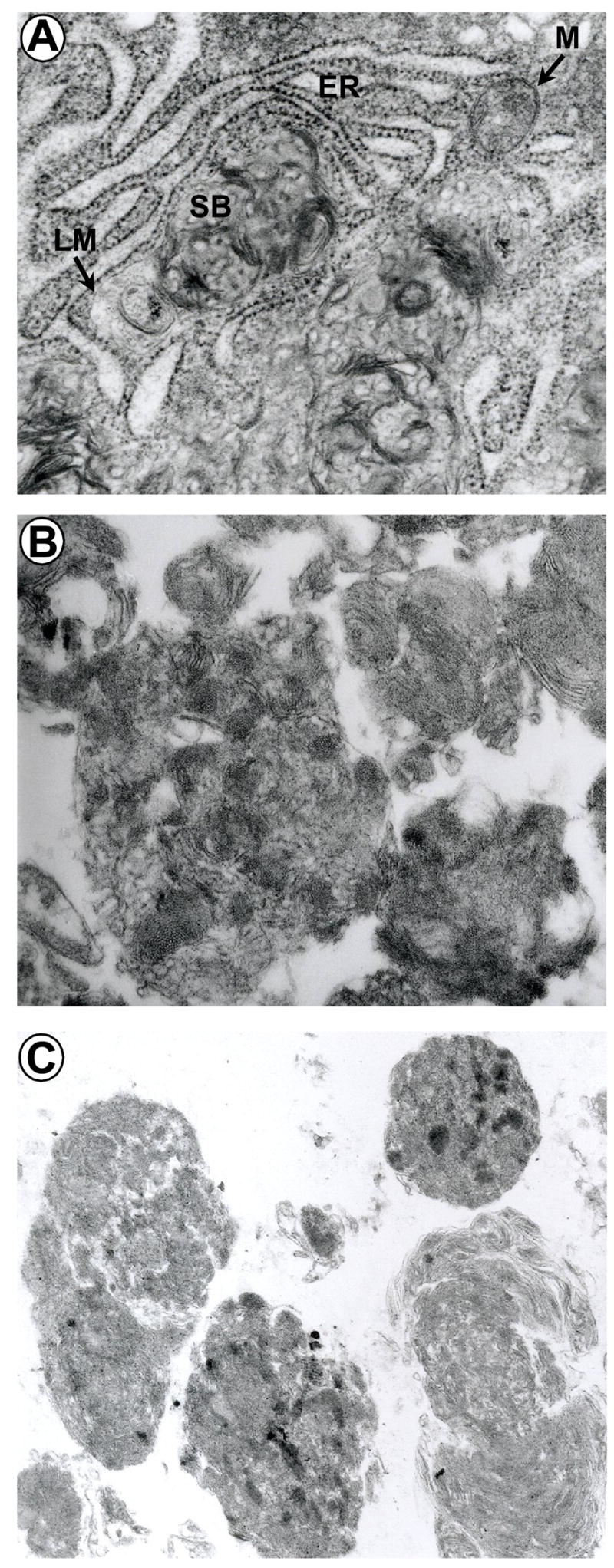
(A) Storage bodies, SB, in the cytoplasm of a pancreatic cell, magnification × 31,800. Note the limiting membrane, LM, defined in some places and the proximity to the rough endoplasmic reticulum, ER, and a mitochondrion, M. (B) Storage bodies isolated from brain, magnification × 29,400 and (C) liver, × 14,000.
Analysis of storage body proteins by gel electrophoresis and Western blotting showed that the major component was a low molecular weight protein, < 10 KDa, that immunostained for subunit c of ATP synthase on Western blots (Fig. 5). Other components were at much lower concentrations and may arise from contamination, except for the faint band at approximately 14 kDa which is likely to be an oligomer of subunit c, as has been observed before (Fearnley et al., 1990).
Fig 5. Storage body proteins.
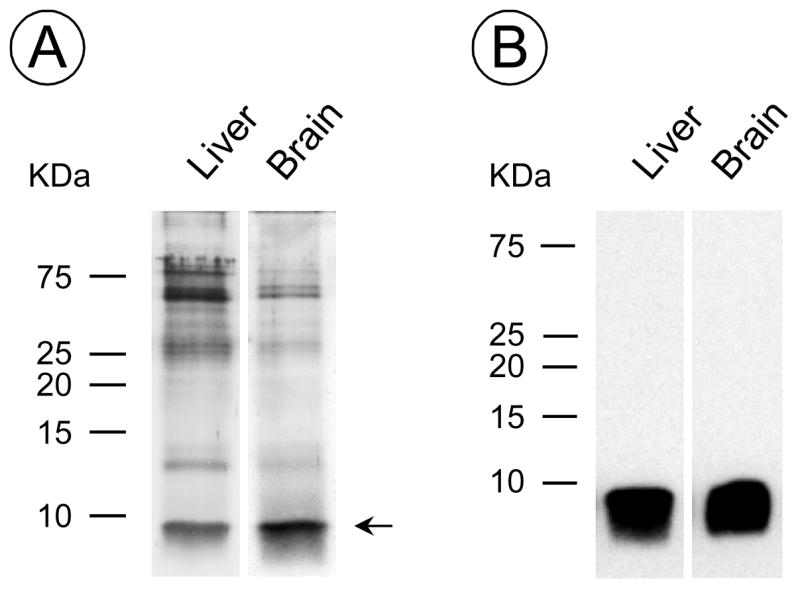
(A) Silver staining of storage body proteins separated by gel electrophoresis and (B) a Western blot of a similar gel immunostained for subunit c of mitochondrial ATP synthase.
Discussion
The diagnosis of an NCL in Borderdale sheep on a New Zealand farm (Jolly et al., 2002) has been followed by the establishment of a colony of sheep for large animal model studies of the CLN5 late infantile variant form of NCL in particular, and more generally of forms involving defective soluble lysosomal proteins. Linkage studies implicating the CLN5 gene locus (Tables 2 and 3) were followed by complete ovine CLN5 cDNA sequencing and determination of the disease-causing mutation, a c.571+1G>A that leads to the splicing out of exon 3 and a shortened putative protein (Figs. 1, 2 and 3).
Alignment of the predicted ovine (NM_001082595), human (NM_006493), bovine (NM_001046299), canine (NM_001011556) and murine (NM_001033242) CLN5 proteins (Fig. 1) revealed a very high degree of homology, 84%, 96%, 90% and 77% respectively. Interspecies variation is almost exclusively concentrated in the initial sequence of the polypeptide, coded for by the GC-rich exon 1. In general promoters and the first exon of eukaryotic genes contain functional elements which play a critical role in the regulation of the transcription, and CpG dinucleotide over-representation is common in this portion of the gene (Kalari et al., 2006; Majewski and Ott, 2002). In this case it proved difficult to sequence and was finally determined only after first establishing the normal sequence by direct sequencing of a BAC clone containing the gene.
Determining the CLN5 sequence confirmed that only one ATG initiation site is present in sheep, as is the case in cattle (Houweling et al., 2006), mice (Holmberg et al., 2004; Kopra et al., 2004) and as indicated in the boxer dog genome submission (Drögemüller et al., 2005). The human CLN5 coding sequence contains four possible ATG initiation sites. Initially it was suggested that the most 5′ upstream was the functional site in humans, providing an open reading frame of 1380 bp coding for a 407-amino acid polypeptide containing two transmembrane domains (Savukoski et al., 1998). Following subsequent protein expression studies and comparison with the mouse gene it was concluded that the most 3′ AUG in humans (AUG62) is the major initiation site, leading to a 38 kDa protein (Holmberg et al., 2004; Isosomppi et al., 2002; Mole et al., 2004).
Recently a fuller alignment of human, canine and bovine CLN5 led to the conclusion that the third human initiation site is that which is evolutionary conserved, being the only one in cattle, mice and dogs (Houweling et al., 2006), as well as in the ovine alignment here (Fig. 1). Using this start codon would lead to a human translation product of 358 amino acids, the same length as the cattle CLN5 protein (Houweling et al., 2006), and slightly shorter than the predicted 361 amino acid ovine protein. Both these later alignments are consistent with the CLN5 product being a soluble lysosomal glycoprotein (Holmberg et al., 2004; Isosomppi et al., 2002; Melville et al., 2005; Vesa and Peltonen, 2002) and a classical mannose-6-phosphate lysosomal targeting motif has been established in proteomic studies of soluble lysosomal proteins (Sleat et al., 2005; Sleat et al., 2006; Sleat et al., 2007), however the role of the CLN5 protein is still unknown.
The c.571+1G>A disease-causing mutation in Borderdale sheep is located at the first nucleotide position in intron 3 coincidental with the conserved intronic donor site of the splicing machinery (Black, 2003). It affects this conserved splice site leading to the excision of exon 3 which in turn results in a frame shift in the coding sequence, a premature stop codon and a putative protein truncated to only 125 amino acids and unlikely to be functional or stable. Transcripts lacking exon 3 are present in normal animals at an extremely low ratio (almost undetectable by RT-PCR), however no full length transcripts are detectable in the affected samples, and therefore there is no template for translation of the full length functional protein.
A similar knock-out of exon 3 causes disease in a knock-out mouse model, typified by loss of vision and fluorescent storage body accumulation but much milder brain atrophy (Kopra et al., 2004). Non-coding mutations are very likely to cause other NCLs, including CLN6 NCL in New Zealand South Hampshire sheep (Tammen et al., 2006), and it is likely that some of the uncharacterised human NCLs (Sharp et al., 2003) are caused by non-coding mutations as well (see www.ucl.ac.uk/ncl/cln6.shtml).
On the other hand all the disease-causing mutations described in the human CLN5 gene to date are in exons: c.225G>A (Savukoski et al., 1998) in exon 1, c.1627G>A (Pineda-Trujillo et al., 2005) and c.335G>C (Bessa et al., 2006) in exon 2, c.565C>T (van Diggelen et al., 2001) and c.669insC (Holmberg et al., 2000) in exon3, c.835G>A (Bessa et al., 2006; Holmberg et al., 2000; Savukoski et al., 1998), c.1175delAT (Holmberg et al., 2000; Savukoski et al., 1998) and c.772T>G (Cannelli et al., 2007) in exon 4. All these mutations in humans and those in animals (Houweling et al., 2006; Melville et al., 2005) lead to the same outcome, a putative shorter protein which probably lacks its biological function.
The storage bodies in situ and isolated storage bodies (Fig. 5) had similar ultrastructures to those found in human CLN5 (Tyynelä et al., 1997) and are typical of the subunit c storing forms of NCL (Goebel et al., 1999). Analysis of storage body proteins showed that this ovine CLN5 is a subunit c storage disease, there being very little of any other proteins stored (Fig. 5), as found in human CLN5 where subunit c storage was demonstrated by direct sequencing (Tyynelä et al., 1997). Leucocyte activity of another soluble lysosomal enzyme implicated in a subunit c storing form of NCL, tripeptidyl peptidase 1 (CLN2) (Palmer et al., 1992; Sleat et al., 1997) was up-regulated two-fold over control values (Table 1) as was that of a major soluble lysosomal protease cathepsin D. However the activities of these enzymes remained modest, control activities being only 10% of those found in human leucocytes (van Diggelen et al., 2001). Control and affected leukocyte activities of palmitoyl protein thioesterase, PPT1, the enzyme implicated in the CLN1 infantile form were similar (Table 1) and equivalent to human leucocytes activities (van Diggelen et al., 2001).
The establishment of a colony providing CLN5 affected sheep adds a valuable large animal model of this disease. The clinical and pathological courses of this ovine CLN5 form and human CLN5 are similar in keeping with the genetic and biochemical similarities noted above. Blindness is the first clinical sign of the disease in both species. This is noticeable in the Borderdales at 10–11 months of age, a few months earlier than in the CLN6 South Hampshires. Observations so far are that the course of the disease is similar to the CLN6 form in South Hampshires with the brain atrophy being more severe (Jolly et al., 2002).
The human-like gyrencephalic brains and physiology of sheep make them useful for modelling interventions. They have been bred for ease of human handling and economy of management. This flock is a valuable complement to the established South Hampshire and Merino CLN6 flocks (Tammen et al., 2006). Whereas the CLN6 gene product is an intracellular membrane protein residing in the endoplasmic reticulum (Heine et al., 2004; Mole et al., 2004) and not amenable to cross cell correction, the CLN5 gene product is a soluble lysosomal protein. In addition to its presence in brain tissue it has been isolated from urine and plasma via binding to the 300 kDa mannose-6-phosphate receptor (Sleat et al., 2005; Sleat et al., 2006; Sleat et al., 2007). This indicates that it is exported from some cells and may be taken up by others via plasma membrane 300 kDa mannose-6-phosphate receptors, and opens the way for extracellular protein correction from sources such as enzyme replacement therapy or colonies of genetically modified cells.
The generality of findings in other forms of Batten disease will also be examined in the Borderdale model. For instance recent studies of preclinical pathology in CLN6 affected South Hampshire sheep revealed no correlation between neurodegeneration and storage body accumulation but a strong correlation with early glial activation (Oswald et al., 2005). These changes became evident within the developing brain (Kay et al., 2006) indicating a central role in pathogenesis of this form of NCL. A similar early inflammation in the Borderdale sheep would indicate that this is a general characteristic of different forms of Batten disease and point the way to possible therapies.
Acknowledgments
This study was supported by the Neurological Foundation of New Zealand (DP, GK, NM), a New Zealand Neurological Foundation Philip Wrightson Fellowship (TF), the Battens Disease Support and Research Association (DP, IT, NM, GK), a F.H. Loxton scholarship (PH), the United States National Institutes of Health grant NS 40297 (DP, GK, NM) and Lysosomal Diseases New Zealand. We especially thank Bruce Sheat for all his active co-operation in sourcing the animals and Nigel Jay and Gretchen McGuire for their expert technical assistance and Ms. Joke Keulemans for her expert help with the enzyme analyses.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bessa C, Teixeira CA, Mangas M, Dias A, Sá Miranda MC, Guimãraes A, Ferreira JC, Canas N, Cabral P, Ribeiro MG. Two novel CLN5 mutations in a Portuguese patient with vLINCL: Insights into molecular mechanisms of CLN5 deficiency. Mol Genet Metab. 2006;89:245–253. doi: 10.1016/j.ymgme.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- Cannelli N, Nardocci N, Cassandrini D, Morbin M, Aiello C, Bugiani M, Criscuolo L, Zara F, Striano P, Granata T, Bertini E, Simonati A, Santorelli FM. Revelation of a novel CLN5 mutation in early juvenile neuronal ceroid lipofuscinosis. Neuropediatrics. 2007;38:46–49. doi: 10.1055/s-2007-981449. [DOI] [PubMed] [Google Scholar]
- Cao Y, Espinola JA, Fossale E, Massey AC, Cuervo AM, MacDonald ME, Cotman SL. Autophagy is disrupted in a knock-in mouse model of juvenile neuronal ceroid lipofuscinosis. J Biol Chem. 2006;281:20483–20493. doi: 10.1074/jbc.M602180200. [DOI] [PubMed] [Google Scholar]
- Chen R, Fearnley IM, Palmer DN, Walker JE. Lysine 43 is trimethylated in subunit c from bovine mitochondrial ATP synthase and in storage bodies associated with Batten disease. J Biol Chem. 2004;279:21883–21887. doi: 10.1074/jbc.M402074200. [DOI] [PubMed] [Google Scholar]
- Cooper JD. Progress towards understanding the neurobiology of Batten disease or neuronal ceroid lipofuscinosis. Curr Opin Neurol. 2003;16:121–128. doi: 10.1097/01.wco.0000063762.15877.9b. [DOI] [PubMed] [Google Scholar]
- Dickson LR, Dopfmer I, Dalefield RR, Graydon RJ, Jolly RD. A method of cerebro-cortical biopsy in lambs. N Z Vet J. 1989;37:21–22. doi: 10.1080/00480169.1989.35541. [DOI] [PubMed] [Google Scholar]
- Drögemüller C, Wöhlke A, Distl O. Characterization of candidate genes for neuronal ceroid lipofuscinosis in dog. J Hered. 2005;96:735–738. doi: 10.1093/jhered/esi088. [DOI] [PubMed] [Google Scholar]
- Ezaki J, Takeda-Ezaki M, Koike M, Ohsawa Y, Taka H, Mineki R, Murayama K, Uchiyama Y, Ueno T, Kominami E. Characterization of Cln3p, the gene product responsible for juvenile neuronal ceroid lipofuscinosis, as a lysosomal integral membrane glycoprotein. J Neurochem. 2003;87:1296–1308. doi: 10.1046/j.1471-4159.2003.02132.x. [DOI] [PubMed] [Google Scholar]
- Fearnley IM, Walker JE, Martinus RD, Jolly RD, Kirkland KB, Shaw GJ, Palmer DN. The sequence of the major protein stored in ovine ceroid lipofuscinosis is identical with that of the dicyclohexylcarbodiimide-reactive proteolipid of mitochondrial ATP synthase. Biochem J. 1990;268:751–758. doi: 10.1042/bj2680751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fossale E, Wolf P, Espinola JA, Lubicz-Nawrocka T, Teed AM, Gao H, Rigamonti D, Cattaneo E, MacDonald ME, Cotman SL. Membrane trafficking and mitochondrial abnormalities precede subunit c deposition in a cerebellar cell model of juvenile neuronal ceroid lipofuscinosis. BMC Neurosci. 2004;5:57. doi: 10.1186/1471-2202-5-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebel HH, Mole SE, Lake BD, editors. The Neuronal Ceroid Lipofuscinoses (Batten Disease) IOS Press; Amsterdam: 1999. [Google Scholar]
- Green P. Construction and comparison of chromosome 21 radiation hybrid and linkage maps using CRI-MAP. Cytogenet Cell Genet. 1992;59:122–124. doi: 10.1159/000133221. [DOI] [PubMed] [Google Scholar]
- Haltia M. The neuronal ceroid-lipofuscinoses. J Neuropathol Exp Neurol. 2003;62:1–13. doi: 10.1093/jnen/62.1.1. [DOI] [PubMed] [Google Scholar]
- Haltia M. The neuronal ceroid-lipofuscinoses: from past to present. Biochim Biophys Acta. 2006;1762:850–856. doi: 10.1016/j.bbadis.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Heine C, Koch B, Storch S, Kohlschütter A, Palmer DN, Braulke T. Defective endoplasmic reticulum-resident membrane protein CLN6 affects lysosomal degradation of endocytosed arylsulfatase A. J Biol Chem. 2004;279:22347–22352. doi: 10.1074/jbc.M400643200. [DOI] [PubMed] [Google Scholar]
- Henry HM, Penty JM, Pierson CA, Crawford AM. Ovine microsatellites at the OarHH35, OarHH41, OarHH44, OarHH47 and OarHH64 loci. Anim Genet. 1993;24:222. doi: 10.1111/j.1365-2052.1993.tb00300.x. [DOI] [PubMed] [Google Scholar]
- Holmberg V, Jalanko A, Isosomppi J, Fabritius AL, Peltonen L, Kopra O. The mouse ortholog of the neuronal ceroid lipofuscinosis CLN5 gene encodes a soluble lysosomal glycoprotein expressed in the developing brain. Neurobiol Dis. 2004;16:29–40. doi: 10.1016/j.nbd.2003.12.019. [DOI] [PubMed] [Google Scholar]
- Holmberg V, Lauronen L, Autti T, Santavuori P, Savukoski M, Uvebrant P, Hofman I, Peltonen L, Järvelä I. Phenotype-genotype correlation in eight patients with Finnish variant late infantile NCL (CLN5) Neurology. 2000;55:579–581. doi: 10.1212/wnl.55.4.579. [DOI] [PubMed] [Google Scholar]
- Houweling PJ, Cavanagh JA, Palmer DN, Frugier T, Mitchell NL, Windsor PA, Raadsma HW, Tammen I. Neuronal ceroid lipofuscinosis in Devon cattle is caused by a single base duplication (c.662dupG) in the bovine CLN5 gene. Biochim Biophys Acta. 2006;1762:890–897. doi: 10.1016/j.bbadis.2006.07.008. [DOI] [PubMed] [Google Scholar]
- Hunot S, Hirsch EC. Neuroinflammatory processes in Parkinson's disease. Ann Neurol. 2003;53:S49–58. doi: 10.1002/ana.10481. [DOI] [PubMed] [Google Scholar]
- Isosomppi J, Vesa J, Jalanko A, Peltonen L. Lysosomal localization of the neuronal ceroid lipofuscinosis CLN5 protein. Hum Mol Genet. 2002;11:885–891. doi: 10.1093/hmg/11.8.885. [DOI] [PubMed] [Google Scholar]
- Jeyakumar M, Thomas R, Elliot-Smith E, Smith DA, van der Spoel AC, d'Azzo A, Perry VH, Butters TD, Dwek RA, Platt FM. Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis. Brain. 2003;126:974–987. doi: 10.1093/brain/awg089. [DOI] [PubMed] [Google Scholar]
- Jolly RD, Arthur DG, Kay GW, Palmer DN. Neuronal ceroid lipofuscinosis in Borderdale sheep. N Z Vet J. 2002;50:199–202. doi: 10.1080/00480169.2002.36311. [DOI] [PubMed] [Google Scholar]
- Kalari KR, Casavant M, Bair TB, Keen HL, Comeron JM, Casavant TL, Scheetz TE. First exons and introns--a survey of GC content and gene structure in the human genome. In Silico Biol. 2006;6:237–42. [PubMed] [Google Scholar]
- Kay GW, Hughes SM, Palmer DN. In vitro culture of neurons from sheep with Batten disease. Mol Genet Metab. 1999;67:83–88. doi: 10.1006/mgme.1999.2849. [DOI] [PubMed] [Google Scholar]
- Kay GW, Palmer DN, Rezaie P, Cooper JD. Activation of nonneuronal cells within the prenatal developing brain of sheep with neuronal ceroid lipofuscinosis. Brain Pathol. 2006;16:110–116. doi: 10.1111/j.1750-3639.2006.00002.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp SJ, Hishida O, Wambugu J, Rink A, Longeri ML, Ma RZ, Da Y, Lewin HA, Barendse W, Teale AJ. A panel of polymorphic bovine, ovine and caprine microsatellite markers. Anim Genet. 1995;26:299–306. doi: 10.1111/j.1365-2052.1995.tb02663.x. [DOI] [PubMed] [Google Scholar]
- Kopra O, Vesa J, von Schantz C, Manninen T, Minye H, Fabritius AL, Rapola J, van Diggelen OP, Saarela J, Jalanko A, Peltonen L. A mouse model for Finnish variant late infantile neuronal ceroid lipofuscinosis, CLN5, reveals neuropathology associated with early aging. Hum Mol Genet. 2004;13:2893–2906. doi: 10.1093/hmg/ddh312. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lonka L, Salonen T, Siintola E, Kopra O, Lehesjoki AE, Jalanko A. Localization of wild-type and mutant neuronal ceroid lipofuscinosis CLN8 proteins in non-neuronal and neuronal cells. J Neurosci Res. 2004;76:862–871. doi: 10.1002/jnr.20133. [DOI] [PubMed] [Google Scholar]
- Majewski J, Ott J. Distribution and characterization of regulatory elements in the human genome. Genome Res. 2002;12:1827–1836. doi: 10.1101/gr.606402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melville SA, Wilson CL, Chiang CS, Studdert VP, Lingaas F, Wilton AN. A mutation in canine CLN5 causes neuronal ceroid lipofuscinosis in Border collie dogs. Genomics. 2005;86:287–294. doi: 10.1016/j.ygeno.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Minagar A, Shapshak P, Fujimura R, Ownby R, Heyes M, Eisdorfer C. The role of macrophage/microglia and astrocytes in the pathogenesis of three neurologic disorders: HIV-associated dementia, Alzheimer disease, and multiple sclerosis. J Neurol Sci. 2002;202:13–23. doi: 10.1016/s0022-510x(02)00207-1. [DOI] [PubMed] [Google Scholar]
- Mole SE, Michaux G, Codlin S, Wheeler RB, Sharp JD, Cutler DF. CLN6, which is associated with a lysosomal storage disease, is an endoplasmic reticulum protein. Exp Cell Res. 2004;298:399–406. doi: 10.1016/j.yexcr.2004.04.042. [DOI] [PubMed] [Google Scholar]
- Montgomery GW, Sise JA. Extraction of DNA from sheep white blood cells. NZ J Agric. 1990;33:437–441. [Google Scholar]
- Neumann H. Control of glial immune function by neurons. Glia. 2001;36:191–199. doi: 10.1002/glia.1108. [DOI] [PubMed] [Google Scholar]
- Nicholas KB, Nicholas HBJ, Deerfield DW. GeneDoc: Analysis and visualization of genetic variation. EMBNEW News. 1997;4:14. [Google Scholar]
- Oetting WS, Lee HK, Flanders DJ, Wiesner GL, Sellers TA, King RA. Linkage analysis with multiplexed short tandem repeat polymorphisms using infrared fluorescence and M13 tailed primers. Genomics. 1995;30:450–458. doi: 10.1006/geno.1995.1264. [DOI] [PubMed] [Google Scholar]
- Ohmi K, Greenberg DS, Rajavel KS, Ryazantsev S, Li HH, Neufeld EF. Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc Natl Acad Sci U S A. 2003;100:1902–1907. doi: 10.1073/pnas.252784899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald MJ, Kay GW, Shemilt SJA, Rezaie P, Cooper JD, Palmer DN. Glial activation spreads from specific cerebral foci and precedes neurodegeneration in presymptomatic ovine neuronal ceroid lipofuscinosis (CLN6) Neurobiol Dis. 2005;20:49–63. doi: 10.1016/j.nbd.2005.01.025. [DOI] [PubMed] [Google Scholar]
- Palmer DN, Bayliss SL, Westlake VJ. Batten disease and the ATP synthase subunit c turnover pathway: raising antibodies to subunit c. Am J Med Genet. 1995;57:260–265. doi: 10.1002/ajmg.1320570230. [DOI] [PubMed] [Google Scholar]
- Palmer DN, Martinus RD, Cooper SM, Midwinter GG, Reid JC, Jolly RD. Ovine ceroid lipofuscinosis. The major lipopigment protein and the lipid-binding subunit of mitochondrial ATP synthase have the same NH2-terminal sequence. J Biol Biochem. 1989;264:5736–5740. [PubMed] [Google Scholar]
- Palmer DN, Fearnley IM, Walker JE, Hall NA, Lake BD, Wolfe LS, Haltia M, Martinus RD, Jolly RD. Mitochondrial ATP synthase subunit c storage in the ceroid-lipofuscinoses (Batten disease) Am J Med Genet. 1992;42:561–567. doi: 10.1002/ajmg.1320420428. [DOI] [PubMed] [Google Scholar]
- Palmer DN, Martinus RD, Barns G, Reeves RD, Jolly RD. Ovine ceroid-lipofuscinosis. I: Lipopigment composition is indicative of a lysosomal proteinosis. Am J Med Genet Suppl. 1988;5:141–158. doi: 10.1002/ajmg.1320310618. [DOI] [PubMed] [Google Scholar]
- Pineda-Trujillo N, Cornejo W, Carrizosa J, Wheeler RB, Munera S, Valencia A, Agudelo-Arango J, Cogollo A, Anderson G, Bedoya G, Mole SE, Ruiz-Linares A. A CLN5 mutation causing an atypical neuronal ceroid lipofuscinosis of juvenile onset. Neurology. 2005;64:740–742. doi: 10.1212/01.WNL.0000151974.44980.F1. [DOI] [PubMed] [Google Scholar]
- Pontikis CC, Cella CV, Parihar N, Lim MJ, Chakrabarti S, Mitchison HM, Mobley WC, Rezaie P, Pearce DA, Cooper JD. Late onset neurodegeneration in the Cln3−/− mouse model of juvenile neuronal ceroid lipofuscinosis is preceded by low level glial activation. Brain Res. 2004;1023:231–242. doi: 10.1016/j.brainres.2004.07.030. [DOI] [PubMed] [Google Scholar]
- Rider JA, Rider DL. Thirty years of Batten disease research: Present status and future goals. Mol Genet Metab. 1999;66:231–233. doi: 10.1006/mgme.1999.2827. [DOI] [PubMed] [Google Scholar]
- Savukoski M, Klockars T, Holmberg V, Santavuori P, Lander ES, Peltonen L. CLN5, a novel gene encoding a putative transmembrane protein mutated in Finnish variant late infantile neuronal ceroid lipofuscinosis. Nat Genet. 1998;19:286–288. doi: 10.1038/975. [DOI] [PubMed] [Google Scholar]
- Sharp JD, Wheeler RB, Parker KA, Gardiner RM, Williams RE, Mole SE. Spectrum of CLN6 mutations in variant late infantile neuronal ceroid lipofuscinosis. Hum Mutat. 2003;22:35–42. doi: 10.1002/humu.10227. [DOI] [PubMed] [Google Scholar]
- Siintola E, Partanen S, Stromme P, Haapanen A, Haltia M, Maehlen J, Lehesjoki AE, Tyynelä J. Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain. 2006;129:1438–1445. doi: 10.1093/brain/awl107. [DOI] [PubMed] [Google Scholar]
- Siintola E, Topcu M, Aula N, Lohi H, Minassian BA, Paterson AD, Liu XQ, Wilson C, Lahtinen U, Anttonen AK, Lehesjoki AE. The novel neuronal ceroid lipofuscinosis gene MFSD8 encodes a putative lysosomal transporter. Am J Hum Genet. 2007;81:136–146. doi: 10.1086/518902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleat DE, Donnelly RJ, Lackland H, Liu CG, Sohar I, Pullarkat RK, Lobel P. Association of mutations in a lysosomal protein with classical late-infantile neuronal ceroid lipofuscinosis. Science. 1997;277:1802–1805. doi: 10.1126/science.277.5333.1802. [DOI] [PubMed] [Google Scholar]
- Sleat DE, Lackland H, Wang Y, Sohar I, Xiao G, Li H, Lobel P. The human brain mannose 6-phosphate glycoproteome: a complex mixture composed of multiple isoforms of many soluble lysosomal proteins. Proteomics. 2005;5:1520–1532. doi: 10.1002/pmic.200401054. [DOI] [PubMed] [Google Scholar]
- Sleat DE, Wang Y, Sohar I, Lackland H, Li Y, Li H, Zheng H, Lobel P. Identification and validation of mannose 6-phosphate glycoproteins in human plasma reveal a wide range of lysosomal and non-lysosomal proteins. Mol Cell Proteomics. 2006;5:1942–1956. doi: 10.1074/mcp.M600030-MCP200. [DOI] [PubMed] [Google Scholar]
- Sleat DE, Zheng H, Lobel P. The human urine mannose 6-phosphate glycoproteome. Biochim Biophys Acta. 2007;1774:368–372. doi: 10.1016/j.bbapap.2006.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll G, Jander S. The role of microglia and macrophages in the pathophysiology of the CNS. Prog Neurobiol. 1999;58:233–247. doi: 10.1016/s0301-0082(98)00083-5. [DOI] [PubMed] [Google Scholar]
- Stone RT, Pulido JC, Duyk GM, Kappes SM, Keele JW, Beattie CW. A small-insert bovine genomic library highly enriched for microsatellite repeat sequences. Mamm Genome. 1995;6:714–724. doi: 10.1007/BF00354294. [DOI] [PubMed] [Google Scholar]
- Tammen I, Houweling PJ, Frugier T, Mitchell NL, Kay GW, Cavanagh JA, Cook RW, Raadsma HW, Palmer DN. A missense mutation (c.184C>T) in ovine CLN6 causes neuronal ceroid lipofuscinosis in Merino sheep whereas affected South Hampshire sheep have reduced levels of CLN6 mRNA. Biochim Biophys Acta. 2006;1762:898–905. doi: 10.1016/j.bbadis.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Tyynelä J, Palmer DN, Baumann M, Haltia M. Storage of saposins A and D in infantile neuronal ceroid-lipofuscinosis. FEBS Lett. 1993;330:8–12. doi: 10.1016/0014-5793(93)80908-d. [DOI] [PubMed] [Google Scholar]
- Tyynelä J, Sohar I, Sleat DE, Gin RM, Donnelly RJ, Baumann M, Haltia M, Lobel P. A mutation in the ovine cathepsin D gene causes a congenital lysosomal storage disease with profound neurodegeneration. EMBO J. 2000;19:2786–2792. doi: 10.1093/emboj/19.12.2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyynelä J, Suopanki J, Santavuori P, Baumann M, Haltia M. Variant late infantile neuronal ceroid-lipofuscinosis: pathology and biochemistry. J Neuropathol Exp Neurol. 1997;56:369–375. doi: 10.1097/00005072-199704000-00005. [DOI] [PubMed] [Google Scholar]
- van Diggelen OP, Thobois S, Tilikete C, Zabot MT, Keulemans JL, van Bunderen PA, Taschner PE, Losekoot M, Voznyi YV. Adult neuronal ceroid lipofuscinosis with palmitoyl-protein thioesterase deficiency: first adult-onset patients of a childhood disease. Ann Neurol. 2001;50:269–272. doi: 10.1002/ana.1103. [DOI] [PubMed] [Google Scholar]
- Vesa J, Hellsten E, Verkruyse LA, Camp LA, Rapola J, Santavuori P, Hofmann SL, Peltonen L. Mutations in the palmitoyl protein thioesterase gene causing infantile neuronal ceroid lipofuscinosis. Nature. 1995;376:584–587. doi: 10.1038/376584a0. [DOI] [PubMed] [Google Scholar]
- Vesa J, Peltonen L. Mutated genes in juvenile and variant late infantile neuronal ceroid lipofuscinoses encode lysosomal proteins. Curr Mol Med. 2002;2:439–444. doi: 10.2174/1566524023362311. [DOI] [PubMed] [Google Scholar]
- Wada R, Tifft CJ, Proia RL. Microglial activation precedes acute neurodegeneration in Sandhoff disease and is suppressed by bone marrow transplantation. Proc Natl Acad Sci U S A. 2000;97:10954–10959. doi: 10.1073/pnas.97.20.10954. [DOI] [PMC free article] [PubMed] [Google Scholar]