

Abstract
The N-terminal 16-kDa domain of Escherichia coli Ada protein (N-Ada16k) repairs DNA methyl phosphotriester lesions by an irreversible methyl transfer to its cysteine residue. Upon the methylation, the sequence-specific DNA binding affinity for the promoter region of the alkylation resistance genes is enhanced by 103-fold. Then, it acts as a transcriptional regulator for the methylation damage. In this paper, we identified the methyl acceptor residue of N-Ada16k and determined the solution structure of the methylated form of N-Ada16k by using NMR and mass spectrometry. The results of a 13C-filtered 1H-13C HMBC experiment and MALDI-TOF MS and MS/MS experiments clearly showed that the methyl acceptor residue is Cys38. The solution structure revealed that it has two distinct subdomains connected by a flexible linker loop: the methyltransferase (MTase) subdomain with the zinc–thiolate center, and the helical subdomain with a helix-turn-helix motif. Interestingly, there is no potential hydrogen bond donor around Cys38, whereas the other three cysteine residues coordinated to a zinc ion have potential donors. Hence, Cys38 could retain its inherent nucleophilicity and react with a methyl phosphotriester. Furthermore, the structure comparison shows that there is no indication of a remarkable conformational change occurring upon the methylation. This implies that the electrostatic repulsion between the negatively charged DNA and the zinc–thiolate center may avoid the contact between the MTase subdomain and the DNA in the nonmethylated form. Thus, after the Cys38 methylation, the MTase subdomain can bind the cognate DNA because the negative charge of the zinc–thiolate center is reduced.
Keywords: N-terminal domain of Ada protein, solution structure, methyl acceptor residue, mass spectrometry, DNA methyltransferase, transcriptional factor, Cys38 methylation
DNA in living organisms is constantly subject to modification by intracellular and extracellular DNA alkylation agents which are cytotoxic, mutagenic, and carcinogenic (Wood et al. 2001; Lindahl 2004). Escherichia coli acquires an inducible response against the alkylation damage in DNA to obtain its cellular resistance to alkylation agents. In this process, termed the adaptive response, Ada protein plays a critical role with a unique functional switch mechanism from a DNA methyltransferase to a transcriptional regulator (Teo et al. 1984).
The 39-kDa Ada protein is comprised of two functional domains linked with the hinge region. The N-terminal 20-kDa domain (N-Ada20k) specifically repairs the Sp diastereomer of DNA methyl phosphotriester lesions by a direct and irreversible methyl transfer to a cysteine residue (Teo et al. 1984; Demple et al. 1985; Nakabeppu et al. 1985; Lindahl et al. 1988; Sedgwick et al. 1988). The C-terminal 19-kDa domain accepts the methyl group from the mutagenic O6-methyl-guanine and O4-methylthymine at Cys321. Methylation of a cysteine residue in N-Ada20k dramatically enhances a sequence-specific DNA binding affinity of the Ada protein for the promoter regions of its own gene, ada, and other alkylation resistance genes, alkA, aidB, and alkB, by 103-fold (Teo et al. 1986; Rebeck et al. 1988; Sakumi and Sekiguchi 1989; Akimaru et al. 1990; Myers et al. 1992; Sakashita et al. 1993; He and Verdine 2002). Further investigations on N-Ada20k indicate that the N-terminal 16-kDa fragment (N-Ada16k, residues 1–146) has a methyltransferase activity and a sequence-specific DNA binding activity with almost the same affinity as that of the native protein and consists of two functionally different subdomains: the methyltransferase subdomain (MTase subdomain, residues 1–73) in the N-terminal region and the helical subdomain (residues 83–146) in the C-terminal region (Sakashita et al. 1993, 1995). The helical subdomain contains a helix-turn-helix (HTH) motif, which is a common and conserved structural motif in a number of transcription factors. The solution structure of the MTase subdomain (N-Ada10k, residues 1–92) has been determined by NMR and revealed a unique folding topology (Myers et al. 1993a; Lin et al. 2001). N-Ada10k has a four-stranded β-sheet and two α-helices. A single zinc ion is tightly bound at the edge of the β-sheet to play an essential role for the correct protein folding. The ligands for the zinc ion are four conserved cysteine residues containing the methyl acceptor residue, Cys38, Cys42, Cys69, and Cys72 (Myers et al. 1993a,b; Ohkubo et al. 1994; Habazettl et al. 1996). It has been shown that the replacement of a zinc ion to a cadmium ion and the mutation of those cysteine residues to an aspartic acid or a histidine residue cause the loss of the Ada’s repair activity (Myers et al. 1995; Sun et al. 2001). Thus, the MTase subdomain of the Ada protein is the first example of the zinc-catalyzed cysteine methylation. However, there has been controversy about the identification of the methyl acceptor residue in N-Ada16k. In 1988, the peptide mapping assay by Sedgwick and coworkers has indicated that the methyl acceptor residue is Cys69 in N-Ada16k (Sedgwick et al. 1988). On the other hand, He and coworkers has recently proposed that Cys38 may be the methyl acceptor residue based on the unpublished results (He and Verdine 2002; He et al. 2003).
In this paper, the methyl acceptor residue of N-Ada16k was identified as Cys38 by NMR and mass spectrometry. Furthermore, the solution structure of the Cys38 methylated form of N-Ada16k (meC38 N-Ada16k) from Escherichia coli was determined by NMR analysis. The DNA binding site of meC38 N-Ada16k was also determined by NMR analysis on the protein/DNA complex. The mechanism of DNA methyl phosphotriester repair via Cys38 and the functional switch in the Ada protein were proposed from these results.
Results
The identification of the methyl acceptor residue by NMR
The 12C-methylated form of uniformly 13C-labeled N-Ada16k was used to identify the methyl acceptor residue by NMR. The 2D 13C-filtered 1H-13C HMBC spectrum of the 12C-methylated form of N-Ada16k contains a single cross-peak at 2.27 ppm (1H)/45.1 ppm (13C) (Fig. 1), corresponding to the correlation between protons of the transferred 12CH3 and a cysteine 13Cβ. Then, the assignment of the cross-peak was made based on the 13C chemical shift value and the 13Cβ chemical shift values of cysteine residues of the methylated form of N-Ada16k (Takinowaki et al. 2004). As shown in Table 1, 13Cβ of Cys38 could be assigned, indicating that the methyl acceptor residue of N-Ada16k for DNA methyl phosphotriester repair is clearly Cys38.
Figure 1.

2D 13C-filtered 1H-13C HMBC spectrum of the methylated form of N-Ada16k.
Table 1.
Comparison of 13C chemical shifts for the identification of the methyl acceptor residue of N-Ada16k
| 13C chemical shifts (ppm) | ||
| Cysteine residues | 13Cβa | The 13C-filtered 1H-13C HMBC spectrum |
| Cys38 | 45.2 | 45.1 |
| Cys42 | 31.1 | — |
| Cys69 | 34.9 | — |
| Cys72 | 32.7 | — |
a These values were cited from the sequence-specific assignment of the methylated form of N-Ada16k deposited in BioMagResBank under accession code 6054.
The identification of the methyl acceptor residue by MALDI-TOF-MS
To confirm the result of NMR analysis, the alternative identification of the methyl acceptor residue of N-Ada16k was performed by mass spectrometry. The iodoacetamide-treated tryptic digests of the nonmethylated and the methylated forms of N-Ada16k were used for this analysis. Comparison of both MS spectra shows that the peak at m/z 1354.6 was shifted to the peak at m/z 1311.6 upon the methylation, corresponding to the peptide fragment of residues 33–43 containing Cys38 and Cys42 (nonmethylated form, T33–43, and methylated form, meT33–43) deduced from the expected mass (Fig. 2A,B). In contrast, the peak at m/z 2303.1 corresponding to the expected mass of the peptide fragment of residues 50–70 containing Cys69 (T50–70) was not shifted upon the methylation (Fig. 2C,D). There is no peak corresponding to the expected mass of the Cys69 methylated T50–70 peptide (2259.0869 Da). Therefore, Cys69 was ruled out and it was considered that either Cys38 or Cys42 is the methyl acceptor residue.
Figure 2.

An expanded view of MALDI-TOF mass spectra of the iodoacetamide-treated tryptic fragments of the nonmethylated and the methylated forms of N-Ada16k. (A) The peak at m/z 1354.6 of the nonmethylated form corresponds to the expected mass (1354.6360 Da) of the T33–43 peptide containing Cys38 and Cys42, which follows the carboxyamidomethylations of two cysteine residues with the carboxyamidomethyl group substitution of the cysteine γ-thiol hydrogen. (B) The peak at m/z 1311.6 of the methylated form corresponds to the expected mass (1311.630 Da) of the meT33–43 peptide, which follows the carboxy-amidomethylation of one cysteine residue and the methylation of the other. (C, D) Each of peaks at m/z 2303.1 corresponds to the expected mass (2302.0927 Da) of the T50–70 peptide containing Cys69 with the carboxyamidomethylation. (E) MS/MS spectrum of the meT33–43 peptide. An arrow indicates a mono-charged parent ion (m/z 1311.6 Da). The matched y-type and b-type fragment ions are displayed. The peptide sequence was identified to TTGIFmeC38RPSC42R, where meC represents Sγ-methyl cysteine.
The position of the methylated cysteine residue for the meT33–43 peptide sequence was identified by tandem MS (MS/MS) analysis, which yields a series of product ions (y-type and b-type ion) by sequential collision-induced dissociation. The meT33–43 peptide sequence was identified by y1, y4, y5, b3, b4, b5, b7, b9, and b10 fragment ions as shown in Figure 2E, indicating that its peptide sequence is not TTGIFC38RPSmeC42R but TTGIFmeC38RPSC42R. Therefore, mass spectrometry also clearly shows that the methyl acceptor residue is Cys38.
Structure determination and overall structure of meC38 N-Ada16k
The solution structure of meC38 N-Ada16k was determined by including the information of the corrected methyl acceptor residue, Cys38. The almost complete assignments for backbone and side chain 1H, 13C, and 15N resonances of meC38 N-Ada16k have already been reported elsewhere (Takinowaki et al. 2004), and are available at the BioMagResBank (http://www.bmrb.wisc.edu) under accession code 6054. The detailed restraint data for the structure calculation of meC38 N-Ada16k were summarized in Table 2. A total of 1735 NOE restraints were employed for this structure calculation, including 710 intraresidue, 427 sequential, 285 medium-range (i − i + 2, i − i + 3, i − i + 4), and 313 long-range restraints. In addition, the 148 dihedral angle restraints were obtained from the program TALOS and the 94 hydrogen bond restraints were observed from H-D exchange experiments. The 14 zinc–thiolate center distance restraints were utilized to enforce a proper tetrahedral coordination to a metal ion. The 17 structures of the lowest total energy structures with no distance restraint violations >0.5 Å and no torsion angle restraint violations above 5° were selected for further analysis. Ramachandran φ/ψ plots for the ensemble of 17 structures indicate that 73.8% of the nonglycine and nonproline residues are found in the most favored region and 21.0% in the additional allowed regions.
Table 2.
Structural statistics for the final 17 structures of meC38 N-Ada16k
| No. of restraints | |
| All | 1991 |
| NOE distance restraints | 1735 |
| Intraresidue | 710 |
| Sequential | 427 |
| Medium range (i − i + 2, i − i + 3, i − i + 4) | 285 |
| Long range | 313 |
| Hydrogen bond distance restraints | 94 |
| Zinc-thiolate center distance restraints | 14 |
| Dihedral angle restraints | 148 |
| Deviations from idealized covalent geometry | |
| Bonds (Å ) | 0.0011 ± 0.00004 |
| Angle (°) | 0.2972 ± 0.0018 |
| Impropers (°) | 0.1539 ± 0.0068 |
| Mean coordinate RMSD from mean structure | |
| Overall structure (residues 9–123) | |
| Backbone heavy atoms | 8.35 ± 1.96 Å |
| All heavy atoms | 8.85 ± 1.98 Å |
| MTase subdomain (residues 9–73) | |
| Backbone heavy atoms | 0.40 ± 0.90 Å |
| All heavy atoms | 1.13 ± 0.17 Å |
| The helical subdomain | |
| Residues 83–123 | |
| Backbone heavy atoms | 0.68 ± 0.19 Å |
| All heavy atoms | 1.19 ± 0.19 Å |
| Residues 83–142 | |
| Backbone heavy atoms | 1.01 ± 0.37 Å |
| All heavy atoms | 1.54 ± 0.30 Å |
The overall structure of meC38 N-Ada16k has two distinct globular subdomains: the MTase subdomain (residues 1–73), and the helical subdomain (residues 83–146). The two subdomains are connected by a flexible linker loop (residues 74–82). A ribbon diagram of a representative lowest energy NMR structure of meC38 N-Ada16k is shown in Figure 3A. There was no NOE observed between the MTase and the helical subdomain, suggesting that they do not directly interact in solution. The 15N relaxation analysis indicates that both subdomains do not tumble together as a unit (Supplemental Fig. S1). Therefore, the relative orientation of two subdomains is assumed not to be fixed. For individual subdomains, however, the superposition of backbone atoms clearly shows that the calculated structures were well converged (Fig. 3B,C). The average RMSD values to the mean structure for backbone atoms in the well-structured regions of the MTase subdomain (residues 9–73) and the helical subdomain (residues 83–123) were 0.40 ± 0.09 Å and 0.68 ± 0.19 Å, respectively. The average RMSD values to the mean structure for all heavy atoms in the well-structured regions of the MTase subdomain and the helical subdomain were 1.13 ± 0.17 Å and 1.19 ± 0.19 Å, respectively.
Figure 3.
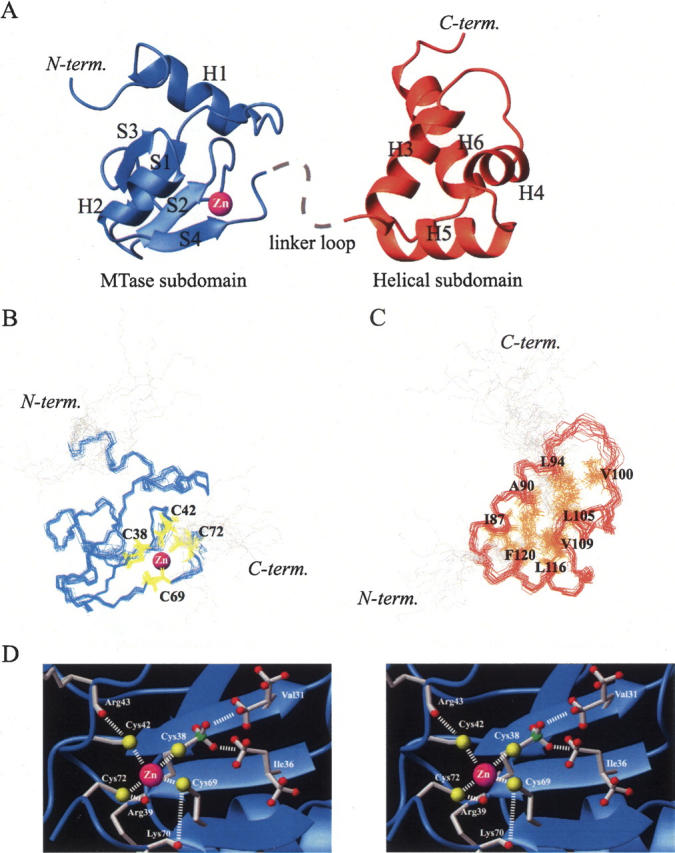
Solution structure of meC38 N-Ada16k. (A) Ribbon representation showing the MTase subdomain (blue) and the helical subdomain (orange). Bound to a protein is the zinc ion (magenta sphere) and the dashed line indicates the linker loop connecting two subdomains. The superposition of 17 lowest energy backbone conformers of (B) the MTase subdomain (blue, residues 9–73) with side chains of Cys38, Cys42, Cys69, and Cys72 (yellow) and (C) the helical subdomain (red, residues 83–123) with side chains of Ile87, Ala90, Leu94, Val100, Leu105, Val109, Leu116, and Phe120 (orange) involved in hydrophobic packing. (D) Zinc–thiolate center of meC38 N-Ada16k in a stereoview. Side chains of Val31, Ile36, Cys38, Arg39, Cys42, Arg43, Cys69, Lys70, and Cys72 are illustrated by stick models. The zinc ion is represented as a magenta sphere. Possible interactions are shown in a dashed line.
The MTase subdomain of meC38 N-Ada16k has one characteristic β-sheet sandwiched between two α-helices. A first long helix, H1 (residues 9–18), is vertically packed to a following β-sheet, which consists of four β-strands, S1 (residues 28–31), S2 (residues 36–38), S3 (residues 52–55), and S4 (residues 67–69). S1, S2, and S3 form an antiparallel β-sheet in centering S1 and S4 stands parallel to S2 to form a four-stranded β-sheet. A typical type I β-turn (residues 32–35) connects S1 and S2 and an unstructured long loop region (residues 39–51) links S2 to S3. A second helix, H2 (residues 58–64), is tightly packed on the face of the four-stranded β-sheet. The four cysteine residues, Sγ-methyl Cys38, Cys42, Cys69, and Cys72, coordinated to the zinc ion, are located on the edge of a β-sheet (Fig. 3B). On the other hand, the structure of the helical sub-domain consists of four α-helices, H3 (residues 83–94), H4 (residues 102–109), H5 (residues 113–123), and H6 (residues 128–142), which form a four-stranded helix bundle. Each helix is aligned at about a 60°, 90°, and 60° angle with the next one. The three helices, H3, H4, and H5, are more tightly packed. These helices form the core structure of the helical subdomain with the extensive hydrophobic interactions among Ile87, Ala90, Leu94, Val100, Leu105, Val109, Leu116, and Phe120 (Fig. 3C). H4 and H5 form a HTH motif, of which the recognition helix is H5. For helix H6, the N-terminal part is packed into other helices but the C-terminal part is exposed to the solvent.
Zn(Cys)4 coordination of meC38 N-Ada16k
Figure 3D shows the zinc–thiolate center of meC38 NAda16k. The Sγ-methyl group of Cys38 is buried in the hydrophobic pocket next to the zinc–thiolate center (see Discussion). A γ-methyl group of Val31 on the S1 strand and a δ-methyl group of Ile36 on the S2 strand are located within a distance of 3.0 Å to the Sγ-methyl group. These three hydrophobic methyl groups form a hydrophobic core. In addition, distances between Sγ of Cys42 and HN of Arg43, and Sγ of Cys72 and HN of Arg39 are below 3.0 Å, indicating the formation of NH···S hydrogen bonds (Adman et al. 1975). A distance between Sγ of Cys69 and HN of Lys70 is about 4.0 Å, which is still within the range of a NH···S hydrogen bond considering the dispersion of NMR structures. Slow hydrogen exchange with an exchange time of >4 h was observed for HN of Arg39. Therefore, HN of Arg39 forms a stable hydrogen bond. In contrast, there is no potential hydrogen bond donor around the Sγ of Cys38 within a distance of 4.0 Å. NH···S hydrogen bonds are often observed in zinc proteins, which play a key role in the structure stabilization (Klein et al. 2000).
The sequence-specific DNA binding regions of meC38 N-Ada16k
To determine the sequence-specific DNA binding sites of the methylated form of the Ada protein, 2D 1H-15N HSQC spectra of meC38 N-Ada16k with DNA fragments containing an ada or alkA promoter sequence were recorded. As shown in Figure 4A, upon the DNA binding, several backbone 15N and 15NH resonances of the protein are perturbed by both DNA fragments. Each perturbed site is mapped onto the primary and tertiary structure of meC38 N-Ada16k (Fig. 4B). For the meC38 N-Ada16k/ada DNA complex, the main perturbed sites are the β-sheet region of the MTase subdomain and the HTH region of the helical subdomain, corresponding to preliminary NMR studies by Sakashita et al. (1995). In addition, the linker loop region connecting two subdomains also exhibits substantial chemical shift perturbations, indicating that the backbone conformation of the flexible linker loop alters upon the sequence-specific DNA binding. The DNA binding by such a flexible subdomain connector has been often found in zinc finger proteins (Wuttke et al. 1997). For the meC38 N-Ada16k/alkA DNA complex, the β-sheet, HTH, and linker loop region also show large chemical shift perturbations. Consequently, the perturbed sites upon the sequence-specific DNA binding are almost the same between ada and alkA promoters, indicating that meC38 N-Ada16k makes contacts to an ada or alkA promoter DNA in the same manner.
Figure 4.

(A) An expanded view of the 2D 1H-15N HSQC spectra of the meC38 N-Ada16k (black), meC38 N-Ada16k/ada DNA complex (left, red), and meC38 N-Ada16k/alkA DNA complex (right, blue). Mapping of NMR signal perturbations on (B) the meC38 N-Ada16k/ada DNA complex (left), meC38 NAda16k/alkA DNA complex (right), and (C) N-Ada16k/ada DNA complex. The N-Ada16k/ada DNA complex, illustrated with the data, was obtained from our previous experiments (Sakashita et al. 1995). Each of perturbed sites is represented by filled boxes in the primary structure and red in the three-dimensional structure. Secondary structure elements are indicated schematically: the α-helices are indicated by a spiral and the β-strands by an arrow.
The DNA binding mode of a HTH motif
To understand the DNA recognition mechanism of the HTH motif, a model of the helical subdomain/DNA complex was constructed by fitting the H3–H5 region (residues 84–94, 103–109, and 113–119) of meC38 N-Ada16k to the corresponding region (residues 147–157, 162–168, and 173–179) of Hin recombinase (Feng et al. 1994). The structure of Hin recombinase in the DNA complex was shown to resemble the structure of the helical subdomain determined here by the Dali search (Holm and Sander 1993) with the RMSD value of 2.77 Å. Then, the relative position of the protein for the DNA was adjusted by avoiding violations of van der Waals radius of atoms. In this model, the major groove of DNA is wide enough to accommodate the recognition helix H5 (Fig. 5). Especially, Phe114, His115, His117, and Arg118 on H5 could make dense contacts to bases or phosphate backbones, namely the aromatic ring of Phe114 shows hydrophobic interaction with bases, positive side chains of His115 and His117 interact with negatively charged phosphate groups of DNA, and the –NH2 groups of the side chain of Arg118 may donate a hydrogen bond to bases. This model is consistent with previous results of mutational studies, which indicate that the replacements of Phe114 to isoleucine and Arg118 to glutamine completely abolishes the binding affinity of meC38 N-Ada16k for the DNA and the replacement of His117 to glutamine drastically decreases the affinity as well (Sakashita et al. 1995).
Figure 5.

The DNA binding model of the HTH motif of meC38 N-Ada16k. Side chains of Phe114, His115, His117, and Arg118 are illustrated by stick models, whose positions are at a distance where the predicted contacts would be possible.
Discussion
DNA methyl phosphotriester repair via Cys38
As described above, both NMR and MS results clearly show that the methyl acceptor residue is Cys38. However, Sedgwick and coworkers have previously reported that Cys69 is the methyl acceptor residue by using the peptide mapping method for tryptic digests of the Ada protein labeled by incubation with [3H] MNU-treated DNA (Sedgwick et al. 1988). They isolated a fraction containing a [3H]-labeled fragment by HPLC chromatography among a mixture of the tryptic digests and analyzed by Edman degradation. As shown in Figure 5 of their paper, the HPLC elution profile was quite crowded, and it seems to be difficult to isolate a single digested fragment. Hence, it might lead to the misidentification of the methyl acceptor residue of N-Ada16k.
N-Ada16k can specifically demethylate the Sp diastereomer of DNA methyl phosphotriester lesions by a direct and irreversible methyl transfer to Cys38 (Lindahl et al. 1988). 113Cd NMR studies revealed that all of the thiols of four cysteine residues are coordinated to the zinc ion both before and after the Cys38 methylation (Ohkubo et al. 1994). In this report, the structure of the zinc–thiolate center of the methylated form (meC38 N-Ada16k) indicates that the three protein backbone amides of Arg43, Lys70, and Arg39 are close to three inactive cysteine residues of Cys42, Cys69, and Cys72 to be candidates of hydrogen bond donors to these cysteine residues, but there is not such a candidate to the methyl acceptor residue, Cys38 (Fig. 3D). In the nonmethylated form (PDB accession code 1EYF), there are also the same potential hydrogen bond donors around Cys42, Cys69, and Cys72. However, it is interesting that there is no potential hydrogen bond donor around the reactive sulfur atom of Cys38 in the nonmethylated form. A recent study on small model compounds containing sulfur atoms coordinated to a zinc ion has shown that a single intramolecular NH···S hydrogen bond can dramatically decrease the reactivity of the sulfur atom by two orders of magnitude (Smith et al. 2005). In the case of zinc proteins, the statistical analysis on electrostatic potential and packing of zinc–thiolate centers has pointed out that hydrogen bonds of sulfur ligands are important factors in reducing the reactivity of the zinc–thiolate center as well as stabilizing the protein folding (Maynard and Covell 2001). Hence, in the Ada protein, the nucleophilicity of the Cys42, Cys69, and Cys72 sulfur atoms might be reduced relative to that of the Cys38 sulfur atom by hydrogen bonds so that only Cys38 can react as a nucleophile.
Figure 6 shows the protein surface around Cys38 on the MTase subdomain of the nonmethylated and the methylated forms. In the nonmethylated form, there is a remarkable hydrophobic pocket next to the zinc–thiolate center, which is formed by Phe29, Val31, and Ile36. The reactive sulfur atom of Cys38 faces this pocket, and is partially exposed to the solvent. The position of Cys38 sulfur atom is suitable to make contacts to a protruded methyl group from the Sp diastereomer of a methyl phosphotriester but not to approach a distal methyl group from the Rp diastereomer. On the contrary, in the methylated form, NMR structure shows that the hydrophobic pocket is completely filled by the transferred methyl group of Cys38. The pocket size seems to be quite adequate to accommodate a methyl group. This finding can explain the previous in vivo adaptive response assays of Bacillus subtilis Ada protein toward various alkylating agents, which have shown that the most effective inducing agents for the adaptive response are methylating agents as well as ethylating ones, whereas more bulky alkylating agents are inactive (Morohoshi and Munakata 1983). The hydrophobicity of the pocket could help the Cys38 sulfur reach the methylated phosphate group and more effectively facilitate the methyl transfer reaction.
Figure 6.

Protein surfaces of the nonmethylated (left, PDB accession code 1EYF) and the methylated (right, PDB accession code 1WPK) forms of the MTase subdomain. The basic residues with and without the chemical shift perturbations upon the DNA binding are colored blue and sky blue, respectively. The hydrophobic residues are colored dark orange. The hydrophobic residues in Figure 3D (Val31 and Ile36) are colored orange red. The methyl acceptor residue (Cys38) and the other three cysteine residues (Cys42, Cys69, and Cys72) are colored green and yellow, respectively. In the methylated form, the Sγ-methyl group of Cys38 is colored pink.
The model of functional switch mechanism
The results of the DNA titration experiments shown in Figure 4B and C have revealed that there are two different binding modes for the methylated and the nonmethylated forms of the Ada protein. For meC38 N-Ada16k, which shows the sequence-specific DNA binding, the interaction seems to occur in two positions because the chemical shift perturbations were detected in both of its subdomains, i.e., on the β-sheet region of the MTase subdomain and the HTH motif of the helical subdomain. On the other hand, for N-Ada16k, which shows the nonspecific DNA binding, the interaction seems to occur only through the HTH motif because the perturbation was localized at the motif region on the helical subdomain (Sakashita et al. 1995). These data clearly show that the MTase subdomain can make contacts to the DNA after the Cys38 methylation. The following scheme of the acquisition of the DNA binding activity has been proposed by Lin et al. (2001). That is, the cysteine methylation triggers a conformational change of the Ada protein, which should induce some rearrangements of the surface of the MTase subdomain. Consequently, a new DNA binding site would be created to provide a strong sequence-specific binding activity. However, the present study clarifies that there is no remarkable conformational change between the nonmethylated and the methylated forms. The superposition of the methylated form for the nonmethylated form exhibits the small RMSD value of 1.8 Å for the backbone atoms (residues 9–73). The transferred methyl group of Cys38 just fits in the hydrophobic pocket without remarkable distortion. In the MTase subdomain, side chains of several basic residues, Arg39, Arg43, Arg67, Lys70, and Arg71, whose backbone chemical shifts are perturbed upon the sequence-specific DNA binding, are located so as to surround the zinc–thiolate center (Fig. 6). These basic residues could facilitate electrostatic interactions with negatively charged phosphate backbones of the target DNA. The comparison of the protein surface between the nonmethylated and the methylated forms shows that the distributions of these positive charges are almost the same in both forms, indicating that there is no additional positive charge which is exposed to the solvent upon the methylation. Based on these observations, the structural change of the Ada protein is not likely to be directly involved in making contacts to the DNA during the sequence-specific binding. Then, a following question arises. Why, even though the DNA binding surface in the MTase subdomain also retains in the nonmethylated form, its surface could not make contacts to the DNA? It is noteworthy that the zinc–thiolate center, consisting of four thiolates coordinated to the zinc ion of the MTase subdomain, is assumed to have the net charge of −2. Therefore, this charge causes a repulsive electrostatic interaction with negatively charged phosphate backbones on the DNA. The methyl transfer toward Cys38 could reduce the negative charge of the zinc–thiolate center to some extent so that the repulsive interaction with DNA would be reduced. Hence, the Ada protein might control its DNA binding affinity by changing the electrostatic repulsion between the protein and the DNA. The electrostatic potentialsof the nonmethylated and the methylated forms show that the negative charge distribution around the zinc–thiolate center is significantly reduced in the methylated form to facilitate the DNA binding (Supplemental Fig. S2). The crystal structure of the complex of 8-oxoguanine DNA glycosylase I with DNA has recently shown that, in the interaction between the protein and the DNA, the negative charge of 8-oxoguanine has an important role to provide the preferential binding affinity of mutagenic guanine compared with normal guanine (Banerjee et al. 2005). Thus, it is concluded that a methyl group transferred to Cys38 even with rather small volume may play a role as an “electrostatic switch” to enhance its sequence-specific DNA binding affinity by 103-fold and acts as a chemosensor for the DNA alkyaltion damage in E. coli.
Materials and methods
Mass spectrometry
Expression and purification of N-Ada16k were performed as previously described (Takinowaki et al. 2004). The methylated form of N-Ada16k was prepared by incubating N-Ada16k with methylnitrosourea (MNU)-treated DNA at 37 °C for 30 min (Nakabeppu and Sekiguchi 1986). The reaction condition was optimized so that the protein and the MNU-treated DNA were combined at a molar ratio of 1:1. As estimated from the peak intensity in the NMR spectrum, >95% of N-Ada16k were methylated in this condition. Samples for mass spectrometry were prepared as a solution with protein concentration of 150 μM in 20 mM Tris-HCl buffer (pH 8.5) with 1 mM EDTA, 20% (v/v) glycerol, and 300 mM NaCl. The in-solution proteolytic digestion protocol was used as follows. Each protein solution of 250 μL was reduced by 5 μL of 1 M dithiothreitol at 37°C for 1 h, alkylated by 1 μL of 1 M iodoacetamide in the dark for 30 min, and digested by 4 μg of trypsin at 37°C for 2 h. The mixture of digests of the nonmethylated or the methylated form of N-Ada16k was separately analyzed by MS and MS/MS experiments to identify the methyl acceptor residue of N-Ada16k. All mass measurements were carried out on a QSTAR Pulsar, hybrid quadropole TOF MS/MS equipped with an o-MALDI ion source (Applied Biosystems/MDS Sciex). A solution of 10 mg/mL 2,5–5-dihydroxybenzoic acid, 33% acetonitrile, and 0.1% trifluoroacetic acid was used as a matrix. A volume of 1 μL of each proteolytic digest was mixed on the target disk with an equal volume of matrix and ionized using MALDI methods (Kaufmann 1995). Argon was used as the collision gas. An external mass calibration was performed with renin. All data for MS and MS/MS spectra were analyzed with Mascot software (Matrix Science).
NMR spectroscopy
Uniformly 15N and 13C-labeled proteins were incubated by the same method above mentioned except the E. coli BL21(DE3) cells in M9 minimal medium, containing 15N ammonium chloride (1 g/L) and/or 13C glucose (2 g/L) as the sole nitrogen and carbon sources. The 12C-methylated form of uniformly 13C-labeled N-Ada16k was prepared by incubating uniformly 13C-labeled N-Ada16k with MNU-treated DNA. The NMR samples were prepared in 50 mM sodium phosphate buffer of D2O or 85% H2O/15% D2O mixture at pH 6.5 with 300 mM NaCl and 5 mM 2-mercaptoethanol. The protein concentration was approximately adjusted to 1 mM in 5-mm microcell NMR tube (Shigemi) for all NMR experiments. All 2D and 3D NMR experiments were performed at 30°C on an INOVA600 spectrometer (Varian) equipped with shielded gradient triple resonance probes. The pulsed-field gradient techniques with a WATERGATE (Piotto et al. 1992) were utilized in all H2O experiments for the solvent suppression. Transmitter frequencies for 1H, 15N, 13Cα, aliphatic 13C, aromatic 13C, and carbonyl 13C were typically set to 4.76, 119.0, 55.0, 43.0, 125.0, and 176.0 ppm, respectively. Sodium 2,2-dimethyl-2-silapentane-5-sulfonate (DSS) was used as an external reference of 1H chemical shifts. 15N and 13C chemical shifts were indirectly calibrated from each gyromagnetic ratio (Wishart et al. 1995). Backbone and side-chain assignments were obtained from 2D 1H-15N HSQC, 1H-13C HSQC, 3D HNCO, HNCA, HN(CO)CA, HNCACB, CBCA(CO)NH, HBHA(CBCA CO)NH, (Hβ)Cβ(CγCδ)Hδ, and HCCH-TOCSY spectra (Bax et al. 1994a; Kay 1995). NOEs were collected from 3D 15N-edited NOESY (75 ms mixing time) and 13C-edited NOESY (75 ms mixing time) spectra (Bax et al. 1994a; Kay 1995). 2D 13C-filtered 1H-13C HMBC spectrum (Bax et al. 1994b), modified to detect the correlation between 12C attached 1H and 13C through 3JHC coupling, was recorded for identification of the methyl acceptor residue of N-Ada16k. Backbone amide groups slowly exchanging with the solvent were identified from a series of 2D 1H-15N HSQC spectra following a rapid buffer exchange to D2O. The sequence-specific DNA binding sites were investigated by 2D 1H-15N HSQC spectra using synthetic DNAs containing a following ada or alkA promoter sequence: 5′-GAAAAATTAAAGCGCAAG ATG-3′, 3′-CTTTTTAATTTCGCGTTCTAC-5′ for an ada sequence and 5′-CCGGAATATGAAAGCAAAGCG-3′, 5′-GGCCTTATACTTTCGTTTCGC-3′ for an alkA sequence. The Ada consensus sequences separated by six base pairs are underlined (Landini and Volkert 1995). The DNA duplex was formed by mixing equimolar amounts of each single-stranded oligonucleotide and annealed by heating 100°C for 1 min and cooling to room temperature gradually. The protein and the DNA were combined at a molar ratio of 1:1. All the NMR data were processed with NMRPipe (Delaglio et al. 1995) and analyzed with the PIPP program (Garrett et al. 1991).
Structure calculation
NOE restrains were classified into four categories: strong, medium, weak, and very weak, corresponding to the distance restraints of 1.8–2.8 Å, 1.8–3.5 Å, 1.8–5.0 Å, and 1.8–6.0 Å, respectively. The φ and ψ torsion angle restraints were evaluated from the 15N, Hα, 13Cα, 13Cβ, and 13C′ chemical shifts using program TALOS (Cornilescu et al. 1999). The restraints deduced from intramolecular hydrogen bonds of protein backbone, which were identified by H-D exchange experiments, were classified into two groups: between the amide proton and the carbonyl oxygen of 1.5–2.5 Å and between the amide nitrogen and the carbonyl oxygen of 2.5–3.5 Å (Wüthrich 1986). The distance restraints for the zinc–thiolate center were incorporated in the structure calculation (Myers et al. 1993a). The initial solution structures were calculated using the distance geometry algorithm in the programs CNS (Brunger et al. 1998). The structure optimization and energy minimization were achieved by simulated annealing algorithm. The final 17 lowest energy structures were analyzed by using the programs MOLMOL (Koradi et al. 1996) and PROCHECK (Laskowski et al. 1996). Structural statistics for the 17 structures are included in Table 2. All of these structures have been deposited in the Protein Data Bank (http://www.rcsb.org/pdb/) under the accession code 1WPK.
Acknowledgments
We thank S. Watanabe and T. Kayamori, Protein Analysis Group, Hitachi Science Systems, Ltd., for supporting the MALDI-TOF-MS and -MS/MS analysis to identify the methyl acceptor residue of N-Ada16k.
Abbreviations
N-Ada16k, the N-terminal 16-kDa domain of the Ada protein
meC38 N-Ada16k, the Cys38 methylated form of N-Ada16k
MTase, methyltransferase
HTH, helix-turn-helix
NMR, nuclear magnetic resonance
MALDI-TOF MS, matrix assisted laser desorption/ionization time of flight mass spectrometry
MNU, methylnitrosourea
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051786306.
Supplemental material: see www.proteinscience.org
References
- Adman, E., Watenpaugh, K.D., and Jensen, L.H. 1975. NH—S hydrogen bonds in Peptococcus aerogenes ferredoxin, Clostridium pasteurianum rubredoxin, and Chromatium high potential iron protein. Proc. Natl. Acad. Sci. 72: 4854–4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akimaru, H., Sakumi, K., Yoshikai, T., Anai, M., and Sekiguchi, M. 1990. Positive and negative regulation of transcription by a cleavage product of Ada protein. J. Mol. Biol. 216: 261–273. [DOI] [PubMed] [Google Scholar]
- Banerjee, A., Yang, W., Karplus, M., and Verdine, G.L. 2005. Structure of a repair enzyme interrogating undamaged DNA elucidates recognition of damaged DNA. Nature 434: 612–618. [DOI] [PubMed] [Google Scholar]
- Bax, A., Delaglio, F., Grzesiek, S., and Vuister, G.W. 1994a. Resonance assignment of methionine methyl groups and χ3 angular information from long-range proton–carbon and carbon–carbon J correlation in a calmodulin–peptide complex. J. Biomol. NMR 4: 787–797. [DOI] [PubMed] [Google Scholar]
- Bax, A., Vuister, G.W., Grzesiek, S., Delaglio, F., Wang, A.C., Tschudin, R., and Zhu, G. 1994b. Measurement of homo- and heteronuclear J couplings from quantitative J correlation. Methods Enzymol. 239: 79–105. [DOI] [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54(Pt 5): 905–921. [DOI] [PubMed] [Google Scholar]
- Cornilescu, G., Delaglio, F., and Bax, A. 1999. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 13: 289–302. [DOI] [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6: 277–293. [DOI] [PubMed] [Google Scholar]
- Demple, B., Sedgwick, B., Robins, P., Totty, N., Waterfield, M.D., and Lindahl, T. 1985. Active site and complete sequence of the suicidal methyltransferase that counters alkylation mutagenesis. Proc. Natl. Acad. Sci. 82: 2688–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, J.A., Johnson, R.C., and Dickerson, R.E. 1994. Hin recombinase bound to DNA: The origin of specificity in major and minor groove interactions. Science 263: 348–355. [DOI] [PubMed] [Google Scholar]
- Garrett, D.S., Powers, R., Gronenborn, A.M., and Clore, G.M. 1991. A common sense approach to peak picking two-, three- and four-dimensional spectra using automatic computer analysis of contour diagrams. J. Magn. Reson. 95: 214–220. [DOI] [PubMed] [Google Scholar]
- Habazettl, J., Myers, L.C., Yuan, F., Verdine, G.L., and Wagner, G. 1996. Backbone dynamics, amide hydrogen exchange, and resonance assignments of the DNA methylphosphotriester repair domain of Escherichia coli Ada using NMR. Biochemistry 35: 9335–9348. [DOI] [PubMed] [Google Scholar]
- He, C. and Verdine, G.L. 2002. Trapping distinct structural states of a protein/DNA interaction through disulfide crosslinking. Chem. Biol. 9: 1297–1303. [DOI] [PubMed] [Google Scholar]
- He, C., Wei, H., and Verdine, G.L. 2003. Converting the sacrificial DNA repair protein N-ada into a catalytic methyl phosphotriester repair enzyme. J. Am. Chem. Soc. 125: 1450–1451. [DOI] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1993. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233: 123–138. [DOI] [PubMed] [Google Scholar]
- Kaufmann, R. 1995. Matrix-assisted laser desorption ionization (MALDI) mass spectrometry: A novel analytical tool in molecular biology and biotechnology. J. Biotechnol. 41: 155–175. [DOI] [PubMed] [Google Scholar]
- Kay, L.E. 1995. Pulsed field gradient multi-dimensional NMR methods for the study of protein structure and dynamics in solution. Prog. Biophys. Mol. Biol. 63: 277–299. [DOI] [PubMed] [Google Scholar]
- Klein, D.J., Johnson, P.E., Zollars, E.S., De Guzman, R.N., and Summers, M.F. 2000. The NMR structure of the nucleocapsid protein from the mouse mammary tumor virus reveals unusual folding of the C-terminal zinc knuckle. Biochemistry 39: 1604–1612. [DOI] [PubMed] [Google Scholar]
- Koradi, R., Billeter, M., and Wüthrich, K. 1996. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 14: 52–55. [DOI] [PubMed] [Google Scholar]
- Landini, P. and Volkert, M.R. 1995. Transcriptional activation of the Escherichia coli adaptive response gene aidB is mediated by binding of methylated Ada protein. Evidence for a new consensus sequence for Ada-binding sites. J. Biol. Chem. 270: 8285–8289. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., Rullmannn, J.A., MacArthur, M.W., Kaptein, R., and Thornton, J.M. 1996. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8: 477–486. [DOI] [PubMed] [Google Scholar]
- Lin, Y., Dotsch, V., Wintner, T., Peariso, K., Myers, L.C., Penner-Hahn, J.E., Verdine, G.L., and Wagner, G. 2001. Structural basis for the functional switch of the E. coli Ada protein. Biochemistry 40: 4261–4271. [DOI] [PubMed] [Google Scholar]
- Lindahl, T. 2004. Molecular biology: Ensuring error-free DNA repair. Nature 427: 598. [DOI] [PubMed] [Google Scholar]
- Lindahl, T., Sedgwick, B., Sekiguchi, M., and Nakabeppu, Y. 1988. Regulation and expression of the adaptive response to alkylating agents. Annu. Rev. Biochem. 57: 133–157. [DOI] [PubMed] [Google Scholar]
- Maynard, A.T. and Covell, D.G. 2001. Reactivity of zinc finger cores: Analysis of protein packing and electrostatic screening. J. Am. Chem. Soc. 123: 1047–1058. [DOI] [PubMed] [Google Scholar]
- Morohoshi, F. and Munakata, N. 1983. Adaptive response to simple alkylating agents in Bacillus subtilis cells. Mutant Res. 110: 23–37. [Google Scholar]
- Myers, L.C., Terranova, M.P., Nash, H.M., Markus, M.A., and Verdine, G.L. 1992. Zinc binding by the methylation signaling domain of the Escherichia coli Ada protein. Biochemistry 31: 4541–4547. [DOI] [PubMed] [Google Scholar]
- Myers, L.C., Verdine, G.L., and Wagner, G. 1993a. Solution structure of the DNA methyl phosphotriester repair domain of Escherichia coli Ada. Biochemistry 32: 14089–14094. [DOI] [PubMed] [Google Scholar]
- Myers, L.C., Terranova, M.P., Ferentz, A.E., Wagner, G., and Verdine, G.L. 1993b. Repair of DNA methylphosphotriesters through a metalloactivated cysteine nucleophile. Science 261: 1164–1167. [DOI] [PubMed] [Google Scholar]
- Myers, L.C., Jackow, F., and Verdine, G.L. 1995. Metal dependence of transcriptional switching in Escherichia coli Ada. J. Biol. Chem. 270: 6664–6670. [DOI] [PubMed] [Google Scholar]
- Nakabeppu, Y. and Sekiguchi, M. 1986. Regulatory mechanisms for induction of synthesis of repair enzymes in response to alkylating agents: Ada protein acts as a transcriptional regulator. Proc. Natl. Acad. Sci. 83: 6297–6301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakabeppu, Y., Kondo, H., Kawabata, S., Iwanaga, S., and Sekiguchi, M. 1985. Purification and structure of the intact Ada regulatory protein of Escherichia coli K12, O6-methylguanine-DNA methyltransferase. J. Biol. Chem. 260: 7281–7288. [PubMed] [Google Scholar]
- Ohkubo, T., Sakashita, H., Sakuma, T., Kainosho, M., Sekiguchi, M., and Morikawa, K. 1994. Methylation dependent functional switch mechanism newly found in the Escherichia coli Ada protein. J. Am. Chem. Soc. 116: 6035–6036. [Google Scholar]
- Piotto, M., Saudek, V., and Sklenar, V. 1992. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 2: 661–665. [DOI] [PubMed] [Google Scholar]
- Rebeck, G.W., Coons, S., Carroll, P., and Samson, L. 1988. A second DNA methyltransferase repair enzyme in Escherichia coli. Proc. Natl. Acad. Sci 85: 3039–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakashita, H., Sakuma, T., Ohkubo, T., Kainosho, M., Sakumi, K., Sekiguchi, M., and Morikawa, K. 1993. Folding topology and DNA binding of the N-terminal fragment of Ada protein. FEBS Lett. 323: 252–256. [DOI] [PubMed] [Google Scholar]
- Sakashita, H., Sakuma, T., Akitomo, Y., Ohkubo, T., Kainosho, M., Sekiguchi, M., and Morikawa, K. 1995. Sequence-specific DNA recognition of the Escherichia coli Ada protein associated with the methylation-dependent functional switch for transcriptional regulation. J. Biochem. (Tokyo) 118: 1184–1191. [DOI] [PubMed] [Google Scholar]
- Sakumi, K. and Sekiguchi, M. 1989. Regulation of expression of the ada gene controlling the adaptive response. Interactions with the ada promoter of the Ada protein and RNA polymerase. J. Mol. Biol. 205: 373–385. [DOI] [PubMed] [Google Scholar]
- Sedgwick, B., Robins, P., Totty, N., and Lindahl, T. 1988. Functional domains and methyl acceptor sites of the Escherichia coli ada protein. J. Biol. Chem. 263: 4430–4433. [PubMed] [Google Scholar]
- Smith, J.N., Hoffman, J.T., Shirin, Z., and Carrano, C.J. 2005. H-bonding interactions and control of thiolate nucleophilicity and specificity in model complexes of zinc metalloproteins. Inorg. Chem. 44: 2012–2017. [DOI] [PubMed] [Google Scholar]
- Sun, L.J., Yim, C.K., and Verdine, G.L. 2001. Chemical communication across the zinc tetrathiolate cluster in Escherichia coli Ada, a metalloactivated DNA repair protein. Biochemistry 40: 11596–11603. [DOI] [PubMed] [Google Scholar]
- Takinowaki, H., Matsuda, Y., Yoshida, T., Kobayashi, Y., and Ohkubo, T. 2004. 1H, 13C and 15N resonance assignments of the N-terminal 16 kDa domain of Escherichia coli Ada protein. J. Biomol. NMR 29: 447–448. [DOI] [PubMed] [Google Scholar]
- Teo, I., Sedgwick, B., Demple, B., Li, B., and Lindahl, T. 1984. Induction of resistance to alkylating agents in E. coli: The ada+ gene product serves both as a regulatory protein and as an enzyme for repair of mutagenic damage. EMBO J. 3: 2151–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo, I., Sedgwick, B., Kilpatrick, M.W., McCarthy, T.V., and Lindahl, T. 1986. The intracellular signal for induction of resistance to alkylating agents in E. coli. Cell 45: 315–324. [DOI] [PubMed] [Google Scholar]
- Wishart, D.S., Bigam, C.G., Yao, J., Abildgaard, F., Dyson, H.J., Oldfield, E., Markley, J.L., and Sykes, B.D. 1995. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR 6: 135–140. [DOI] [PubMed] [Google Scholar]
- Wood, R.D., Mitchell, M., Sgouros, J., and Lindahl, T. 2001. Human DNA repair genes. Science 291: 1284–1289. [DOI] [PubMed] [Google Scholar]
- Wüthrich, K.1986. NMR of proteins and nucleic acids. Wiley, New York.
- Wuttke, D.S., Foster, M.P., Case, D.A., Gottesfeld, J.M., and Wright, P.E. 1997. Solution structure of the first three zinc fingers of TFIIIA bound to the cognate DNA sequence: Determinants of affinity and sequence specificity. J. Mol. Biol. 273: 183–206. [DOI] [PubMed] [Google Scholar]