

Abstract
Caspase recruitment domains (CARDs) are small helical protein domains that adopt the Greek key fold. For the two CARDs studied to date, RICK-CARD and caspase-1-CARD (CP1-CARD), the proteins unfold by an apparent two-state process at equilibrium. However, the folding kinetics are complex for both proteins and may contain kinetically trapped species on the folding pathway. In the case of RICK-CARD, the time constants of the slow refolding phases are consistent with proline isomerism. RICK-CARD contains three prolines, P47 in turn 3, and P85 and P87. The latter two prolines constitute a nonconserved PxP motif in helix 6. To examine the role of the prolines in the complex folding kinetics of RICK-CARD, we generated seven proline-to-alanine mutants, including three single mutants, three double mutants, and one triple mutant. We examined the spectroscopic properties, equilibrium folding, binding to CP1-CARD, and folding kinetics. The results show that P85 is critical for maintaining the function of the protein and that all mutations decrease the stability. Results from single mixing and sequential mixing stopped-flow studies strongly suggest the presence of parallel folding pathways consisting of at least two unfolded populations. The mutations affect the distribution of the two unfolded species, thereby affecting the population that folds through each channel. The two conformations also are present in the triple mutant, demonstrating that interconversion between them is not due to prolyl isomerism. Overall, the data show that the complex folding pathway, especially formation of kinetically trapped species, is not due to prolyl isomerism.
Keywords: caspase recruitment domain, RICK, Greek key, folding kinetics, equilibrium folding, folding intermediates, kinetic traps
Caspase recruitment domains (CARDs) play important roles in apoptosis, transcription activation, and inflammation (Li and Yuan 1999; Martin 2001). CARDs are members of the death domain (DD) superfamily, which includes the DD, the CARD, the pyrin domain (PyD), and the death effector domain (DED) (Hofmann 1999; Fairbrother et al. 2001; Weber and Vincenz 2001). These protein domains consist of ~100 amino acid residues that form six α-helices folded into a Greek key topology (Vaughn et al. 1999). The biological function of the domain is to convey signals to downstream effectors (Hofmann 1999; Weber and Vincenz 2001; Shi 2002; Shiozaki et al. 2002). CARDs are found as part of selected protein kinases, procaspases, and adaptor proteins. For example, RICK (CARDIAK, RIP2; RIP-like interacting CLARP kinase) contains an N-terminal serine/threonine kinase catalytic domain and a C-terminal CARD (Thome et al. 1998). The CARD of RICK (RICK-CARD) is involved in CD95 (also called Fas)-initiated apoptosis, the activation of JNK (Jun N-terminal kinase) and NF-κB, and initiation of inflammatory responses. RICK-CARD interacts with the CARD of procaspase-1 (CP1-CARD), which results in caspase activation and subsequent cleavage of interleukin-1β and interleukin-18 (Inohara et al. 1998; McCarthy et al. 1998; Thome et al. 1998). RICK also is considered a key therapeutic target for bacterial-induced inflammation (Jeyaseelan 2002).
The members of the CARD family have an identical topology, a unique α-helical Greek key fold, and similar three-dimensional structures (Day et al. 1999; Vaughn et al. 1999; Zhou et al. 1999), although the amino acid sequence identity is low in general (Druilhe et al. 2001; Lee et al. 2001; Weber and Vincenz 2001). While they fold into a complex topology, the Greek key, CARDs are amenable to examination by a number of biochemical and biophysical techniques (Vaughn et al. 1999; Zhou et al. 1999; Chen and Clark 2003 Chen and Clark 2004), and we have suggested that CARDs are good models for studies of the folding of homologous proteins (Chen and Clark 2004). In these studies, the goals are to examine the roles of amino acid sequence identity and structural topology on the conservation of folding pathways (Plaxco et al. 1998, 2000; Gunasekaran et al. 2001; Mirny and Shakhnovich 2001). We showed that the folding of the CARDs of RICK (Chen and Clark 2003) and of procaspase-1 (CP1-CARD) (Chen and Clark 2004) are described by an apparent two-state process at equilibrium, while the folding and unfolding kinetics are quite complex. The folding rate is several orders of magnitude slower than predicted by the contact order (Plaxco et al. 1998) or the long-range order (Gromiha and Selvaraj 2001), and this is due, in part, to the apparent formation of kinetically trapped species during folding.
RICK-CARD consists of 95 amino acids (Mr = 10,917 Da) and contains one tryptophan (helix 1), two tyrosines (helices 3 and 6), and a single phenylalanine (helix 5) but no disulfide bonds or prosthetic groups. While the equilibrium unfolding occurs without apparent intermediates, the conformational free energy of RICK-CARD is only ~3 kcal/mol (10 mM Tris-HCl at pH 8, 1 mM DTT, 25°C). Kinetically, RICK-CARD refolds through several intermediate states, and the relaxation times (τ1–τ3) of the observed kinetic phases are 32 msec, 1 sec, and 83 sec, respectively (Chen and Clark 2003). In addition, the kinetics of unfolding in 4 M urea-containing buffers demonstrate four phases (burst, τ1 = 1 sec, τ2 = 12 sec, τ3 = 71 sec). The burst phase intermediate in unfolding retains ~70% of the conformational free energy of the native ensemble, and the fast phases of refolding (32 sec−1) and of unfolding (0.13 sec−1) in the absence of denaturant represent the major folding reactions in terms of burial of hydrophobic surface area. Otherwise, the nature of the intermediates is unknown currently. We speculated that proline isomerism might be the source of the slow reactions observed in refolding and in unfolding since the apparent rates of the slow phases are independent of the final urea concentration, and the relaxation times are consistent with proline isomerism (Chen and Clark 2003). RICK-CARD contains three prolines (P47, P85, P87), including a nonconserved PxP motif in helix 6 (Fig. 1). Although the proline residues are predicated to be in trans in homology models of the protein structure (Chen and Clark 2003), we cannot rule out the possibility that one or more of the proline residues reside in the cis configuration. While the role of cis/trans proline isomerism in kinetic complexity has been well established for other proteins (Brandts et al. 1975; Schmid 1983; Kiefhaber et al. 1990a; Bhuyan and Udgaonkar 1999; Reader et al. 2001), the specific role of the proline residues in the kinetic complexity of RICK-CARD is unknown. Our previous results showed that the native ensemble is not directly accessible from the unfolded ensemble (Chen and Clark 2003, 2004); thus, the protein unfolds and refolds through at least one intermediate conformation. We suggested that one or more of the intermediatesmay be kinetically trapped species. To examine these issues, we generated seven proline-to-alanine mutants of RICK-CARD, consisting of three single mutants (P47A, P85A, P87A), three double mutants (P47A/P85A, P47A/P87A, P85A/P87A), and one triple mutant (P47A/P85A/P87A).
Figure 1.

Homology model of RICK-CARD (Chen and Clark 2003). The three prolines, P47, P85, and P87, are indicated, and helices 1–6 are labeled. The single tryptophan side-chain (helix 1) is shown. The figure was generated with PyMOL (Delano Scientific).
Results and Discussion
General spectroscopic properties of the RICK-CARD mutants
The fluorescence emission spectra of the mutants are either unchanged or slightly red-shifted compared with that of the wild type (data not shown). In general, the emission maximum is ~328 nm when the protein is excited at 280 nm and is ~330 nm when excited at 295 nm. The mutations of P85 to alanine and of P47/P87 to alanine had the largest effects on the fluorescence emission, as the maximum was red-shifted to ~335 nm when the samples were excited at 280 nm (data not shown), indicating that the aromatic residues are more exposed to solvent. This is consistent with the near-UV circular dichroism (CD) spectra described below.
The near-UV and far-UV CD spectra of the seven mutants are shown in Figure 2 and are compared to those of wild-type RICK-CARD (dashed lines in each panel). In general, the far-UV CD signals decreased for all mutants, demonstrating that the mutations were structurally perturbing. In the far-UV, P87A (Fig. 2C) was the least disruptive to the secondary structure, whereas the triple mutant (Fig. 2G) was the most disruptive. Note that the triple mutant, P47A/P85A/P87A, is referred to as P3A.
Figure 2.
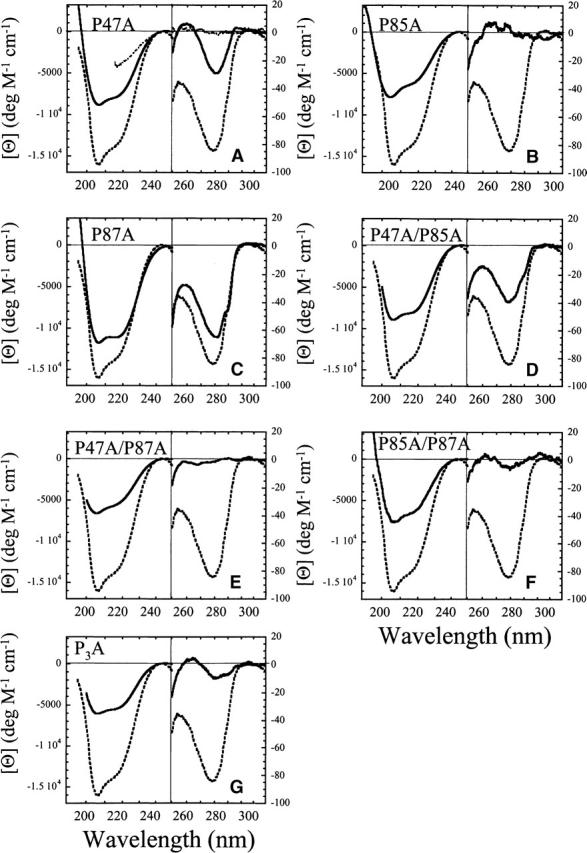
Far- and near-UV CD spectra of RICK-CARD and proline mutants. In all panels, the spectra of wild-type RICK-CARD (dashed lines) are compared with the RICK-CARD mutants (solid lines). (A) Spectra for RICK-CARD unfolded in 4 M urea-containing buffer (short dashed lines) are shown for comparison. The following spectra are shown: (A) P47A, (B) P85A, (C) P87A, (D) P47A/P85A, (E) P47A/P87A, (F) P85A/P87A, and (G) P3A.
In the near-UV, P87A was again the least structurally perturbing (Fig. 2C). In contrast, the removal of P85 (Fig. 2B) resulted in the loss of near-UV signal such that it was similar to that of unfolded wild-type RICK-CARD (cf. with short dashed line in Fig. 2A). This was true also of the double mutant P47A/P87A (Fig. 2E). The double mutant P85A/P87A (Fig. 2F) and the triple mutant, P3A (Fig. 2G), each retained only residual signals in the near-UV. Overall, the data show that removal of any proline is structurally perturbing, although the structure is perturbed to different degrees. In the single mutants, P85A is the most disruptive to the structure, whereas in the double mutants the combination of P87A with either P47A or P85A results in large changes in near-UV CD signal. It is interesting to note that replacing the PxP motif in helix 6 with a sequence that is more conducive to forming a helix, AxA, results in significant disruption of the asymmetric environment of the aromatic residues, which presumably correlates with a disruption of the tertiary structure. This suggests that the PxP motif in helix 6 is important for maintaining tertiary contacts that stabilize interactions between helix 6 and helix 1 and/or helix 5 (Fig. 1).
Effects of proline substitutions on binding to CP1-CARD
We previously characterized the interaction between wild-type RICK-CARD and CP1-CARD by monitoring changes in tryptophanyl fluorescence emission upon complex formation, as CP1-CARD lacks tryptophans (Chen and Clark 2004). In those studies we obtained an equilibrium dissociation constant of 1.4 μM, which is consistent with that determined for the interactions of Tube with Pelle, two CARD-containing proteins (Schiffmann et al. 1999). Using the same binding assay, we determined that all of the variants were able to bind CP1-CARD except P85A and P85A/P87A (data not shown).
Stability of the RICK-CARD mutants
To determine the effects of the substitutions on protein stability, we examined the urea-induced equilibrium unfolding of the RICK-CARD mutants by monitoring changes in fluorescence emission and far-UV CD in the presence of urea-containing buffers (Fig. 3). Two probes were monitored in the case of fluorescence emission. Samples were excited at 280 nm to monitor the fluorescence emission of tryptophanyl and tyrosinyl residues, or samples were excited at 295 nm to monitor tryptophanyl fluorescence emission. It has been shown that folding intermediates may show different fluorescence emission properties with use of these two probes (Clark et al. 1993; Bose and Clark 2001). The equilibrium folding of wild-type RICK-CARD is well described by an apparent two-state folding mechanism (ΔGH2O = 3.0 ± 0.1 kcal mol−1, m = 1.27 ± 0.06 kcal mol−1 M−1 at pH 8 and 25°C) (Chen and Clark 2003), and representative data are shown in Figure 3 for comparison with the mutants. The results show that two mutants (P87A and P85A/P87A) are described by an apparent two-state equilibrium folding process. In agreement with the CD spectra (Fig. 2), which show a loss in secondary structure, the proteins are less stable than is wild-type RICK-CARD. The mutation of P87 to alanine (Fig. 3C) was the least perturbing (ΔGH2O = 2.4 ± 0.1 kcal mol−1, m = 1.59 ± 0.08 kcal mol−1 M−1 at pH 8 and 25°C), although the m-value is significantly larger than that of wild-type RICK-CARD. Overall, the data for this mutant are consistent with a well-formed native structure in the absence of denaturant. This agrees with the far-UV CD spectra (Fig. 2) that showed the P87A substitution to be the least perturbing. In addition, a two-state process best described the data for the double mutant, P85A/P87A (ΔGH2O = 1.2 ± 0.1 kcal mol−1, m = 0.62 ± 0.08 kcal mol−1 M−1 at pH 8 and 25°C) (Fig. 3F); however, the large urea-dependent baseline in the pretransition suggests that the native ensemble consists of a mixture of native and non-native conformations. This also agrees with the CD spectra (Fig. 2), which show a decrease in secondary and tertiary structure for this mutant.
Figure 3.
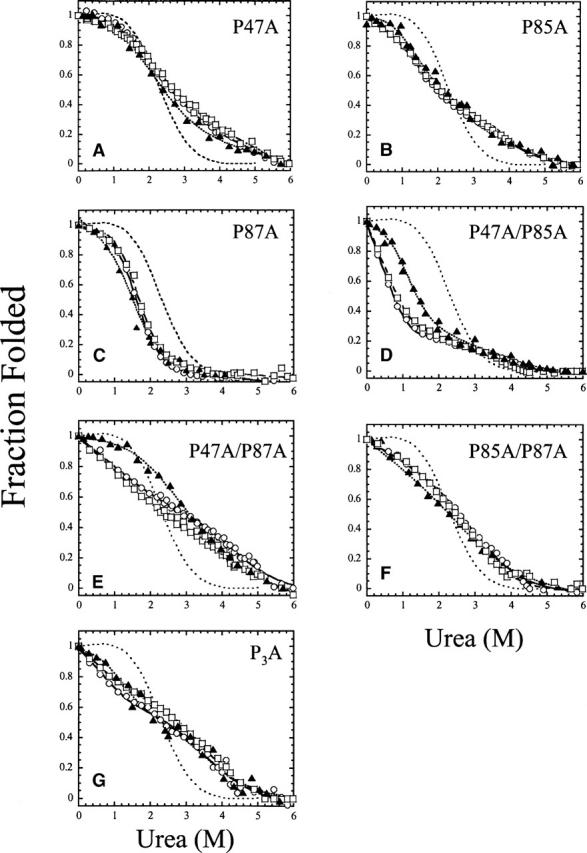
Equilibrium unfolding of the proline mutants of RICK-CARD. The fluorescence emission (excitation at 280 [○] or 295 nm [□]) and CD (220 nm; ▴) are plotted versus urea concentration. The data were fit globally as described in Materials and Methods, and the results of the fits are shown as the solid line (fluorescence emission, excitation 280 nm), dashed line (fluorescence emission, excitation 295 nm), or dotted line (CD, 220 nm). The intermediate dashed line in each panel represents wild-type RICK-CARD, as described previously (Chen and Clark 2003). Parameters obtained from the global fits are described in the text. (A) P47A, (B) P85A, (C) P87A, (D) P47A/P85A, (E) P47A/P87A, (F) P85A/P87A, and (G) P3A.
The data for the remaining mutants are not well described by a two-state folding process. A pretransition baseline is not observed for the P85A single mutant (Fig. 3B), the P47A/P85A (Fig. 3D) and P47A/P87A (Fig. 3E) double mutants, or the P3A triple mutant (Fig. 3G). This suggests that the native structure is not well formed and that partially folded structures exist in the absence of urea. In addition, an inflection is observed at ~2.5 M urea, demonstrating the formation of a stable intermediate. While the results can be fit to a three-state equilibrium folding model (N  I U), it is difficult to determine with any accuracy the conformational free energy of the first transition (N I). As described by Eftink (1994), for cases with a strongly urea-dependent slope to the pretransition baseline, assuming that the initial signal is due entirely to native protein, generally leads to an overestimation of the conformational free energy and m-value. Determining the stability of these mutants would require quantifying the population of non-native species as well as other biophysical parameters, such as the quantum yield, since the fluorescence emission is proportional to the population of macro-states. Qualitatively, however, the presence of non-native structure is in agreement with the changes in fluorescence emission described above, where a red-shift in the emission spectrum was observed. The loss of secondary and tertiary structure observed by CD (Fig. 2) as well as the results of the equilibrium unfolding data (Fig. 3) show that these mutations are destabilizing.
I U), it is difficult to determine with any accuracy the conformational free energy of the first transition (N I). As described by Eftink (1994), for cases with a strongly urea-dependent slope to the pretransition baseline, assuming that the initial signal is due entirely to native protein, generally leads to an overestimation of the conformational free energy and m-value. Determining the stability of these mutants would require quantifying the population of non-native species as well as other biophysical parameters, such as the quantum yield, since the fluorescence emission is proportional to the population of macro-states. Qualitatively, however, the presence of non-native structure is in agreement with the changes in fluorescence emission described above, where a red-shift in the emission spectrum was observed. The loss of secondary and tertiary structure observed by CD (Fig. 2) as well as the results of the equilibrium unfolding data (Fig. 3) show that these mutations are destabilizing.
The proline substitutions affect the kinetics of refolding and unfolding
As described previously, four kinetic phases are observed during the unfolding of wild-type RICK-CARD (Chen and Clark 2003). A burst phase represents formation of an intermediate that retains ~70% of the native stability and buried hydrophobic surface. Following the burst phase, three kinetic phases are observed with time constants of 1 sec, 12 sec, and 71 sec. The two slower phases are independent of the final urea concentration, and as described below, the fast phase represents the major phase of unfolding. While the results have been described elsewhere (Chen and Clark 2003), the data are shown in Figure 4H for comparison with the RICK-CARD mutants. Overall, the data for the mutants show that unfolding appears to be complete within the dead-time for mixing (Fig. 4). In all cases, upon mixing with either 4 M or 6 M urea-containing buffers, the signals match those of the unfolded protein controls within ~2 msec and do not change beyond this initial signal. This shows that regardless of the proline substitution, the intermediates that occur during unfolding of wild-type RICK-CARD have been destabilized so that unfolding of the mutants occurs rapidly. It should be noted, however, that the single tryptophan in helix 1 is partially exposed to solvent and may report on the rapid unfolding of helix 1 and/or helix 6. We also note that the proteins were excited at 280 nm in the fluorescence assays, which excites the tryptophanyl and tyrosinyl residues (helices 3 and 6), so the probes should report on the global unfolding of the protein. At this point, however, we cannot rule out the possibility that the probes report on the rapid unfolding of local structures.
Figure 4.
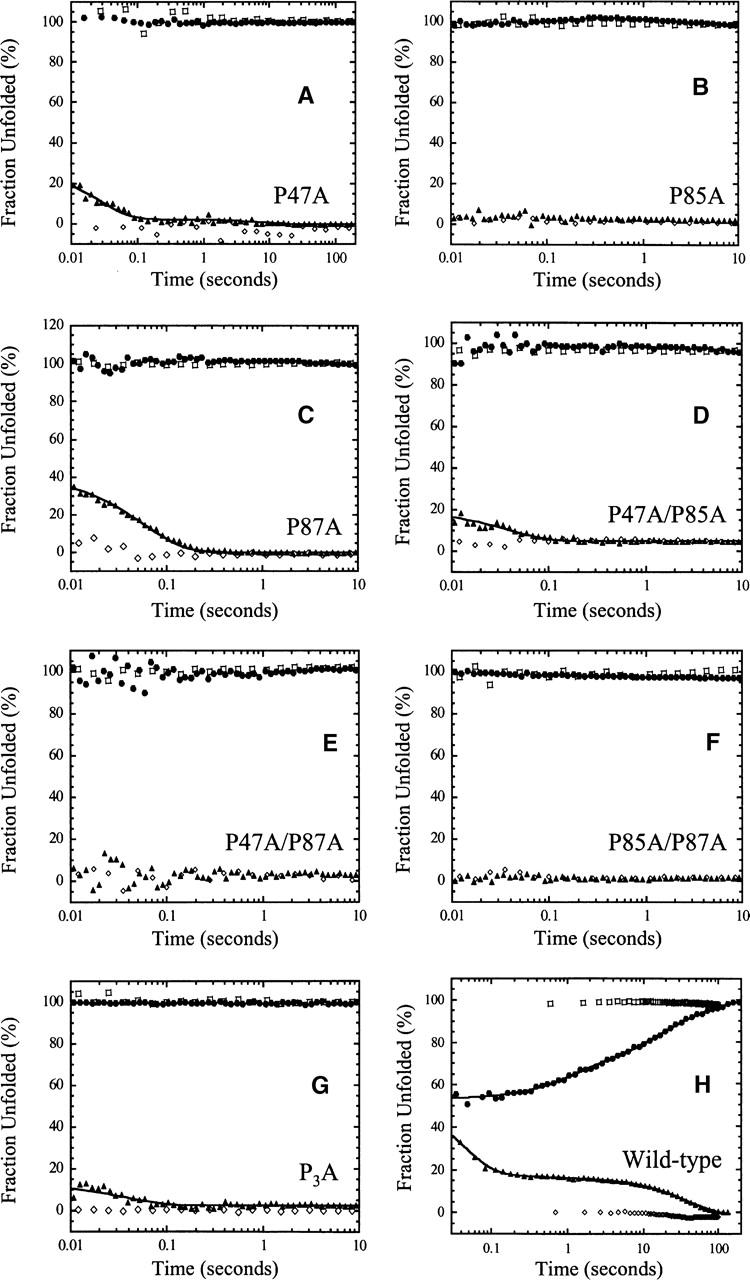
Single mixing stopped-flow refolding and unfolding traces of (A) P47A, (B) P85A, (C) P87A, (D) P47A/P85A, (E) P47A/P87A, (F) P85A/P87A, (G) P3A, and (H) wild-type RICK-CARD. In all panels, the native control (⋄) and the unfolded control (□) are shown. For each protein, the unfolding traces are shown as solid circles (•), and the refolding traces are shown as solid triangles (▴). The solid lines represent fits to a sum of exponentials, as described in the text. Results from the fits are shown in Table 1.
Under strongly native conditions, four phases are observed in the refolding of wild-type RICK-CARD. The data are shown here for comparison with the RICK-CARD mutants (Fig. 4H), although they have been described elsewhere (Chen and Clark 2003). A burst phase is observed within the dead-time for mixing, and the amplitude encompasses 65% of the total change in fluorescence emission. Based on a linear response of the burst phase amplitude to final urea concentration, it was inferred that the burst phase does not correspond to formation of a folding intermediate. As described below, however, the proline mutants suggest that an intermediate does form in the burst phase. Following the burst phase, three kinetic phases are observed with time constants (τ1–τ3) of 32 msec, 1 sec, and 83 sec (see Table 1). The first of these three phases represents the major folding phase in terms of burial of hydrophobic surface. While the rate constant of the second phase has a strong negative dependence on the final urea concentration and is not observed at urea concentrations >1.2 M, the rate constant of the third phase is independent of the final urea concentration (Chen and Clark 2003). The relative amplitudes of the three phases are 16%, 2%, and 17%, respectively (Table 1).
Table 1.
Time constants and amplitudes from refolding kinetics of RICK-CARD proline mutants
| Aburst (% total) | A1 (% total) | τ1 (sec) | A2 (% total) | τ2 (sec) | A3 (% total) | τ3 (sec) | τUN (sec) | mU-TS (kcal mol−1 M−1) | |
| Wild type | 65 | 16 ± 0.2 | 0.032 ± 0.0005 | 2 ± 0.3 | 1.0 ± 0.13 | 17 ± 0.1 | 83 ± 1 | 0.031 ± 0.006 | 0.90 ± 0.03 |
| Category I | |||||||||
| P47A | 75 | 23 ± 0.2 | 0.029 ± 0.0007 | 2 ± 0.1 | 8.3 ± 1.2 | 0.017 ± 0.003 | 0.81 ± 0.06 | ||
| P87A | 65 | 35 ± 0.3 | 0.039 ± 0.002 | 0.037 ± 0.003 | 0.79 ± 0.2 | ||||
| P47A/P85A | 85 | 15 ± 0.1 | 0.037 ± 0.0008 | 0.018 ± 0.002 | 0.85 ± 0.02 | ||||
| P3A | 89 | 11 ± 0.2 | 0.035 ± 0.002 | 0.016 ± 0.008 | 0.84 ± 0.1 | ||||
| Category II | |||||||||
| P85A | 100 | ||||||||
| P47A/P87A | 100 | ||||||||
| P85A/P87A | 100 | ||||||||
The data in Figure 4 were fit to a sum of one, two, or three exponentials, as described in the text. The data for wild-type RICK-CARD were reported previously (Chen and Clark 2003).
Based on their refolding properties, the RICK-CARD proline mutants can be grouped into two categories. In the first category, the burst phase is observed and is followed by at least one additional kinetic phase (see Table 1). The mutants in this category are P47A (Fig. 4A), P87A (Fig. 4C), P47A/P85A (Fig. 4D), and P3A (Fig. 4G). In the second category, only the burst phase is observed during refolding. Themutants in this category are P85A (Fig. 4B), P47A/P87A (Fig. 4E), and P85A/P87A (Fig. 4F). In category I, the refolding of P47A (Fig. 4A) is characterized by a burst phase and two observable phases. For this mutant, the burst phase encompasses ~75% of the total change in fluorescence emission and is followed by the fast phase (A1 = 23%, τ1 = 29 msec) and the intermediate phase (A2 = 2%, τ2 = 8.3 sec) (Table 1). The difference in the time constant for the intermediate phase compared with that of wild-type RICK-CARD (8.3 sec vs. 1 sec) likely results from the strong negative urea dependence of this phase and the difference in final urea concentrations for the two experiments (0.54 M vs. 0.36 M). Removal of P47 results in the loss of the slow phase of refolding; however, this is true for all of the RICK-CARD mutants (Fig. 4; Table 1). For the other proteins in category I (P87A, P47A/P85A, and P3A), the burst phase is followed by the fast phase, and the intermediate and slow phases are not observed (Table 1). It should be noted, however, that the amplitude of the intermediate phase is small (2%–3% of the total fluorescence change), so it would be difficult to discern this phase if the amplitude were decreased in the mutants. For P87A, the amplitude of the burst phase is similar to that of wild-type RICK-CARD (~65%), and the remaining amplitude is accounted for in the fast phase. For the double mutant P47A/P85A and the triple mutant, the burst phase accounts for 85%–90% of the total change in fluorescence emission, with the fast phase accounting for the remaining amplitude. In category II, refolding appears to be complete within the dead-time for mixing as the burst phase encompasses 100% of the total change in fluorescence emission (Fig. 4; Table 1). There were no further changes in fluorescence emission from ~2 msec to 100 sec of refolding.
The amplitudes of the burst phases of refolding and of unfolding were monitored as a function of final urea concentration, and the results are shown in Figure 5. The signals obtained after 10 msec of refolding or of unfolding are plotted with the final signals from both experiments. The kinetic data also are compared to the results from the equilibrium unfolding experiments (the experiments are shown in Fig. 3 and summarized by the solid lines in Fig. 5). The data show that the burst phase amplitudes are distinct from those of the final signal only for P47A (Fig. 5A) and P87A (Fig. 5C). In general, if the burst phase amplitude was greater than ~85% of the total change in fluorescence emission, then it is difficult to distinguish between the initial and final signals of refolding. For the two single mutants, P47A (Fig. 5A) and P87A (Fig. 5C), the response of the burst phase amplitude to increasing urea concentration is linear, as it is for the wild-type protein, except that it clearly is not a continuation of the unfolded baseline. This suggests that the burst phase represents the formation of a partially folded conformation that has marginal stability and low cooperativity of unfolding. The results for the other mutants suggest that equilibrium occurs rapidly since the signals obtained from the kinetic studies are similar to those from the equilibrium folding data. The kinetic data suggest that the reactions have reached equilibrium within ~100 msec; however, as we show later, the reactions reach a true equilibrium after ~20 sec. In these experiments the data were collected for a maximum of 10 sec, which may explain why in some cases the kinetic amplitudes do not overlay with the equilibrium data at intermediate urea concentrations (for example, see Fig. 5D).
Figure 5.
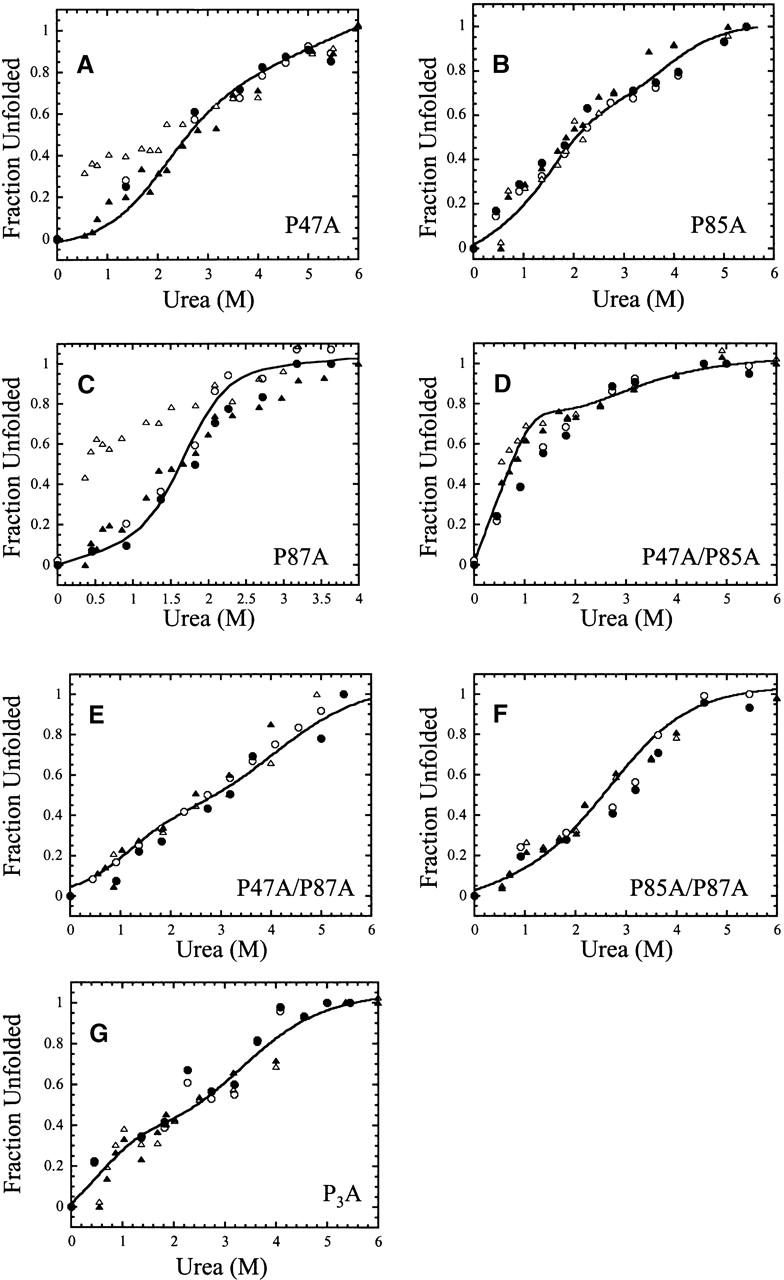
Signals of unfolding and of refolding versus final urea concentration: (A) P47A, (B) P85A, (C) P87A, (D) P47A/P85A, (E) P47A/P87A, (F) P85A/P87A, and (G) P3A. The initial signals (10 msec) of unfolding (○) and of refolding (▵) and the final signals (10 sec) of unfolding (•) and of refolding (▴) are plotted. The solid lines represent fits of the equilibrium unfolding data shown in Figure 3.
In addition to the burst phase amplitudes, the rate constant of the fast phase for refolding was determined at several final urea concentrations for the category I proteins, and the results are summarized in Table 1. The data were fit as described (Jackson and Fersht 1991; Ferguson et al. 1999) to determine the rate of refolding in the absence of urea (kUN) and mU-TS, which reflects the change in solvent accessible surface area between the unfolded and transition state ensembles. The results show that the values of mU-TS are similar, from 0.8 to 0.9 kcal mol−1 M−1 (Table 1). This suggests that, as for the wild-type RICK-CARD, the fast phase represents the major refolding phase in terms of burial of hydrophobic surface.
Sequential mixing experiments
We employed sequential mixing (double jump) refolding experiments to monitor the appearance of slow-folding species under denaturing conditions (Brandts et al. 1975; Kiefhaber et al. 1990b). In these experiments, the native protein is unfolded in urea-containing buffer for various times (delay time), and then the protein is returned to buffer and allowed to refold (N→U (delay)→N) (Schmid 1986). The refolding trace following the second mix is monitored experimentally (U→N). For wild-type RICK-CARD, the burst, fast and slow phases observed in the single mixing experiments (Fig. 4) could be monitored following return of the protein to native conditions, whereas for P87A RICK-CARD, the burst and fast phases were monitored. Representative data are shown in Figure 6.
Figure 6.
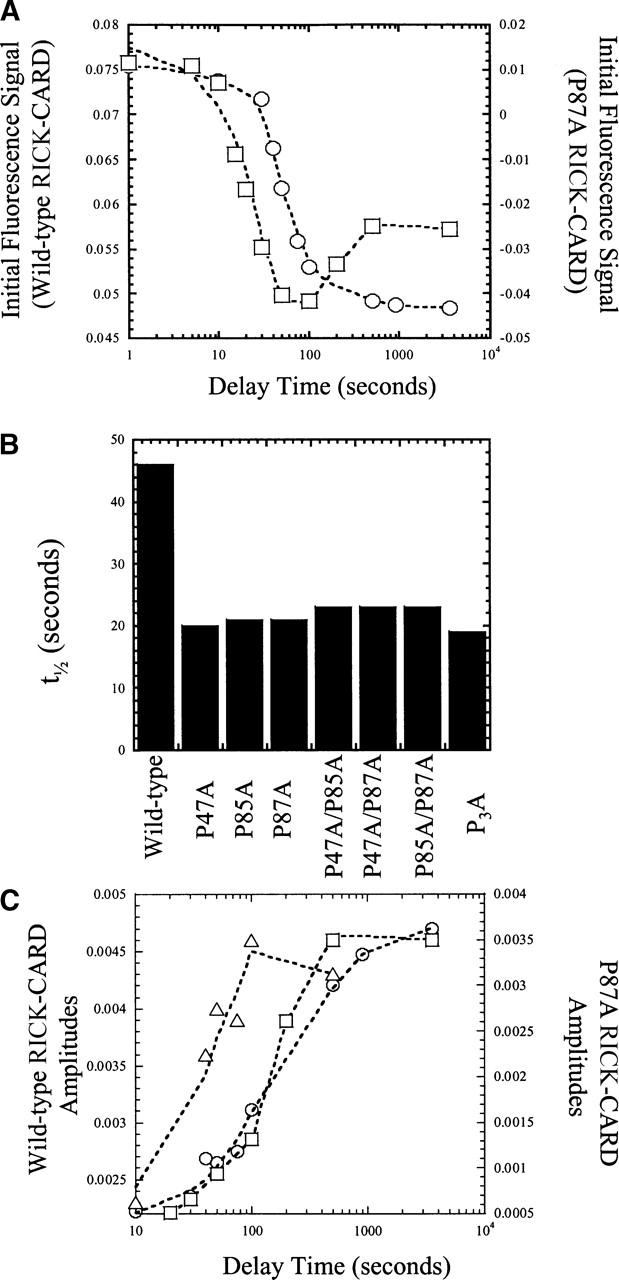
Sequential mixing stopped flow studies. (A) Fluorescence signal versus delay time in sequential mixing double jump experiments for wild-type (○) and P87A (□) RICK-CARDs. (B) Half-times of the burst phase transitions for the proline mutants and for wild-type RICK-CARD. The half-times shown were estimated from the first transition obtained from data such as those shown in A. (C) Amplitudes of the fast (○) and slow (▵) refolding phases versus unfolding delay time for wild-type RICK-CARD. Amplitudes of the fast (□) refolding phase versus unfolding delay time for P87A RICK-CARD. The lines through the data are of no theoretical value, and are presented to guide the eye.
One should note that the data in Figure 6A are represented as the fluorescence emission signal, whereas those in Figure 6C are the amplitudes of the fast or slow phases. That is, the raw traces were fit to one or more exponentials, and the amplitudes obtained from the fits are plotted versus unfolding delay time in Figure 6C. The fluorescence signals in Figure 6A change as the burst phase signal goes from that of the unfolded protein toward that of the native protein. The data shown in Figure 4H, plotted as fraction unfolded (%), show that the refolding burst phase encompasses ~65% of the total signal change (see also Table 1), and the amplitudes of the fast and slow phases encompass 16% and 17%, respectively, of the total signal change. The signals shown in Figure 6 are consistent with these values. For example, using the maximum amplitudes for the fast and slow phases (0.0045) and the change in fluorescence emission of the burst phase (0.075 − 0.048 = 0.027) yields a total change in fluorescence emission of 0.036 (0.027+ 0.0045+0.0045). Thus, under these conditions, the burst phase encompasses 75% of the total change in fluorescence emission, and the fast and slow phase amplitudes each represent ~13% of the total change in fluorescence emission.
No burst phase amplitudes were observed for P87A or wild-type RICK-CARD unless the proteins were unfolded for >10 sec (Fig. 6A). For both proteins, the results show that at short unfolding delay times, the signal remains that of the unfolded protein when the protein is shifted to native conditions. The longer the protein is unfolded, the more the burst signal goes to the native amplitude upon refolding. This occurs with a half-time of ~20 sec for P87A or ~45 sec for wild-type RICK-CARD (Fig. 6B). While the single mixing studies (Fig. 4) suggest that P87A unfolds rapidly (<2 msec), the results shown here demonstrate that the protein cannot refold unless it is unfolded for >10 sec. Following this time, the burst phase amplitude appears and reaches a maximum in ~50 sec (>200 sec for wild-type RICK-CARD). In addition, during the short unfolding times (delay times ≤20 sec), the fast phase in refolding is not observed for either protein (Fig. 6C); however, the slow refolding phase is observed for wild-type RICK-CARD (Fig. 6C) and appears with a half-time of 30–40 sec. Thus, for wild-type RICK-CARD, at delay times of 20–30 sec, only the burst and slow refolding phases are observed upon refolding. For P87A, only the burst phase is observed.
For P87A, longer times of unfolding (>50 sec) result in a second phase upon refolding in which the burst phase signal decreases. This occurs with a half-time of ~200 sec (Fig. 6A). These results strongly suggest that the unfolded ensemble consists of two slowly interconverting forms. As the second conformation is populated, the amplitude of the burst phase decreases. Similar results were obtained for P47A, P47A/P85A, and P3A except that the change in amplitude of the second phase was smaller than that for P87A (data not shown).
As shown in Figure 6C, the appearance of the fast phase of refolding correlates with the second amplitude change observed in the burst phase. At unfolding delay times >30 sec, the fast refolding phase is observed when the protein is shifted to native conditions, and the amplitude reaches a maximum when delay times are >500 sec (t1/2~200 sec). This shows that the second population of the unfolded ensemble is associated with the fast refolding phase. The same is true for wild-type RICK-CARD since the fast phase is observed only after extended times of unfolding. A similar analysis could not be performed with the other category I proteins because the amplitudes of the fast phase were too small to be observed in this experiment.
In the case of the category II mutants, changes in the burst phase signals were similar to that of P87A, except that the second transition was not observed (data not shown). The half-times for the transition (or the first phase in the case of the category I proteins) are shown in Figure 6B.
These results are not expected when the native conformation is directly accessible from the unfolded ensemble (N U) nor are they observed when proline isomerism occurs in the unfolded state (N U1 U2). In the latter case, results of the double jump experiment show a large population of native protein (N) at the short delay times and a decreasing population of N at longer delay times due to equilibration between U1 and U2 (Odefey et al. 1995; Mayr et al. 1996; Kiefhaber et al. 1997; Pappenberger et al. 2001).
Overall, the data shown in Figure 6 suggest that the proteins fold through parallel channels in which at least two unfolded species slowly interconvert (t1/2~200 sec in 4.8 M urea). Furthermore, the results suggest that the mutations affect the partitioning of the unfolded ensemble, which in turn affects the concentrations of species that fold through each channel. The results indicate that the fast and slow refolding phases, observed in single mixing studies, are each associated with one channel.
In addition to the double jump experiments, we also performed sequential mixing unfolding experiments, also known as interrupted refolding (Schmid 1983; Pappenberger et al. 2001). In these studies, protein that had been unfolded in urea-containing buffer is rapidly mixed with buffer in order to return the protein to conditions favoring the native conformation. Following various times of refolding (delay time), the protein is returned to urea, and the signal is monitored (U→N (delay)→U). Overall, this technique provides details on the refolding pathway because it is assumed that the native conformation represents the thermodynamically most stable conformation. If this is the case, then intermediates that form during refolding will be unstable and will unfold faster than the population of native protein that has formed (Pappenberger et al. 2001). In this experiment, the amplitudes should represent the population of native protein, and the signal traces will be similar to those of the single mixing experiments only when the native conformation is formed. It was shown in Figure 4 that unfolding appeared to be very rapid for each of the mutants as the signal overlays that of the unfolded control within 10 msec of unfolding. Because of this, formation of the native conformation could be observed only by monitoring the burst phase signal as a function of the refolding time since there were no observable kinetic phases. Similar results have been described for CP1-CARD, where it was shown that the protein unfolds very rapidly (Chen and Clark 2004). For the wild-type protein, one should be able to monitor four phases in unfolding, as described above. In the experiments described below, we were able to monitor three of the four phases. We note that the two slower rates differ by less than a factor of ten, so the slow phase observed in this experiment may represent both phases. The interrupted refolding experiments were performed with P87A and P3A, as representative of the mutants, and the results were compared to those of wild-type RICK-CARD (Fig. 7).
Figure 7.
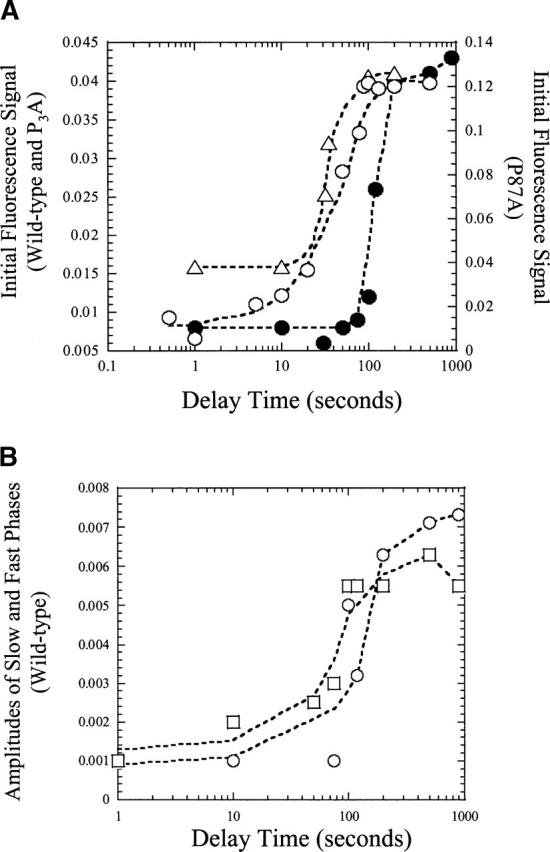
Interrupted refolding studies. (A) Fluorescence signal versus delay time for wild-type (•), P3A (▵), and P87A (○) RICK-CARDs. (B) Amplitudes of fast (○) and slow (□) phases versus delay time for wild-type RICK-CARD. The lines through the data are of no theoretical value, and are presented to guide the eye.
As shown in Figure 7A, at short refolding delay times the signal remains that of the native protein when the protein is shifted to unfolded conditions. The longer the protein is refolded, the more the burst signal goes to the unfolded amplitude upon unfolding. For wild-type RICK-CARD, no native protein is observed unless the protein has been refolded for ~100 sec, and as shown in Figure 7, A and B, the three observable phases appear at the same time. In comparison, the mutants must be refolded for ~10 sec before the native conformation is attained. Surprisingly, following short delay times, the signal did not return to that of the unfolded conformation when the protein was returned to the urea-containing buffer. As described previously for RICK-CARD (Chen and Clark 2003) and CP1-CARD (Chen and Clark 2004), the results suggest that an alternate native-like species forms at short refolding delay times that slowly interconverts to the correct native conformation at long refolding delay times. This correct native conformation unfolds as predicted from the data seen in Figure 4. Overall, the results show that the wild-type protein is fully folded after ~200 sec (t1/2~150 sec), whereas the mutants are fully folded after ~80 sec (t1/2~30 sec).
The single mixing experiments (Fig. 4) show fully native wild-type RICK-CARD after 100 sec of folding, whereas the interrupted refolding studies show that only ~20% of the protein is native at 100 sec of refolding. For the mutants, the results in Figure 4 suggest that the native conformation is reached after 1 sec of refolding, whereas the results in Figure 7 show that there is little native conformation populated at refolding times <10 sec. Together, these data suggest that there may be two native conformers of the proteins and that the interconversion between the conformers is slow.
Proposed model for folding of RICK-CARD
We previously suggested a sequential folding model for RICK-CARD (Chen and Clark 2003). In that model, the unfolded protein folds through two intermediates prior to forming the native conformation (U→I1→I2→N). The fast refolding phase (Table 1, τ1), where most of the hydrophobic surface area is buried, is represented by the U→I1 transition; the intermediate phase (Table 1, τ2) is represented by the I1→I2 transition; and the slow phase (Table 1, τ3) is represented by the I2→N transition. In light of the data presented here for the seven proline-to-alanine substitution mutants of RICK-CARD, the results are difficult to reconcile with a sequential folding model. They are, however, consistent with folding via parallel channels. The results suggest that the unfolded ensemble consists of at least two populations, U1 and U2. The results indicate that the two unfolded conformations interconvert slowly, with a half-time of ~200 sec in 4.8 M urea (Fig. 6), although folding occurs rapidly from both unfolded forms to yield non-native species. Results from the double jump experiments (Fig. 6) suggest that the fast and slow refolding phases for wild-type RICK-CARD, which are observed in the single mixing studies (Fig. 4), occur in separate channels since the slow phase appears at shorter delay times. From our data it is not clear how many non-native species reside in each channel. For example, the intermediate refolding phase (τ = 1 sec) is not observed in the double jump experiment, likely because the amplitude of the phase is very small. This phase could represent the transition of an intermediate to the native conformation, or it could represent a transition between the two channels. It is clear, however, that neither unfolded species folds directly to the native conformation. Thus, at present we cannot assign the intermediate or slow phases to specific transitions in the folding pathway.
The presence of two native conformers is suggested by the interrupted refolding studies (Fig. 7), where it was observed that the native conformation formed at times much longer than those suggested by the single mixing experiments (Fig. 4). Based on results from sequential mixing stopped-flow studies, we suggested that an intermediate that forms during refolding represents a misfolded, or kinetically trapped species (Chen and Clark 2003). That is, when RICK-CARD is refolded for a short time, it forms a native-like species that slowly isomerizes to the correct native conformation. It is not yet clear whether the second native conformer suggested by the interrupted refolding studies (Fig. 7) is the native-like kinetic trap described previously.
The proline mutations appear to affect the distribution of unfolded conformers, which, in turn, affects the observed folding kinetics in the single mixing studies. For example, the single mutant P85A and the double mutants P47A/P87A and P85A/P87A, that is, the category II proteins, primarily populate one unfolded conformation because the fast refolding phase is not observed for these mutants. The category I proteins, P47A, P87A, P47A/P85A, and P3A, are similar to wild-type RICK-CARD in that both unfolded conformers are populated, and folding occurs from both channels. Consequently, the fast refolding phase is observed for these proteins. The mutations affect the distribution of the populations of the unfolded conformers to varying degrees, and this affects the population of protein that folds through each channel. For example, for P87A a relatively larger population folds through the channel that contains the fast refolding phase compared to P3A, since there is a larger amplitude for this phase in P87A.
Other models may also be invoked to explain the sequential mixing studies and cannot be ruled out at this point. For example, the loss of secondary and tertiary structure resulting from the mutations (Fig. 2), the lower stability (Fig. 3) and the stabilization of partially folded intermediates at equilibrium (Fig. 3) suggest that some amount of non-native, and possibly unfolded, protein species is present under native-like conditions. If this were true, then the loss of kinetic phases during refolding, as observed in the category II proteins, may be due to the inability of these mutants to fully form the native conformation.
Conclusions
The following general conclusions can be drawn from the data presented here. First, P85, in helix 6, is critical for maintaining the function of the protein. At present, it is not clear how this region of helix 6 affects the binding site for CP1-CARD since this site consists primarily of helices 2 and 3 of RICK-CARD. Second, all of the mutations destabilize the protein. Third, the mutations increase the rate of unfolding. Intermediates that are observed during unfolding of wild-type RICK-CARD are not observed for the mutants. Fourth, the results suggest the presence of parallel folding channels consisting of at least two conformations in the unfolded ensemble. The mutations affect the distribution of populations of unfolded species. Fifth, the mutations affect the rate of refolding. The results show that the native conformation forms two to three times faster in the mutants than in wild-type RICK-CARD. Overall, the data presented here demonstrate that the interconversions between the two unfolded or the two native conformers are not due to proline isomerism. In addition, the data show that the kinetic complexity in forming the α-helical Greek key is not due to prolyl isomerism, at least in the case of RICK-CARD. This is shown by the triple mutant, P3A, which lacks prolines but retains the two folding channels. At present, it is not clear what causes the two unfolded or native populations or the slow isomerizations that occur during folding and unfolding.
Materials and methods
Reagents
Ultra-pure urea was purchased from Nacalai Tesque Inc. Dithiothreitol (DTT) was either from Sigma or Denvil. Trizma-Base (Tris), sodium chloride (NaCl), isopropyl β-D-1-thiogalactopyranoside (IPTG), DEAE Sepharose, Q-Sepharose, and ampicillin were purchased from Sigma. All buffers were filtered through either 0.45- or 0.22-μm filter membranes. Urea stock solutions (10 M) were prepared as described previously (Pace et al. 1989) in a buffer of 10 mM Tris-HCl (pH 8) and 1 mM DTT. The urea concentration of each stock solution was calculated by weight and by refractive index, and the solution was used only if the two values were within ± 1%.
Plasmid construction
Single proline mutants of RICK-CARD (P47A, P85A, and P87A) were generated by site-directed mutagenesis (Stratagene) using pCARD-RICK (Chen and Clark 2003) as the template in PCR amplification. The PCR primers used for mutagenesis were the following. The primer sequence for P47A was 5′-CTTGTTAGTACTAAGGCTACAAGGA CCTC-3′. In this mutant, a unique ScaI restriction site was created (underlined in the above sequence). The resulting gene contains a silent mutation at the third base for the codon of threonine 45, which is shown in bold. The mutated base for P47A is shown in italics. The primer sequence for P85A is 5′-CAAATGGGTCTGCAGGCTTACCCG-3′. A unique PstI restriction site was created (underlined in the above sequence). A silent mutation at the third base of leucine 83 is generated and shown in bold. The mutated base for proline 85 to alanine is shown in italics. The primer sequence for P87A is 5′-GGGTCTCAGCCATATGCGGAAATACTTGTGG-3′. Two silent mutations (in bold) were made in order to incorporate an NdeI restriction site (underlined) that was used for screening clones. The mutated base for proline 87 to alanine is shown in italics.
Double mutants of RICK-CARD include P47A/P85A, P47A/P87A, and P85A/P87A. The plasmid P47A/P85A was produced by using the primers for P85A (shown above) and the plasmid for the single mutant P47A as template for site-directed mutagenesis. In the resulting double mutant, two unique sites, ScaI and PstI, were generated. The plasmid P47A/P87A was generated by using the primers for P87A and the plasmid of the single mutant P47A as template. Two unique sites, ScaI and NdeI, were generated. The plasmid P85A/P87A was generated by using the primer 5′-GGGTCTGCAGGCATATGC GGAAATACTTGTGG-3′ for site-directed mutagenesis and plasmid pP87A as the template. Two unique sites, PstI and NdeI, were generated.
The triple mutant, P47A/P85A/P87A, or P3A, was constructed by using plasmid P85A/P87A as template and the primers for P47A in site-directed mutagenesis. Again, the ScaI site was used for screening. All constructs were sequenced (both strands) to confirm the sequences.
Protein purification
Escherichia coli BL21(DE3)pLysS cells were transformed with the plasmids of the mutants individually and used for protein purification. The mutants P47A, P85A, and P85A/P87A were purified using protocols described previously for RICK-CARD (Chen and Clark 2003). The P47A/P85A, P47A/P87A, and P3A proteins were purified using protocols described for RICK-CARD (Chen and Clark 2003) but with the following modifications. Cells were grown in LB media at 37°C until the absorbance at 600 nm reached 0.6. The cultures were then induced with 0.8 mM IPTG and grown overnight at 20°C. The proteins were present in inclusion bodies. P87A was purified using the protocol described previously (Chen and Clark 2003) except for the following modifications. After washing and dissolving the insoluble fraction in ~8 M urea-containing buffer A (10 mM Tris-HCl at pH 8, 1 mM DTT), the dissolved protein was centrifuged at 14,000 rpm, 4°C for 30 min, and the supernatant was filtered through 0.45-μm filter membrane. The protein solution was loaded onto a Q-Sepharose column (3.2 × 14 cm) equilibrated with 4 M urea-containing buffer A. A linear gradient of 0–400 mM NaCl in 4 M urea-containing buffer A (400 mL total volume) was used to elute the protein. The flow rate was ~4 mL/min. P87A eluted at ~100 mM NaCl. The pure protein fractions were pooled and concentrated. P87A was refolded in ice-cold buffer A containing 0.1 M NaCl at ~0.5 mg/mL by either the rapid dilution described for RICK-CARD (Chen and Clark 2003) or by dialysis.
Fluorescence emission and CD spectroscopy
Fluorescence emission spectra of the mutants of RICK-CARD were obtained by exciting the proteins (3 μM) at 280 or 295 nm, and the emission spectra were measured from 300 to 400 nm (PTI C-61 spectrofluorometer, Photon Technology International). All spectra were corrected for buffer signals. The experiments were maintained at a constant temperature of 25°C using a circulating water bath. CD spectra were obtained with a J600A spectropolarimeter (Jasco Inc.) at a constant temperature of 25°C using a circulating water bath. The path length of the CD cells used was 0.1 cm (far-UV) or 1 cm (near-UV). All spectra were corrected for buffer signals. For far and near-UV CD, the protein concentrations of P47A, P85A, P87A, P47A/P85A, P47A/P87A, P85A/P87A, and P3A were 23.7, 17.3, 38.9, 19.2, 45.4, 10.5, and 28 μM, respectively. All proteins were in buffer A.
Equilibrium folding/unfolding
Equilibrium folding/unfolding studies were performed either with a titrator (Olis Inc.) or by manual mixing. Stock solutions of native RICK-CARD mutants, 3 or 6 μM, were prepared in buffer A for both fluorescence and CD measurements. Stock solutions of unfolded protein were prepared in the same buffer containing 6 M urea. Both stocks (native or unfolded protein) contained the same protein concentration and were incubated at room temperature for at least 30 min prior to the experiments. A titrator (Olis Inc.) was used for equilibrium folding studies by fluorescence emission, and an incubation time >30 min was used after each titration. It was determined that this time is sufficient to allow for equilibration. Samples were excited at 280 or 295 nm, and fluorescence emission was monitored from 300 to 400 nm. All samples were incubated at 25°C.
The emission spectrum at each urea concentration was examined as described previously (Royer et al. 1993) to determine the average emission wavelength, 〈λ〉, using Equation 1,
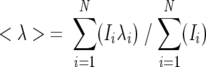 |
[1] |
where Ii is the fluorescence emission signal at wavelength λi. For the CD experiments, titrations were performed manually by mixing unfolded protein with the native protein. A quartz cuvette with 1-cm path length was used in these experiments, where the signal at 220 nm was measured for 120 sec, and the data were averaged and corrected for background signals.
The normalized average emission wavelengths and the normalized CD data were plotted versus urea concentration, and the data were fit globally either to a two-state equilibrium process (N U), described by Santoro and Bolen (1988), or to a three-state model (N I U) (Fu and Liang 2002). The program IgorPro (Wavemetrics Inc.) was used in the global fits, and the uncertainties described in the text derive from the fits. For data that exhibited a strongly urea-dependent pretransition baseline, indicating the presence of non-native structure in the absence of urea, the native baseline slope was fixed to that of the wild-type protein. All other parameters were allowed to vary in the fits.
Single-mixing stopped-flow fluorescence experiments
Single-mixing kinetic experiments were performed using a stopped-flow spectrofluorometer (SX.18MV, Applied Photophysics). The temperature was controlled at 25°C using a circulating water bath. The samples were excited at 280 nm, and the fluorescence emission was measured using a cutoff filter of 305 nm.
Refolding experiments were performed by mixing stock protein solutions (33 μM) that had been prepared in buffer A containing either 4 M urea for P87A or 6 M urea for the other mutants. The final protein concentration was 3 μM, and a mixing ratio of 1:10 was used. For measures of the burst phase amplitude, stock protein (33 μM) in either 4 M or 6 M urea-containing buffer A was mixed with buffer A containing urea between 0 and 4 M, as shown in the figures. For unfolding experiments, stock protein (33 μM) in buffer A was mixed with buffer A containing urea between 0 and 4 M. The final protein concentration was 3 μM, and a mixing ratio of 1:10 was used. The final urea concentrations are shown in the figures. The refolding traces were fit to a sum of exponentials (one to three), as described previously (Chen and Clark 2003).
Double jump stopped-flow experiments
Sequential mixing double jump experiments were performed using the sequential mixing mode of the stopped-flow spectrofluorometer, as described previously (Chen and Clark 2003). The temperature was held constant at 25°C. In general, the first jump was performed with a mixing ratio of 1:1 using two buffer solutions, described below. After a specified delay time, the solution from the first jump was rapidly mixed with a third solution using a mixing ratio of 1:5. Incubation (delay) times from 0.1 to 900 sec were used after the first jump.
Double jump experiments of the RICK-CARD mutants, except P87A, were carried out as described previously (Chen and Clark 2003) with the following modifications. Native protein (36 μM) in buffer A was mixed with buffer A containing 9.6 M urea for the first jump. After various delay times, as described in the text, the protein solution was mixed with buffer A for the second jump. The signal trace of refolding from the second jump was monitored. The final protein concentration was 3 μM, and the final urea concentration was 0.8 M. The control signal of the native protein was determined from a solution containing protein at 3 μM in 0.8 M urea-containing buffer A. The control signal of the unfolded protein was obtained by a solution containing protein at 3 μM in 4.8 M urea-containing buffer A. Conditions for the double jump experiments for P87A were the same as those described previously (Chen and Clark 2003).
Interrupted refolding experiments
Conditions for interrupted refolding experiments for the RICK-CARD mutants, except P87A, were similar to those described previously for wild-type RICK-CARD (Chen and Clark 2003). The first jump was performed with a 1:10 mixing ratio, and the second jump used a mixing ratio of 1:5. Unfolded protein (99 μM) in buffer A containing 5 M urea was mixed with buffer A for the first jump. After various delay times, as shown in the figures, the protein solution was mixed with buffer A containing 6 M urea. After the first jump, the urea concentration is 0.454 M. After the second jump, the urea concentration was 5 M, and the final protein concentration was 1.5 μM. The signal for the unfolded protein was obtained from the final signal of the experiment that used a 100 second delay time. To obtain the signal of the native protein, the third solution was changed to buffer A, as described (Chen and Clark 2003). Conditions for the interrupted refolding experiments for P87A were the same as those described previously (Chen and Clark 2003).
Acknowledgments
We thank Mr. Brett Feeney for performing the site-directed mutagenesis procedure, and Dr. Carla Mattos for helpful discussions concerning structural aspects of the homology model of RICK-CARD. This work was supported by a grant from the National Institutes of Health (GM065970).
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051943006.
References
- Bhuyan, A.K. and Udgaonkar, J.B. 1999. Observation of multistate kinetics during the slow folding and unfolding of barstar. Biochemistry 38: 9158–9168. [DOI] [PubMed] [Google Scholar]
- Bose, K. and Clark, A.C. 2001. Dimeric procaspase-3 unfolds via a four-state equilibrium process. Biochemistry 40: 14236–14242. [DOI] [PubMed] [Google Scholar]
- Brandts, J.F., Halvorson, H.R., and Brennan, M. 1975. Consideration of the possibility that the slow step in protein denaturation reactions is due to cis-trans isomerism of proline residues. Biochemistry 14: 4953–4963. [DOI] [PubMed] [Google Scholar]
- Chen, Y.-R. and Clark, A.C. 2003. Equilibrium and kinetic folding of a α-helical Greek key protein domain: Caspase recruitment domain (CARD) of RICK. Biochemistry 42: 6310–6320. [DOI] [PubMed] [Google Scholar]
- ———. 2004. Kinetic traps in the folding/unfolding of procaspase-1 CARD domain. Protein Sci. 13: 2196–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, A.C., Sinclair, J.F., and Baldwin, T.O. 1993. Folding of bacterial luciferase involves a non-native heterodimeric intermediate in equilibrium with the native enzyme and the unfolded subunits. J. Biol. Chem. 268: 10773–10779. [PubMed] [Google Scholar]
- Day, C.L., Dupont, C., Lackmann, M., Vaux, D.L., and Hinds, M.G. 1999. Solution structure and mutagenesis of the caspase recruitment domain (CARD) from Apaf-1. Cell Death Differ. 6: 1125–1132. [DOI] [PubMed] [Google Scholar]
- Druilhe, A., Srinivasula, S.M., Razmara, M., Ahmad, M., and Alnemri, E.S. 2001. Regulation of IL-1β generation by Pseudo-ICE and ICE-BERG, two dominant negative caspase recruitment domain proteins. Cell Death Differ. 8: 649–657. [DOI] [PubMed] [Google Scholar]
- Eftink, M.R. 1994. The use of fluorescence methods to monitor unfolding transitions in proteins. Biophys. J. 66: 482–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbrother, W.J., Gordon, N.C., Humke, E.W., O’Rourke, K.M., Starovasnik, M.A., Yin, J.P., and Dixit, V.M. 2001. The PYRIN domain: A member of the death domain-fold superfamily. Protein Sci. 10: 1911–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson, N., Capaldi, A.P., James, R., Kleanthous, C., and Radford, S.E. 1999. Rapid folding with and without populated intermediates in the homologous four-helix proteins Im7 and Im9. J. Mol. Biol. 286: 1597–1608. [DOI] [PubMed] [Google Scholar]
- Fu, L. and Liang, J.J.-N. 2002. Unfolding of human lens recombinant βB2- and γC-crystallins. J. Struc. Biol. 139: 191–198. [DOI] [PubMed] [Google Scholar]
- Gromiha, M.M. and Selvaraj, S. 2001. Comparison between long-range interactions and contact order in determining the folding rate of two-state proteins: Application of long-range order to folding rate prediction. J. Mol. Biol. 310: 27–32. [DOI] [PubMed] [Google Scholar]
- Gunasekaran, K., Eyles, S.J., Hagler, A.T., and Gierasch, L.M. 2001. Keeping it in the family: Folding studies of related proteins. Curr. Opin. Struct. Biol. 11: 83–93. [DOI] [PubMed] [Google Scholar]
- Hofmann, K. 1999. The modular nature of apoptotic signaling proteins. Cell Mol. Life Sci. 55: 1113–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inohara, N., del Peso, L., Koseki, T., Chen, S., and Nunez, G. 1998. RICK, a novel protein kinase containing a caspase recruitment domain, interacts with CLARP and regulates CD95-mediated apoptosis. J. Biol. Chem. 273: 12296–12300. [DOI] [PubMed] [Google Scholar]
- Jackson, S.E. and Fersht, A.R. 1991. Folding of chymotrypsin inhibitor 2, 1: Evidence for a two-state transition. Biochemistry 30: 10428–10435. [DOI] [PubMed] [Google Scholar]
- Jeyaseelan, S. 2002. Rip2: A novel therapeutic target for bacteria-induced inflammation? Trends Microbiol. 10: 356. [DOI] [PubMed] [Google Scholar]
- Kiefhaber, T., Grunert, H.-P., Hahn, U., and Schmid, F.X. 1990a. Replacement of cis proline simplifies the mechanism of ribonuclease T1 folding. Biochemistry 29: 6475–6480. [DOI] [PubMed] [Google Scholar]
- Kiefhaber, T., Quaas, R., Hahn, U., and Schmid, F.X. 1990b. Folding of ribonuclease T1. 1. Existence of multiple unfolded states created by proline isomerization. Biochemistry 29: 3053–3061. [DOI] [PubMed] [Google Scholar]
- Kiefhaber, T., Wagner, C., Bachmann, A., and Wildegger, G. 1997. Direct measurement of nucleation and growth rates in lysozyme folding. Biochemistry 36: 5108–5112. [DOI] [PubMed] [Google Scholar]
- Lee, S.H., Stehlik, C., and Reed, J.C. 2001. Cop, a caspase recruitment domain-containing protein and inhibitor of caspase-1 activation processing. J. Biol. Chem. 276: 34495–34500. [DOI] [PubMed] [Google Scholar]
- Li, H. and Yuan, J. 1999. Deciphering the pathways of life and death. Curr. Opin. Cell Biol. 11: 261–266. [DOI] [PubMed] [Google Scholar]
- Martin, S.J. 2001. Dealing the CARDs between life and death. Trends Cell Biol. 11: 188–189. [DOI] [PubMed] [Google Scholar]
- Mayr, L.M., Odefey, C., Schutkowski, M., and Schmid, F.X. 1996. Kinetic analysis of the unfolding and refolding of ribonuclease T1 by a stopped-flow double-mixing technique. Biochemistry 35: 5550–5561. [DOI] [PubMed] [Google Scholar]
- McCarthy, J.V., Ni, J., and Dixit, V.M. 1998. RIP2 is a novel NF-κB-activating and cell death-inducing kinase. J. Biol. Chem. 273: 16968–16975. [DOI] [PubMed] [Google Scholar]
- Mirny, L. and Shakhnovich, E. 2001. Evolutionary conservation of the folding nucleus. J. Mol. Biol. 308: 123–129. [DOI] [PubMed] [Google Scholar]
- Odefey, C., Mayr, L.M., and Schmid, F.X. 1995. Non-prolyl cis-trans peptide bond isomerization as a rate-determining step in protein unfolding and refolding. J. Mol. Biol. 245: 69–78. [DOI] [PubMed] [Google Scholar]
- Pace, C.N., Shirley, B.A., and Thomson, J.A.1989. Measuring the conformational stability of a protein. In Protein structure and function: A practical approach. (ed. T. Creighton), pp. 311–329. IRL Press, New York.
- Pappenberger, G., Aygun, H., Engels, J.W., Reimer, U., Fischer, G., and Kiefhaber, T. 2001. Nonprolyl cis peptide bonds in unfolded proteins cause complex folding kinetics. Nat. Struct. Biol. 8: 452–458. [DOI] [PubMed] [Google Scholar]
- Plaxco, K.W., Simons, K.T., and Baker, D. 1998. Contact order, transition state placement and the refolding rates of single domain proteins. J. Mol. Biol. 277: 985–994. [DOI] [PubMed] [Google Scholar]
- Plaxco, K.W., Larson, S., Ruczinski, I., Riddle, D.S., Thayer, E.C., Buchwitz, B., Davidson, A.R., and Baker, D. 2000. Evolutionary conservation in protein folding kinetics. J. Mol. Biol. 298: 303–312. [DOI] [PubMed] [Google Scholar]
- Reader, J.S., Van Nuland, N.A.J., Thompson, G.S., Ferguson, S.J., Dobson, C.M., and Radford, S.E. 2001. A partially folded intermediate species of the β-sheet protein apo-pseudoazurin is trapped during proline-limited folding. Protein Sci. 10: 1216–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer, C.A., Mann, C.J., and Matthews, C.R. 1993. Resolution of the fluorescence equilibrium unfolding profile of trp aporepressor using single tryptophan mutants. Protein Sci. 2: 1844–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro, M.M. and Bolen, D.W. 1988. Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl α-chymotrypsin using different denaturants. Biochemistry 27: 8063–8068. [DOI] [PubMed] [Google Scholar]
- Schiffmann, D.A., White, J.H., Cooper, A., Nutley, M.A., Harding, S.E., Jumel, K., Solari, R., Ray, K.P., and Gay, N.J. 1999. Formation and biochemical characterization of tube/pelle death domain complexes: Critical regulators of postreceptor signaling by the Drosophila toll receptor. Biochemistry 38: 11722–11733. [DOI] [PubMed] [Google Scholar]
- Schmid, F.X. 1983. Mechanism of folding of ribonuclease A: Slow refolding is a sequential reaction via structural intermediates. Biochemistry 22: 4690–4696. [DOI] [PubMed] [Google Scholar]
- ———. 1986. Fast-folding and slow-folding forms of unfolded proteins. Meth. Enzymol. 131: 70–82. [DOI] [PubMed] [Google Scholar]
- Shi, Y. 2002. Mechanisms of caspase activation and inhibition during apoptosis. Mol. Cell 9: 459–470. [DOI] [PubMed] [Google Scholar]
- Shiozaki, E.N., Chai, J., and Shi, Y. 2002. Oligomerization and activation of caspase-9, induced by Apaf-1 CARD. Proc. Natl. Acad. Sci. 99: 4197–4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thome, M., Hofmann, K., Burns, K., Martinon, F., Bodmer, J.L., Mattmann, C., and Tschopp, J. 1998. Identification of CARDIAK, a RIP-like kinase that associates with caspase-1. Curr. Biol. 8: 885–888. [DOI] [PubMed] [Google Scholar]
- Vaughn, D.E., Rodriguez, J., Lazebnik, Y., and Joshua-Tor, L. 1999. Crystal structure of Apaf-1 caspase recruitment domain: An α-helical Greek key fold for apoptotic signaling. J. Mol. Biol. 293: 439–447. [DOI] [PubMed] [Google Scholar]
- Weber, C.H. and Vincenz, C. 2001. The death domain superfamily: A tale of two interfaces? Trends Biochem. Sci. 26: 475–481. [DOI] [PubMed] [Google Scholar]
- Zhou, P., Chou, J., Olea, R.S., Yuan, J., and Wagner, G. 1999. Solution structure of Apaf-1 CARD and its interaction with caspase-9 CARD: A structural basis for specific adaptor/caspase interaction. Proc. Natl. Acad. Sci. 96: 11265–11270. [DOI] [PMC free article] [PubMed] [Google Scholar]