

Abstract
The techniques of phage-displayed homolog shotgun scanning, oligomer complementation, NMR secondary structure analysis, and computational docking provide a complementary suite of tools for dissecting protein–protein interactions. Focusing these tools on the interaction between the catalytic sub-unit of protein kinase A (PKAcat) and caveolin-1 scaffolding domain (CSD) reveals the first structural model for the interaction. Homolog shotgun scanning varied each CSD residue as either a wild-type or a homologous amino acid. Wild-type to homolog ratios from 116 different homologous CSD variants identified side-chain functional groups responsible for precise contacts with PKAcat. Structural analysis by NMR assigned an α-helical conformation to the central residues 84– 97 of CSD. The extensive mutagenesis data and NMR secondary structure information provided constraints for developing a model for the PKAcat–CSD interaction. Addition of synthetic CSD to phage-displayed CSD resulted in oligomer complementation, or enhanced binding to PKAcat. Together with previous experiments examining the interaction between CSD and endothelial nitric oxide synthase (eNOS), the results suggest a general oligomerization-dependent enhancement of binding between signal transducing enzymes and caveolin-1.
Keywords: caveolin, phage display, mutagenesis, protein kinase A, molecular recognition, stability and mutagenesis, specificity, structure/function studies, membrane-associated proteins, NMR spectroscopy, docking proteins, computational modeling
Caveolin-1 (cav-1), a multifunctional membrane protein, oligomerizes to form a coat protein for flask-shaped invaginations on the cell surface, called caveolae. Cav-1 plays a role in lipid transport, membrane trafficking, and signal transduction (Liu et al. 2002). Cav-1 and its homolog cav-2 sequester signaling proteins to abet communication between proteins in endothelial cells, whereas cav-3 functions in muscle cells (Venema et al. 1997; Schlegel et al. 1998; Quest et al. 2004; Williams and Lisanti 2004 Williams and Lisanti 2005). Cav-1 interacts with many members of the cAMP-dependent signal transduction pathway including G-proteins, adenylyl cyclase, protein kinase A catalytic subunit (PKAcat), and endothelial nitric oxide synthase (eNOS) (Garcia-Cardena et al. 1996; Song et al. 1996; Engelman et al. 1999; Liu et al. 2002; Cohen et al. 2004). Acting as a modulator of cell signaling, residues 82–101 of cav-1 termed the caveolin scaffolding domain (CSD) are sufficient to bind and inhibit enzymatic activity of the various cav-1 ligands (Li et al. 1995, 1996; Garcia-Cardena et al. 1996; Ju et al. 1997; Venema et al. 1997; Schlegel et al. 1998; Razani et al. 1999; Rybin et al. 2000; Schreiber et al. 2000; Razani and Lisanti 2001; Heijnen et al. 2004; Chun et al. 2005).
The mammalian cell signaling protein PKA regulates many cellular activities including gene expression, development, and metabolism (Slice and Taylor 1989; Taylor et al. 2004; Kim et al. 2005). cAMP binds the two regulatory subunits of PKA holoenzyme, causing the regulatory subunits to release two active catalytic subunits (Taylor et al. 2004). In addition to cav-1, at least seven other proteins can inhibit PKAcat, including four PKA regulatory sub-unit isoforms and three protein kinase inhibitor (PKI) isoforms (Knighton et al. 1991a,b; Zheng et al. 1993). Each inhibitor mimics a natural substrate through binding to the active site (site 1), but the PKA regulatory subunit RIα also binds PKAcat at two other sites. C-terminal to the inhibitor site, PKAcat site 2, includes many hydrophobic residues from residues L198 to E248. Site 3 presents a smaller binding surface at a loop in PKAcat. Excluded from caveolae (Rybin et al. 2000), the regulatory subunit RIα binds with subnanomolar Kd to PKAcat through interactions with sites 1, 2, and 3 (Kim et al. 2005).
Crystal structures of PKAcat bound to the PKI isoforms, and hydrogen/deuterium (H/2H) exchange data have revealed detailed portraits of the binding interfaces between PKAcat and its ligands (Anand et al. 2002, 2003; Hamuro et al. 2004). However, neither crystal nor NMR structures of PKAcat binding to CSD have been described. The CSD region of full-length cav-1 is responsible for oligomerization of individual cav-1 monomers (Song et al. 1997; Schlegel and Lisanti 2000; Pany et al. 2004). Circular dichroism suggests an α-helical CSD conformation (Fernandez et al. 2002), though the exact helix-forming residues were not defined. As observed by electron microscopy, full-length cav-1 monomers spontaneously arrange into building block rings of approximately seven subunits (Sargiacomo et al. 1995; Fernandez et al. 2002). By ultra centrifugation experiments, the heptameric rings further oligomerize to form a >400-kDa complex. However, in the absence of a receptor such as PKAcat, the cav-1 oligomer dissociates under high pressure. Recently, CSD oligomerization has been linked to gains in affinity for binding to eNOS (K. Murase, A.M. Levin, M. Flinspach, T.L. Poulos, and G.A. Weiss, in prep.).
In previous studies of the interaction between CSD and PKAcat, traditional alanine scanning demonstrated that four of the five residues critical for inhibition of PKAcat were aromatic (F89, F92, W98, F99) (Razani et al. 1999; Razani and Lisanti 2001). Thus, CSD could bind and inhibit PKAcat through π-stacking or other specific hydrophobic interactions. Diverse phage-displayed libraries, ranging from short peptides to full-length proteins, have been used extensively for examining protein–protein interactions. Phage-displayed libraries together with combinatorial alanine and homolog mutagenesis have identified key residues for protein function (Smith 1985; Avrantinis et al. 2002; Sidhu et al. 2003; Ladner et al. 2004; Sato et al. 2004). This combinatorial approach to interface scanning has been applied to examination of the human growth hormone (hGH) binding protein-hGH, streptavidin–biotin, and elongation factor Tu–peptide interactions (Weiss et al. 2000; Avrantinis et al. 2002; Murase et al. 2003). Selections for functional alanine-substituted proteins can rapidly assess the gross importance of each residue in a protein–ligand interaction. The tolerance of each position to alanine substitutions can also quantitatively indicate the preference for wild-type side-chain functionalities (Weiss et al. 2000). Analogous to alanine shotgun scanning, homolog shotgun scanning explores libraries with homologous and wild-type substitutions (e.g., Phe to Tyr). Homolog shotgun scanning potentially offers a more detailed appraisal of individual functionalities and has previously identified protein variants with higher affinity than the wild-type ligand (Murase et al. 2003). Phage-displayed studies of the PKAcat-CSD inter-action presented here support an oligomerization and deoligomerization process for regulation of PKAcat inhibition by CSD. We apply homolog shotgun scanning data and preliminary NMR studies to guide PKAcat-CSD docking studies, and propose the first model of CSD binding to a cell signaling protein. Models presented here account for resistance to even subtle homologous mutations by key CSD residues.
Results and Discussion
Enhancement of PKAcat–CSD binding by addition of synthetic CSD
Enhanced binding to PKAcat was observed upon addition of synthetic CSD to a constant concentration of phage-displayed CSD (Fig. 1A). CSD fails to bind to phage lacking displayed CSD (data not shown). To further demonstrate oligomer complementation by exogenous CSD, serial dilutions of phage-displayed CSD binding to PKAcat with and without addition of a constant concentration (5 μM) of synthetic CSD resulted in enhanced phage-displayed CSD binding to PKAcat (Fig. 1B). In the oligomer complementation experiments, the ratio of synthetic CSD to phage-displayed CSD was likely much >1:1. A very high CSD display level, such as 5% on the phage surface, would present ≈135 copies of CSD per phage. At 5 nM phage concentration, the average, effective concentration of phage-displayed CSD is 0.68 μM. Thus, with addition of 5 μM synthetic CSD, the ratio of synthetic to phage-displayed CSD is >7:1. This excess of synthetic CSD can allow saturation of oligomerizing sites on the phage-displayed CSD.
Figure 1.
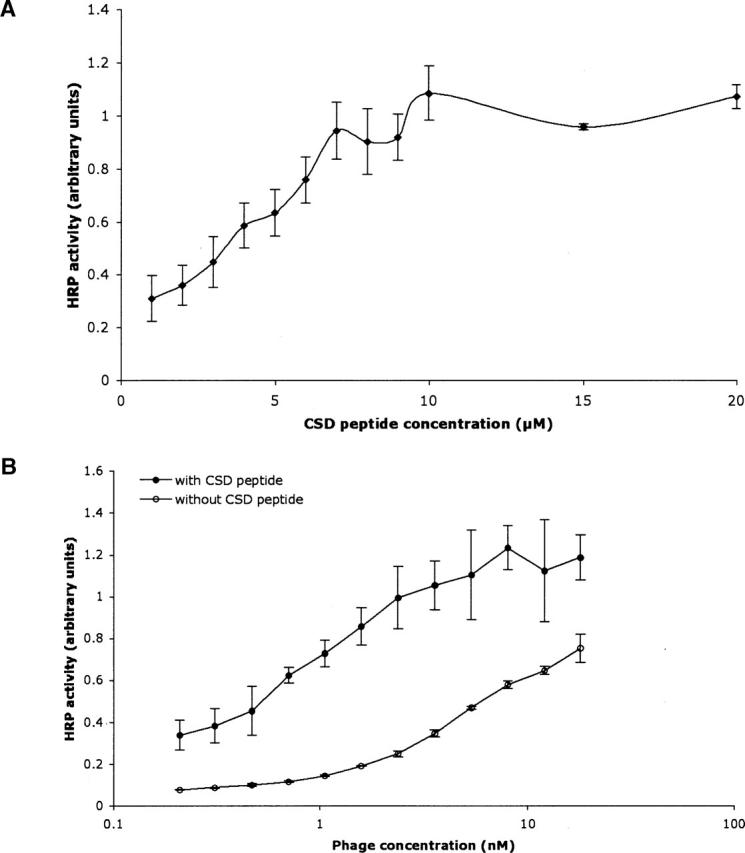
Oligomer complementation of the PKAcat–CSD interaction demonstrated by phage ELISA. (A) Phage-displayed CSD solutions (1 nM) were mixed with the indicated concentrations of synthetic CSD, and incubated with immobilized PKAcat. (B) Serial dilutions of phage-displayed CSD were assayed with or without the addition of 5 μM synthetic CSD, and incubated in wells containing immobilized PKAcat. Anti-phage antibody conjugated to HRP was used for quantification as usual. Error bars depict standard deviation.
Similar results from oligomer complementation experiments were recently observed for the interaction between eNOS and CSD (K. Murase and G.A. Weiss, in prep.). This boost in apparent affinity can be ascribed to oligomerization by the phage-displayed CSD with the added synthetic CSD. Oligomeric CSD could bind with higher affinity to PKAcat due to avidity effects, conformational rigidification, or both. Thus, enhanced binding through CSD oligomerization provides a general mechanism for CSD interaction with different members of the mammalian cell signaling pathway. Similar results with other proteins inhibited by CSD could also be expected.
Phage-displayed homolog shotgun scanning of the caveolin scaffolding domain
As demonstrated by oligomer complementation (Fig. 1), both oligomeric and nonoligomeric phage-displayed CSD bind well to PKAcat. Thus, phage-displayed shotgun scanning can examine the interface between PKAcat and monomeric, phage-displayed CSD. A homolog shotgun scanning library targeting every CSD residue was subjected to five rounds of selection for binding to PKAcat, aspreviously described for analysis of the highest and lowest affinity selectants from the library (A.M. Levin and G.A. Weiss, in prep.). Here, we leverage the sequences of all CSD variants obtained to provide a framework for modeling the PKAcat–CSD interaction. Of 240 total phage clones tested for PKAcat binding by ELISA with comparison to a negative control (binding to the blocking agent, BSA), 132 demonstrated PKAcat binding at least twofold above background binding. Of these, 116 consisted of a unique CSD homolog sequence. The 116 unique sequences were used to derive ratios for side-chain preferences reported here.
High ratios of wild-type to homolog can identify key side-chain functionalities for protein function (Vajdos et al. 2002). Homolog shotgun scanning of CSD revealed only two residues with wild-type-to-homolog ratios >10. For example, the wild-type residue K86 was conserved nearly 12:1 over a homologous K86R substitution. K86 could function as either a solvating hydrophilic residue, with an aliphatic chain capped by an amine group, or participate in a hydrogen bond or salt bridge. If K86 contributes a key hydrogen bond, then K86 must provide the optimal distance. The K86R substitution could also provide a hydrogen bond or salt bridge, but the ideal bonding distance would likely be slightly farther away from the CSD backbone.
F89, the most strongly conserved wild-type residue, preferred wild-type phenylalanine-to-tyrosine with a nearly 22:1 ratio. This strong preference suggests that a phenolic hydroxyl strongly disrupts binding, possibly by extending too far into a hydrophobic pocket. Though both phenylalanine and tyrosine provide bulky, aromatic rings, only F89 could offer a purely hydrophobic side chain ideal for close interaction with hydrophobic residues. Consistent with this observation, an F89A substitution was previously shown to disrupt CSD inhibition of PKAcat (Razani et al. 1999). Residue F89, closely packed against hydrophobic PKAcat side chains, would provide significant binding energy through hydrophobic and van der Waals interactions.
NMR characterization and molecular modeling of CSD
To fit the CSD mutagenesis data to a structural model requires both NMR analysis and molecular modeling, as no structure of CSD bound or unbound to any receptor was available. TOCSY and DQF-COSY NMR spectra were used to assign peaks by amino acid type. Several residues were assigned unambiguously, as they are unique amino acids within the peptide sequence; these include Ile84CSD, Ala87CSD, Ser88CSD, and Val94CSD. Inspection of backbone amide signals shows NH–NH NOEs consistent with a canonical α-helix between residues Ile84CSD and Tyr97CSD (Fig. 2). Helical secondary structure is also supported by circular dichroism measurements on these samples and in previous studies (Fernandez et al. 2002). We found that at below 33% TFE, the NMR results were still consistent with a helical conformation; however, the signals were quite broad indicating aggregation. Thirty-three percent of TFE was the minimum concentration that afforded an NMR spectrum amenable to detailed analysis, but giving identical CD spectra (helical content) to that of the protein in a purely aqueous solution. Further assignment and structure determination are in progress.
Figure 2.

NMR analysis of CSD. This downfield section of a CSD NOESY map (τ m = 400 msec; 20° C, 33% d-TFE, 25 mM d-sodium acetate) shows cross-peaks between back bone amide protons that are consistent with α-helical secondary structure. The major species was found to populate a helical conformation between residues Ile84CSD and Tyr97CSD.
We next generated a model of an α-helical CSD using ICM-Pro v3.3 (Fernandez-Recio et al. 2002). This structure can provide context for analyzing the muta-genesis data from the 116 unique CSD variants. This analysis hypothesizes that CSD bound to PKAcat remains largely α-helical. First, an ideal α-helix was modeled using the CSD protein sequence. Second, the backbone was relaxed to provide a more realistic helix in the context of the CSD sequence. After CSD backbone relaxation, introduction of flexibility-optimized, intra-molecular side-chain interactions yielded the final CSD model. After side-chain optimization, several residues interact with each other, including Trp85CSD with Phe89CSD and Phe99CSD with Tyr100CSD by π-stacking or hydrophobic interactions. Such intramolecular interactions are expected, as short helices often require intra-helix stabilization.
Molecular modeling of CSD binding to PKAcat
With a model of the CSD peptide based upon both NMR data and computational modeling, the program ICM-Pro was used for rigid body docking of the model CSD to PKAcat. ICM-Pro was used to suggest the lowest energy fit between an X-ray crystal structure of PKAcat and the model of CSD. From nearly 300 solutions, the lowest energy fit of CSD bound to the active site of PKAcat. Since all known PKAcat inhibitors mimic natural substrates through binding to the active site (Kim et al. 2005), this model appeared promising. In addition, PKAcat does not undergo any major conformational changes when binding to other proteins. Therefore, in addition to the suggestions from computational modeling, allosteric inhibition outside the active site can be considered a less likely possibility.
Active site docked structures were next screened for plausible positioning of the two residues most highly conserved during homolog shotgun scanning, Lys86CSD and Phe89CSD. For example, docked structures with either Lys86CSD or Phe89CSD extended into solution were considered less likely candidates and were discarded. This initial screen of the docking data assumes that CSD is displayed as a monomer on the phage surface, and any residues conserved 10:1 over a homologous substitution contribute important PKAcat-CSD contacts. Based upon the oligomer complementation data presented here and previous shotgun scanning studies, respectively, these are reasonable assumptions. One structure best fit the homolog shotgun scanning data, for reasons detailed below. This rigid body-docked structure then underwent a grid refinement with PKAcat fixed as a grid representation and the CSD side chains and backbone position refined to the lowest energy (Fig. 4A, below). After grid refinement, the entire docked structure underwent a global side-chain refinement using molecular mechanics, in which the side chains of both PKAcat and CSD were conformationally optimized, while the backbones of both proteins remained fixed (Fig. 3B). The resultant structural model (Fig. 3A) was used for further analysis of the CSD mutagenesis data.
Figure 4.
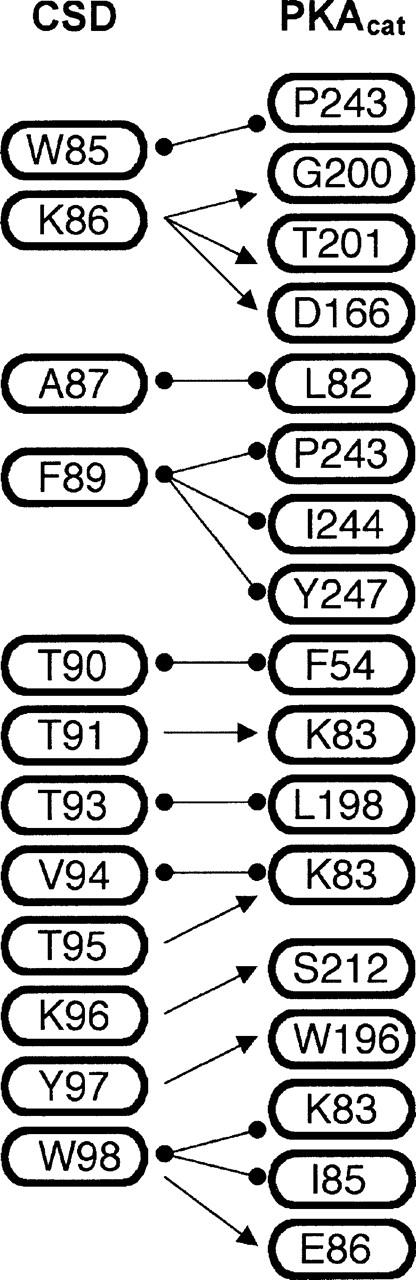
Interactions between CSD (left) and PKAcat (right) from protein–protein docking. Hydrogen bonds are denoted as arrows, and van der Waals interactions are denoted as circles.
Figure 3.

Model of the PKAcat–CSD interaction from computational modeling, mutagenesis, and NMR analysis. (A) Docking structure of CSD (red) spanning across PKAcat sites 1 (yellow), 2 (green), and 3 (orange). The N and C termini of CSD are labeled. (B) Residues Trp98CSD and Phe99CSD before (yellow) and after (red) side-chain refinement. (C) Strongly conserved CSD residue K86 in close contact with PKAcat residues D166, G200, and T201. An arginine substitution in this position could disrupt hydrogen bonds or salt bridges. (D) Strongly conserved CSD residue F89 pointing into PKAcat hydrophobic residues P243, I244, and Y247. Substitution of a hydroxyl at the position marked with an * (resulting from a F89Y mutation) could prevent this close interaction. (E) CSD residue W85 in close proximity to Pro243PKA and Phe89CSD. (F) CSD residue W98 packing against hydrophobic PKAcat residues K86, I85, and possibly hydrogen bonding to E86. CSD residues are labeled in bold. PKAcat residues are labeled in italics.
A PKAcat-CSD structural model
In the structural model of the PKAcat-CSD interaction, CSD extends across sites 1, 2, and 3 of PKAcat (Fig. 3A). CSD forms the fewest contacts with site 1, the active site of PKAcat, but partially occludes access. CSD does not appear to contact any of the PKAcat active site acidic residues (E127, E170, E230), which bind to natural substrates. One of the most conserved CSD wild-type residues, Lys86CSD (Lys:Arg of ≈12:1) can contribute hydrogen bonds to Thr201PKA, Asp166PKA, and the backbone carbonyl of Gly200PKA (Fig. 3C).
The wild-type CSD residue most conserved by homolog shotgun scanning was Phe89CSD (Phe:Tyr of ≈22:1). Accordingly, this residue guided choice of docked structures. Conserved quite strongly over a tyrosine substitution, Phe89CSD required a position in which a tyrosine hydroxyl group would clash with surrounding hydrophobic residues. In the docked structure, a cleft of three residues, Pro243PKA, Ile244PKA, and Tyr247PKA nestles against Phe89CSD, and blocks extension of the phenyl ring (Fig. 3D). This hydro-phobic cluster includes several additional PKAcat hydro-phobic residues, and has been demonstrated as critical for regulatory subunit RIα binding to PKAcat (Kim et al. 2005). RIα residue Ile98RIα associates with the extended hydrophobic cluster through Tyr247PKA, a residue likely providing a π-stacking position for the highly conserved Phe89CSD.
Several other CSD residues conserved as wild-type during homolog shotgun scanning can provide key contacts in the PKAcat-CSD structural model (Fig. 4). For example, Trp85CSD fits neatly against Pro243PKA and possibly Phe89CSD. This interaction can buttress the hydrophobic cluster surrounding Phe89CSD (Fig. 3E), and additionally can stabilize the helical conformation of CSD. Trp98CSD packs against the aliphatic sections of the Lys83PKA and Ile85PKA side chains (Fig. 3F). Also, the nitrogen in the Trp ring can hydrogen-bond to Glu86PKA. A series of CSD threonines, Thr90CSD, Thr91CSD, Thr93CSD, and Thr95CSD, all preferred a Ser substitution (Ser:Thr of ≈4:1). Analysis of the structural model reveals that serine substitutions might allow greater conformational flexibility at those sites for hydrogen binding to Gln84PKA, Lys83PKA, and the backbone of Leu198PKA.
Protein–protein docking in conjunction with empirical data can be useful for situations in which obtaining a crystal structure is difficult (Anand et al. 2003). In the current study, we present a structural model of CSD binding to PKAcat based on phage-displayed homolog shotgun scanning results and NMR data. This first model for CSD structure and binding to its cellular receptor suggests packing against three known PKAcat binding sites. Binding to the PKAcat active site and surrounding residues would inhibit enzymatic activity. Although in the model CSD does not bind to the main acidic active site residues of PKAcat, docking near the active site should block natural substrates from entering the active site. As demonstrated here, the extensive mutagenesis data obtained from techniques like homolog shotgun scanning can provide structural constraints for computational docking. Additionally, the PKAcat–CSD binding interaction, like eNOS–CSD, was enhanced by addition of synthetic CSD to phage-displayed CSD. This experiment, termed “oligomer complementation,” provides a technique for identifying oligomerization-enhanced receptor–ligand interactions. This oligomer-based binding enhancement mechanism could prove typical for many CSD ligands.
Materials and methods
Materials
The following reagents were purchased commercially: reagents for dideoxynucleotide sequencing and peptide synthesis resin (ABI/PE Biosciences); enzymes (New England Biolabs), excluding Taq polymerase (Continental Lab Products); Maxisorp immunoplates (NUNC); anti-M13/horseradish peroxidase conjugate (Amersham Pharmacia); amino acid derivatives, HOBt and HBTU (Novabiochem AG). PKAcat was kindly provided by Professor Susan S. Taylor (Howard Hughes Medical Institute, U.C. San Diego).
Oligonucleotides
DNA degeneracies are represented in the IUB code (K = G/T, M = A/C, N = A/C/G/T, R = A/G, S = G/C, W = A/T, Y = C/T). Mutations or insertions encoded by the oligonucleotides below are highlighted in bold.
M13-F1 primer: 5′-TGTAAAACGACGGCCAGT-3′
SAV-F1 primer: 5′-TGTAAAACGACGGCCAGTCGAGCA CTTCACCAACAA-3′
SAV-R2 primer: 5′-CAGGAAACAGCTATGACGACAACA ACCATCGCCC-3′
Homolog shotgun scanning: 5′-TTGCTACAAATGCCTATG CAGAMGSTRTTTKGARGKCTKCCTWCASCASCTWC ASCRTTASCARGTWCTKGTWCTWCARGGGTGGAG GATCCGGAGG-3′
wt-CSD: 5′-TTGCTACAAATGCCTATGCAGACGGAATA TGGAAAGCAAGCTTCACAACATTCACAGTAACAAA ATACTGGTTCTACAGAGGTGGAGGATCCGGAGG-3′
Peptide synthesis
The CSD peptide was synthesized manually on a 0.4-mmol scale. Fmoc-MBHA resin was used to install a carboxyamide at the C terminus of the synthesized peptide. Side-chain protection groups were removed, and the peptide was simultaneously cleaved from the resin with 95% trifluoroacetic acid (TFA), 2.5% water, and 2.5% triisopropylsilane applied for 4 h. Peptide purification was performed by automated preparative LC/MS (Waters) with a water/acetonitrile gradient containing 0.1% TFA. The purity and identity of each peptide was determined by analytical LC/MS. The observed mass (m/z) for synthetic CSD matched expected values within 0.05% as follows: 1259.4 (MH2+) (calculated mass 1259.5) and 840.2 (MH3+) (calculated mass 840.3).
Construction of phage-displayed CSD and the homolog shotgun scanning library
Phage-displayed CSD and CSD homolog library were constructed using a previously described method with phagemid pM1165a as the template DNA (Sato et al. 2004). pM1165a is identical to a previously described phagemid designed to display peptide libraries (Vajdos et al. 2002) with the following exception: The sequence encoding the displayed peptide was changed to a series of four stop codons, TAA TAA TGA TGA.
Biopanning for phage-displayed CSD variants with PKAcat binding affinity
Phage libraries were cycled through serial rounds of selection for binding to PKAcat. Ninety-six-well Maxisorp plates were coated at 4° C with PKAcat diluted to 10 μg/mL in PKA buffer (100 mM KHPO4, 100 mM KCl at pH 7.3). After 2 h (or overnight) incubation, the coating solution was removed, and the plates were blocked for 30 min with a 0.2% solution of either bovine serum albumin (BSA) or casein in PKA buffer. After the plates were washed four times with wash buffer (0.1% Tween-20 in PKA buffer), 100 μL of the phage library in sample buffer (PKA buffer plus 0.2% BSA or casein, 0.05% Tween-20) was transferred to 24 of the coated wells. After 1 h, the plate was washed eight times with wash buffer (10, 12, 14, or 16 times in subsequent rounds). Phage were eluted by adding 100 μL of 100 mM HCl, and shaken vigorously for 5 min. The eluted phage were immediately neutralized with 33 μL of 1.0 M Tris-HCl (pH 8.0). Any phage remaining on the plate were eluted by first quenching with 100 μL of PBS and then by adding 200 μL log phase XL-1 Blue cells (OD550 0.5–1.0) for 40 min. Half of the HCl-eluted phage solution was used to infect 5 mL of XL1-Blue cells (OD550 0.5–1.0), and the other half was stored at 4° C. After 20 min of shaking at 37° C, the infected cells were combined with the cells incubating on the plate and transferred to 30 mL 2 × YT supplemented with 50 μg/mL carbenicillin and 1010 phage/mL M13-K07 helper phage. After 16 h of incubation at 37° C, cells were removed by centrifugation for 20 min at 8000g, the supernatant was transferred to a new tube containing 6 mL of PEG/NaCl (20% polyethylene glycol 8000, 2.5 M NaCl), mixed well, and incubated on ice for 20 min. Phage were harvested from the culture supernatant by centrifugation (15 min at 17,000g) and resuspended in 200 μL of PKA buffer. After the first round of selection, biopanning was conducted at 1/12th of the scale described above and repeated four times.
DNA sequencing
The phage supernatant (0.2 μL) was added to 23 μL of pre-mixed PCR cocktail A (19.7 μL water, 2.5 μL 10 × PCR buffer, 0.25 μL 25 mM dNTPs, 0.25 μL 330 ng/μL SAV-F1 primer, 0.25 μL 330 ng/μL SAV-R2 primer, and 0.125 μL [5 units/μL] Taq polymerase), and thermocycled (94° C for 3 min, 25× [94° C for 0.5 min, 50° C for 0.5 min, 72° C for 1 min], 72° C for 7 min, and stored at 4° C). PCR product (5 μL) was added to 2 μL of premixed PCR cocktail B (0.25 μL exonuclease-1, 0.25 μL shrimp alkaline phosphatase, 0.20 μL 10 × PCR buffer, 1.30 μL water), and heated to 37° C for 30 min and 80° C for 15 min. The following cocktail was prepared for each sample: 4 μL 0.8 pmol/μL M13-F1 primer, 4 μL sequencing dilution buffer (200 mM Tris at pH 9.0, 5 mM MgCl2), 5.5 μL water, 4 μL Terminator Ready Reaction Mix (ABI). Then, samples were thermocycled as follows: 25 times (96° C for 10 sec, 50° C for 5 sec, 60° C for 4 min), and stored at 4° C. DNA was isolated by precipitation with 80 μL of 75% 2-propanol. Sequencing was performed by the UCI DNA Core facility using an ABI Prism 3700 Capillary Sequencer.
PKA binding assay
A phage-based ELISA assay examined binding between PKAcat and phage-displayed CSD variants. Ninety-six-well microtiter plates were coated with 100 μL of protein solution diluted to 10 μg/mL in PKA buffer. After 2 h (or overnight) incubation at 4° C, the coating solution was removed, and the plates were blocked for 30 min with a 0.2% solution of BSA in PKA buffer. Negative control wells were coated only with the 0.2% BSA solution. After the plates were rinsed with wash buffer (0.05% Tween-20 in PKA buffer) eight times, a solution of phage-displayed peptide (resuspended in PKA buffer, then diluted in half with a solution of 0.05% Tween-20 in 2YT) grown from a single colony was added to both PKAcat-coated and BSA control wells on the same plate. Then, the plates were incubated with gentle shaking for 1 h. After the plates were washed eight times with wash buffer, the plates were incubated 30 min with HRP-conjugated anti-M13 antibody (1:5000) dissolved in PKA buffer containing 0.05% Tween–20, 0.2% BSA. The plate was washed four times with wash buffer and twice with PKA buffer, before incubation for 10 min with 1 mg/mL o-phenyl-enediamine dihydrochloride/0.02% H2O2 solution in citric acid buffer (50 mM citric acid, 50 mM Na2HPO4 at pH 5.0). The absorbance of the unquenched solution was measured at 450 nm by microtiter plate reader (μQuant, Bio-Tek). All solutions of phage-displayed peptides were prepared from overnight cultures grown in parallel under identical conditions, and the relative binding affinities were compared to data obtained on the same ELISA plate.
For the CSD oligomerization assay, different concentrations of chemically synthesized CSD were added to the each phage solutions immediately prior to transfer to the PKAcat-coated plates.
Computational protein modeling and docking
The initial CSD peptide was constructed using Pymol (http://www.pymol.org) with the secondary structure defined as an α-helix. This starting structure was then imported into ICM-Pro 3.3 for refinement by the program’s regularization and side-chain optimization functions. PKAcat-CSD docking was performed essentially as described (Fernandez-Recio et al. 2002), with PKAcat (Kim et al. 2005) as the receptor and the CSD peptide model as the ligand. Initial solutions were obtained by allowing CSD to dock to any epitope of PKAcat, from which we selected conformations (as described in the text) for further grid refinement. The final model was obtained by a full ICM molecular mechanics minimization, in which all PKAcat and CSD side chains were optimized while holding the backbones fixed.
Nuclear magnetic resonance
For NMR analysis, the 3 mM concentration of synthetic CSD required the addition of TFE (33% [v/v] d-TFE in 25 mM d-sodium acetate buffer at pH 5.0). NOESY (τ m = 250 and 400 msec), TOCSY (τ m = 75 msec), and DQF-COSY experiments were performed on a Varian Inova 800 MHz NMR spectrometer at 20° and 25° C. The water peak was eliminated using WATERGATE (Piotto et al. 1992). All spectra were collected using 1024/256 complex points. Data were processed using NMRPipe (Delaglio et al. 1995) and analyzed using Sparky (http://www.cgl.ucsf.edu/home/sparky).
Acknowledgments
We thank Professor Susan S. Taylor (HHMI, UCSD) and Dr. Michael Deal (HHMI, UCSD) for providing the PKAcat used in this study, and E.J. Pone for assistance with the NMR studies. This research was largely supported by a Young Investigator Award from the Arnold and Mabel Beckman Foundation (to G.A.W.) and a pre-doctoral NIH training grant (GMT3207311) (to A.M.L.), and partially by the National Science Foundation (EF-0404057).
Abbreviations
CSD, caveolin-1 scaffolding domain
DQF-COSY, double quantum filtered correlation spectroscopy
eNOS, endothelial nitric oxide synthase
NOESY, nuclear Overhauser effect spectroscopy
NOE, nuclear Overhauser effect
PKAcat, protein kinase A catalytic subunit
PKI, protein kinase inhibitor
TOCSY, total correlations spectroscopy
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051911706.
References
- Anand, G.S., Hughes, C.A., Jones, J.M., Taylor, S.S., and Komives, E.A. 2002. Amide H/2H exchange reveals communication between the cAMP and catalytic subunit-binding sites in the R(I)α subunit of protein kinase A. J. Mol. Biol. 323: 377–386. [DOI] [PubMed] [Google Scholar]
- Anand, G.S., Law, D., Mandell, J.G., Snead, A.N., Tsigelny, I., Taylor, S.S., Ten Eyck, L.F., and Komives, E.A. 2003. Identification of the protein kinase A regulatory RIα-catalytic subunit interface by amide H/2H exchange and protein docking. Proc. Natl. Acad. Sci. 100: 13264–13269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avrantinis, S.K., Stafford, R.L., Tian, X., and Weiss, G.A. 2002. Dissecting the streptavidin–biotin interaction by phage-displayed shotgun scanning. Chembiochem 3: 1229–1234. [DOI] [PubMed] [Google Scholar]
- Chun, J., Kwon, T., Lee, E.J., Hyun, S., Hong, S.K., and Kang, S.S. 2005. The subcellular localization of 3-phosphoinositide-dependent protein kinase is controlled by caveolin-1 binding. Biochem. Biophys. Res. Commun. 326: 136–146. [DOI] [PubMed] [Google Scholar]
- Cohen, A.W., Razani, B., Schubert, W., Williams, T.M., Wang, X.B., Iyengar, P., Brasaemle, D.L., Scherer, P.E., and Lisanti, M.P. 2004. Role of caveolin-1 in the modulation of lipolysis and lipid droplet formation. Diabetes 53: 1261–1270. [DOI] [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6: 277–293. [DOI] [PubMed] [Google Scholar]
- Engelman, J.A., Zhang, X.L., Razani, B., Pestell, R.G., and Lisanti, M.P. 1999. p42/44 MAP kinase-dependent and -independent signaling pathways regulate caveolin-1 gene expression. Activation of Ras-MAP kinase and protein kinase A signaling cascades transcriptionally down-regulates caveolin-1 promoter activity. J. Biol. Chem. 274: 32333–32341. [DOI] [PubMed] [Google Scholar]
- Fernandez, I., Ying, Y., Albanesi, J., and Anderson, R.G. 2002. Mechanism of caveolin filament assembly. Proc. Natl. Acad. Sci. 99: 11193–11198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Recio, J., Totrov, M., and Abagyan, R. 2002. Soft protein–protein docking in internal coordinates. Protein Sci. 11: 280–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Cardena, G., Fan, R., Stern, D.F., Liu, J., and Sessa, W.C. 1996. Endothelial nitric oxide synthase is regulated by tyrosine phosphorylation and interacts with caveolin-1. J. Biol. Chem. 271: 27237–27240. [DOI] [PubMed] [Google Scholar]
- Hamuro, Y., Anand, G.S., Kim, J.S., Juliano, C., Stranz, D.D., Taylor, S.S., and Woods Jr., V.L. 2004. Mapping intersubunit interactions of the regulatory subunit (RIα ) in the type I holoenzyme of protein kinase A by amide hydrogen/deuterium exchange mass spectrometry (DXMS). J. Mol. Biol. 340: 1185–1196. [DOI] [PubMed] [Google Scholar]
- Heijnen, H.F., Waaijenborg, S., Crapo, J.D., Bowler, R.P., Akkerman, J.W., and Slot, J.W. 2004. Colocalization of eNOS and the catalytic subunit of PKA in endothelial cell junctions: A clue for regulated NO production. J. Histochem. Cytochem. 52: 1277–1285. [DOI] [PubMed] [Google Scholar]
- Ju, H., Zou, R., Venema, V.J., and Venema, R.C. 1997. Direct interaction of endothelial nitric-oxide synthase and caveolin-1 inhibits synthase activity. J. Biol. Chem. 272: 18522–18525. [DOI] [PubMed] [Google Scholar]
- Kim, C., Xuong, N.H., and Taylor, S.S. 2005. Crystal structure of a complex between the catalytic and regulatory (RIα ) subunits of PKA. Science 307: 690–696. [DOI] [PubMed] [Google Scholar]
- Knighton, D.R., Zheng, J.H., Ten Eyck, L.F., Ashford, V.A., Xuong, N.H., Taylor, S.S., and Sowadski, J.M. 1991a. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 253: 407–414. [DOI] [PubMed] [Google Scholar]
- Knighton, D.R., Zheng, J.H., Ten Eyck, L.F., Xuong, N.H., Taylor, S.S., and Sowadski, J.M. 1991b. Structure of a peptide inhibitor bound to the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 253: 414–420. [DOI] [PubMed] [Google Scholar]
- Ladner, R.C., Sato, A.K., Gorzelany, J., and de Souza, M. 2004. Phage display-derived peptides as therapeutic alternatives to antibodies. Drug Discov. Today 9: 525–529. [DOI] [PubMed] [Google Scholar]
- Li, S., Okamoto, T., Chun, M., Sargiacomo, M., Casanova, J.E., Hansen, S.H., Nishimoto, I., and Lisanti, M.P. 1995. Evidence for a regulated interaction between heterotrimeric G proteins and caveolin. J. Biol. Chem. 270: 15693–15701. [DOI] [PubMed] [Google Scholar]
- Li, S., Couet, J., and Lisanti, M.P. 1996. Src tyrosine kinases, Gα subunits, and H-Ras share a common membrane-anchored scaffolding protein, caveolin. Caveolin binding negatively regulates the auto-activation of Src tyrosine kinases. J. Biol. Chem. 271: 29182–29190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, P., Rudick, M., and Anderson, R.G. 2002. Multiple functions of caveolin-1. J. Biol. Chem. 277: 41295–41298. [DOI] [PubMed] [Google Scholar]
- Murase, K., Morrison, K.L., Tam, P.Y., Stafford, R.L., Jurnak, F., and Weiss, G.A. 2003. EF-Tu binding peptides identified, dissected, and affinity optimized by phage display. Chem. Biol. 10: 161–168. [DOI] [PubMed] [Google Scholar]
- Pany, S., Vijayvargia, R., and Krishnasastry, M.V. 2004. Caveolin-1 binding motif of α-hemolysin: Its role in stability and pore formation. Biochem. Biophys. Res. Commun. 322: 29–36. [DOI] [PubMed] [Google Scholar]
- Piotto, M., Saudek, V., and Sklenar, V. 1992. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Bio-mol. NMR 2: 661–665. [DOI] [PubMed] [Google Scholar]
- Quest, A.F., Leyton, L., and Parraga, M. 2004. Caveolins, caveolae, and lipid rafts in cellular transport, signaling, and disease. Biochem. Cell Biol. 82: 129–144. [DOI] [PubMed] [Google Scholar]
- Razani, B. and Lisanti, M.P. 2001. Two distinct caveolin-1 domains mediate the functional interaction of caveolin-1 with protein kinase A. Am. J. Physiol. 281: C1241–C1250. [DOI] [PubMed] [Google Scholar]
- Razani, B., Rubin, C.S., and Lisanti, M.P. 1999. Regulation of cAMP-mediated signal transduction via interaction of caveolins with the catalytic subunit of protein kinase A. J. Biol. Chem. 274: 26353–26360. [DOI] [PubMed] [Google Scholar]
- Rybin, V.O., Xu, X., Lisanti, M.P., and Steinberg, S.F. 2000. Differential targeting of β-adrenergic receptor subtypes and adenylyl cyclase to cardiomyocyte caveolae. A mechanism to functionally regulate the cAMP signaling pathway. J. Biol. Chem. 275: 41447–41457. [DOI] [PubMed] [Google Scholar]
- Sargiacomo, M., Scherer, P.E., Tang, Z., Kubler, E., Song, K.S., Sanders, M.C., and Lisanti, M.P. 1995. Oligomeric structure of caveolin: Implications for caveolae membrane organization. Proc. Natl. Acad. Sci. 92: 9407–9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato, K., Simon, M.D., Levin, A.M., Shokat, K.M., and Weiss, G.A. 2004. Dissecting the Engrailed homeodomain–DNA interaction by phage-displayed shotgun scanning. Chem. Biol. 11: 1017–1023. [DOI] [PubMed] [Google Scholar]
- Schlegel, A. and Lisanti, M.P. 2000. A molecular dissection of caveolin-1 membrane attachment and oligomerization. Two separate regions of the caveolin-1 C-terminal domain mediate membrane binding and oligomer/oligomer interactions in vivo. J. Biol. Chem. 275: 21605–21617. [DOI] [PubMed] [Google Scholar]
- Schlegel, A., Volonte, D., Engelman, J.A., Galbiati, F., Mehta, P., Zhang, X.L., Scherer, P.E., and Lisanti, M.P. 1998. Crowded little caves: Structure and function of caveolae. Cell Signal. 10: 457–463. [DOI] [PubMed] [Google Scholar]
- Schreiber, S., Fleischer, J., Breer, H., and Boekhoff, I. 2000. A possible role for caveolin as a signaling organizer in olfactory sensory membranes. J. Biol. Chem. 275: 24115–24123. [DOI] [PubMed] [Google Scholar]
- Sidhu, S.S., Fairbrother, W.J., and Deshayes, K. 2003. Exploring protein–protein interactions with phage display. Chembiochem 4: 14–25. [DOI] [PubMed] [Google Scholar]
- Slice, L.W. and Taylor, S.S. 1989. Expression of the catalytic subunit of cAMP-dependent protein kinase in Escherichia coli. J. Biol. Chem. 264: 20940–20946. [PubMed] [Google Scholar]
- Smith, G.P. 1985. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 228: 1315–1317. [DOI] [PubMed] [Google Scholar]
- Song, K.S., Li, S., Okamoto, T., Quilliam, L.A., Sargiacomo, M., and Lisanti, M.P. 1996. Co-purification and direct interaction of Ras with caveolin, an integral membrane protein of caveolae microdomains. Detergent-free purification of caveolae microdomains. J. Biol. Chem. 271: 9690–9697. [DOI] [PubMed] [Google Scholar]
- Song, K.S., Tang, Z., Li, S., and Lisanti, M.P. 1997. Mutational analysis of the properties of caveolin-1. A novel role for the C-terminal domain in mediating homotypic caveolin-caveolin interactions. J. Biol. Chem. 272: 4398–4403. [DOI] [PubMed] [Google Scholar]
- Taylor, S.S., Yang, J., Wu, J., Haste, N.M., Radzio-Andzelm, E., and Anand, G. 2004. PKA: A portrait of protein kinase dynamics. Biochim. Biophys. Acta 1697: 259–269. [DOI] [PubMed] [Google Scholar]
- Vajdos, F.F., Adams, C.W., Breece, T.N., Presta, L.G., de Vos, A.M., and Sidhu, S.S. 2002. Comprehensive functional maps of the antigen-binding site of an anti-ErbB2 antibody obtained with shotgun scanning mutagenesis. J. Mol. Biol. 320: 415–428. [DOI] [PubMed] [Google Scholar]
- Venema, V.J., Ju, H., Zou, R., and Venema, R.C. 1997. Interaction of neuronal nitric-oxide synthase with caveolin-3 in skeletal muscle. Identification of a novel caveolin scaffolding/inhibitory domain. J. Biol. Chem. 272: 28187–28190. [DOI] [PubMed] [Google Scholar]
- Weiss, G.A., Watanabe, C.K., Zhong, A., Goddard, A., and Sidhu, S.S. 2000. Rapid mapping of protein functional epitopes by combinatorial alanine scanning. Proc. Natl. Acad. Sci. 97: 8950–8954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, T.M. and Lisanti, M.P. 2004. The caveolin proteins. Genome Biol. 5: 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2005. Caveolin-1 in oncogenic transformation, cancer, and metastasis. Am. J. Physiol. 288: C494–C506. [DOI] [PubMed] [Google Scholar]
- Zheng, J., Knighton, D.R., ten Eyck, L.F., Karlsson, R., Xuong, N., Taylor, S.S. and Sowadski, J.M. 1993. Crystal structure of the catalytic subunit of cAMP-dependent protein kinase complexed with MgATP and peptide inhibitor. Biochemistry 32: 2154–2161. [DOI] [PubMed] [Google Scholar]