

Abstract
The major heat shock protein (Hsp) chaperones Hsp70 and Hsp90 both bind the co-chaperone Hop (Hsp70/Hsp90 organizing protein), which coordinates Hsp actions in folding protein substrates. Hop contains three tetratricopeptide repeat (TPR) domains that have binding sites for the conserved EEVD C termini of Hsp70 and Hsp90. Crystallographic studies have shown that EEVD interacts with positively charged amino acids in Hop TPR-binding pockets (called carboxylate clamps), and point mutations of these carboxylate clamp positions can disrupt Hsp binding. In this report, we use circular dichroism to assess the effects of point mutations and Hsp70/Hsp90 peptide binding on Hop conformation. Our results show that Hop global conformation is destabilized by single point mutations in carboxylate clamp positions at pH 5, while the structure of individual TPR domains is unaffected. Binding of peptides corresponding to the C termini of Hsp70 and Hsp90 alters the global conformation of wild-type Hop, whereas peptide binding does not alter conformation of individual TPR domains. These results provide biophysical evidence that Hop-binding pockets are directly involved with domain:domain interactions, both influencing Hop global conformation and Hsp binding, and contributing to proper coordination of Hsp70 and Hsp90 interactions with protein substrates.
Keywords: protein structure/folding, chaperonins, circular dichroism, tetratricopeptide repeat
The dynamic assembly and functional maturation of steroid receptor complexes involves the major molecular chaperones and heat shock proteins Hsp70 and Hsp90 as well as several Hsp-binding co-chaperones. Free receptor undergoes sequential association with Hsp40, Hsp70, the dual co-chaperone Hop, and ultimately Hsp90; only when Hsp90 directly binds receptor is high-affinity hormone binding established (Scherrer et al. 1990; Smith 1993). The progression of chaperone interactions observed with steroid receptors is representative of Hsp70 and Hsp90 interactions with a variety of Hsp90 client proteins, so there likely are common mechanisms underlying the manner in which Hsp90/client protein interactions are established (Wegele et al. 2004). Hop provides an important link between Hsp70 and Hsp90 since it can simultaneously bind both chaperones and effectively targets Hsp90 to pre-existing Hsp70–client complexes (Chen and Smith 1998). That Hop preferentially associates with the ADP-bound form of both chaperones likely serves to coordinate Hsp actions at appropriate stages of each chaperone’s ATPase cycle (Wegele et al. 2004). Additional studies suggest that Hop plays an active role in the functional maturation of Hsp/client complexes apart from assembly and release of Hsp from the complex (Odunuga et al. 2004; Carrigan et al. 2005). Much remains to be learned about how Hop binds to either Hsp, how binding of one Hsp could influence Hop binding or release from the second Hsp, and how Hop ultimately influences Hsp interactions with client proteins.
As with several other Hsp70- or Hsp90-binding co-chaperones, tetratricopeptide repeat (TPR) domains of Hop mediate Hsp binding. Co-chaperone TPR domains typically are composed of three tandem repeats of a loosely conserved 34–amino acid sequence motif (Smith 2004). Each motif favors formation of two anti-parallel α-helices, and the core TPR domain consists of six total α-helices that form a saddle-like structure. The concave surface of the domain provides an interaction site that can accommodate specific peptide binding (Scheufler et al. 2000).
Hop is composed of three distinct TPR domains (TPR1, TPR2a, TPR2b) and two small domains containing a characteristic aspartic acid–proline (DP) repeat motif arranged as TPR1-DP1-TPR2a-TPR2b-DP2 (Prapapanich et al. 1998; Nelson et al. 2003). TPR2a is necessary and sufficient for Hsp90 binding (Chen et al. 1996; Lassle et al. 1997) and specifically binds the peptide corresponding to the C terminus of Hsp90 (MEEVD) (Chen et al. 1998; Scheufler et al. 2000; Odunuga et al. 2003). An X-ray crystallographic structure was solved for a co-crystal of TPR2a plus the MEEVD pentapeptide (Scheufler et al. 2000); this structure revealed how basic side chains of TPR2a that project into the binding pocket form a so-called carboxylate clamp that establishes salt bridges with acidic side chains on the peptide ligand. Point mutation of carboxylate clamp positions in TPR2a disrupts Hsp90 binding (Carrigan et al. 2004); conversely, point mutation of Hsp90 MEEVD readily disrupts binding to Hop (Chen et al. 1998). The carboxylate clamp basic amino acid positions are conserved and functionally important in the TPR domains of other Hsp90-binding co-chaperones (Russell et al. 1999; Ward et al. 2002; Cheung-Flynn et al. 2003), and there are corresponding basic amino acids in the TPR1 and TPR2b domains of Hop that, as discussed below, are also functionally important.
Interactions between Hsp70 and Hop are seemingly more complex than those between Hsp90 and Hop. Similar to TPR2a, a co-crystal structure was obtained for TPR1 bound to the heptapeptide PTIEEVD (Scheufler et al. 2000), which corresponds to the C terminus of Hsp70. Co-crystal structures as well as mutagenic and peptide combinatorial approaches (Brinker et al. 2002) have helped us to understand interactions that distinguish TPR1 and TPR2a interactions with EEVD-containing peptides. Blatch and colleagues (Odunuga et al. 2003) addressed the specificity of TPR–Hsp interactions by successfully engineering a TPR1 mutant that switches from Hsp70 to Hsp90 binding; conversely, they were unable to engineer a TPR2a mutant that gained binding to Hsp70, lending support to the notion that Hsp70 binding to Hop is more complex than Hsp90 binding. Consistent with a role for the Hsp70 EEVD motif in Hop binding, mapping studies have localized Hop binding ability to the C-terminal half of Hsp70 (Gebauer et al. 1997; Demand et al. 1998). On the other hand, C-terminal truncation of the EEVD motif or up to 40 total amino acids failed to disrupt binding to Hop (Carrigan et al. 2004), underscoring the conclusion of Hartl and colleagues (Brinker et al. 2002) based on peptide-binding studies that additional Hsp70 sites must participate in Hop binding. As of yet, these sites in Hsp70 have not been identified. Nonetheless, results showing that TPR1 truncation (Chen et al. 1996; Lassle et al. 1997; Chen and Smith 1998) and TPR1 point mutation (Van Der Spuy et al. 2000; Odunuga et al. 2003; Flom et al. 2005; Song and Masison 2005) block Hsp70 binding affirm the critical importance of Hop TPR1 for binding Hsp70.
Other Hop domains also influence Hsp70 binding. For example, carboxylate clamp point mutation in either TPR2a or TPR2b diminishes Hsp70 binding (Carrigan et al. 2004), which was interpreted as evidence for domain:domain interactions within Hop that influence Hsp70 binding. Point mutation of the C-terminal DP2 minidomain efficiently blocks Hsp70 binding (Chen and Smith 1998; Flom et al. 2005; Song and Masison 2005) and induces alterations in the proteolytic digestion pattern of full-length Hop (Nelson et al. 2003), implying a global conformational change that likely relates to domain:domain interactions. Additionally, a TPR1 double point mutation that disrupts Hsp70 binding induces a conformational change in the full-length protein without altering TPR1 conformation (Odunuga et al. 2003). Finally, genetic evidence in yeast favors a functional interaction between TPR1 and TPR2b (Flom et al. 2005) and between TPR2a and TPR2b (Song and Masison 2005).
These observations underscore the need to better understand structure/function relationships that influence Hsp70 and Hsp90 binding to Hop. In this report, we use circular dichroism spectroscopy (CD) measurements to assess changes in the conformational state of purified Hop, both full-length protein and individual TPR domains, related to carboxylate clamp point mutation or binding to peptides corresponding to the C termini of Hsp70 and Hsp90.
Results
Co-crystal structures and mutagenic analyses indicate that carboxylate clamp positions in Hop TPR domains are important for interactions with peptides corresponding to the C terminus of Hsp70 or Hsp90. Based on crystallographic structures of isolated TPR domains that lack bound peptide, the carboxylate clamp side chains projected into solvent space appear unlikely to significantly influence Hop TPR domain conformation. However, published experimental results suggest that Hop TPR domains and carboxylate clamp residues might be involved in domain:domain interactions that can influence the global conformation of full-length Hop (Odunuga et al. 2003; Carrigan et al. 2004). To directly address the influence of carboxylate clamp residues on Hop conformational states, point mutations were introduced into each of the TPR domains, and the mutant proteins were structurally compared with wild-type Hop (wtHop). We used site-directed mutagenesis to alter a carboxylate clamp position in each TPR domain. In TPR1, lysine 73, which interacts with Hsp70 EEVD side chains and is necessary for Hsp70 binding, was substituted either with glutamic acid (K73E) or alanine (K73A). In TPR2a, arginine 305, which interacts with Hsp90 EEVD side chains and is necessary for Hsp90 binding, was similarly substituted with glutamic acid (R305E) or alanine (R305A). Finally, based on homology with TPR1 and TPR2a, since lysine 429 in TPR2b is predicted to contribute to a putative carboxylate clamp in this Hop TPR domain, thus lysine 429 was similarly substituted with glutamic acid (K429E) or alanine (K429A). Recombinant proteins were generated in bacteria and purified by multistep column chromatography. Each protein was analyzed by Far UV-CD under different pH and temperature conditions as a measure of protein secondary structure and stability (Fig. 1). Except where indicated in later figures, alanine substitutions gave results similar to glutamic acid mutants, thus only data for glutamic acid mutants are shown.
Figure 1.
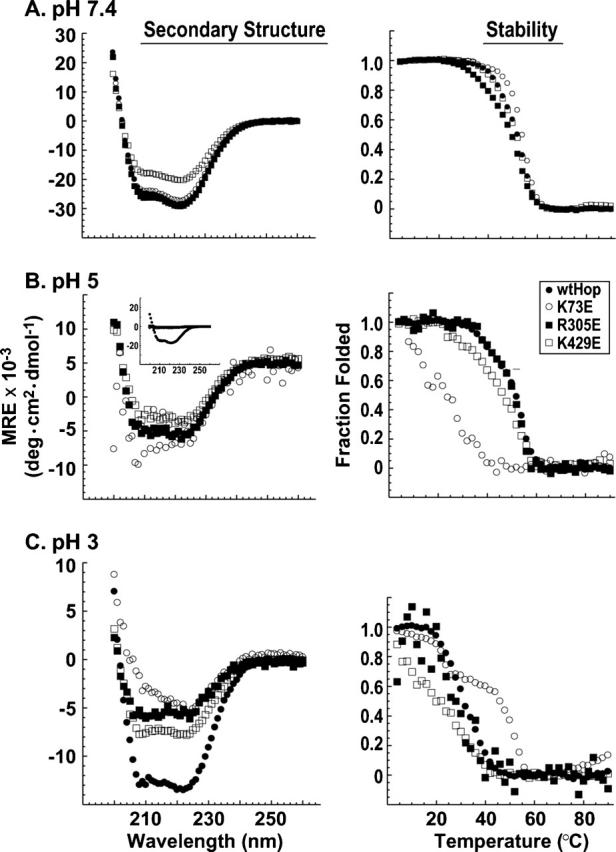
Secondary structure and stability of wtHop and TPR point mutants. Far UV-CD spectra (left-hand panels) and thermal denaturation curves (right-hand panels) were generated at pH 7.4 (A), pH 5 (B), and pH 3 (C) using purified recombinant human Hop forms: wtHop (solid circles), the TPR1 point mutant K73E (open circles), the TPR2a point mutant R305E (solid squares), or the TPR2b point mutant K429E (open squares). Representative data from one of three replicate data sets are shown. For secondary structure measurements, mean residue ellipticity (MRE) was calculated and plotted vs. wavelength. At pH 5 (B), unlike at higher or lower pH, the α-helical content of all mutants was much reduced compared to wtHop (inset panel). In order to discern differences between point mutants, the MRE scale was expanded to enhance the mutant spectra. For stability measurements, the fraction folded was calculated from ellipticity at 222 nm and plotted vs. temperature. At least 60% renaturation was observed upon cooling of protein samples, and aggregation was not observed.
At pH 7.4 and 4°C, CD spectra for wtHop and all mutant forms displayed the strong minima at 211 and 222 nm characteristic of proteins with large α-helical content (Fig. 1A, left). K429 mutants showed reduced helical structure compared to other Hop mutants, but the thermal denaturation profile for all proteins was similar at pH 7.4 (Fig. 1A, right). At pH 5, which would increase the amount of positive charged carboxylate clamp side chains and therefore neutralize glutamic acid mutants, each of the mutants had much reduced helical content compared to wtHop (Fig. 1B, inset on left), but if mutant spectra are expanded (large view), it is clear that each still retains α-helical content. In comparing temperature-dependent stability at pH 5 or below, one must bear in mind that the mutant Hop forms have reduced α-helical content, thus thermal transitions will initiate from a different point in the unfolding process than wtHop. As expected, the different starting points are evident in the Mean Residue Ellipticity (MRE) data for thermal denaturation (data not shown). Alternatively, it is clear from representing these data as fraction folded (Fig. 1B, right) that continued denaturation occurs at lower temperatures for K73E and, to a lesser extent, for K429E than for either wtHop or R305E. These findings suggest either that TPR1 and TPR2b structures are destabilized by carboxylate clamp point mutation or that global Hop stability is reduced by these point mutations. At pH 3, below the theoretical pKa value (4.4) for aspartic and glutamic acid amino acids, all Hop forms display much-reduced helical structure and stability, although wtHop retains more α-helical content than the mutants. For the most part, carboxylate clamp substitution mutants containing glutamic acid (shown) or alanine (not shown) yielded essentially identical CD spectra. An exception is K73E versus K73A. If ellipticity at pH 3 taken from the 211 nm minimum is plotted against temperature (Fig. 2), it is apparent that reversing the carboxylate clamp charge in TPR1 (K73E) has a much greater effect on stability than neutralizing the charge (K73A). Also from Figure 2, it appears that each of the Hop forms, whether wild-type or mutant, retains some structure at high temperature. K73A might have some effect on this residual structure, since both wtHop and K73A have common ellipticities at low temperatures but differ at high temperature. K73E has less initial ellipticity and less final ellipticity but still displays a minor degree of residual structure.
Figure 2.

Structural stability differs with TPR1 point mutations. Far UV-CD measurements were made for equivalent concentrations of wtHop (solid circles), K73E (open circles), and K73A (solid square) at pH 3 over a range of temperatures. The values plotted are the average (n = 3) measured ellipticity (in millidegrees) at the 222 nm peak vs. temperature for each of the Hop forms. Similar patterns were obtained if ellipticity was taken from the 210 nm peak.
The results with full-length proteins demonstrate that all carboxylate clamp mutations affect Hop conformational state, with a more significant effect at pH ≤ 5.0. To distinguish whether mutations are influencing the conformation of independent TPR domains or affecting Hop global conformation, TPR domains were generated in bacteria and purified by multistep column chromatography. Boundaries for the truncated proteins were based on the crystallographic structures for TPR1 and TPR2a (Scheufler et al. 2000) and, in the case of TPR2b, on sequence homologies with the other TPR domains. Far UV-CD spectra were obtained for each of the individual TPR domains (Fig. 3). At pH 7.4, minor differences are observed between wild-type and mutant TPR1 (Fig. 3A, left) and between the TPR2a pair (Fig. 3B, left), but no differences are observed in TPR2b forms (Fig. 3C, left) or between any of the wild-type/mutant pairs at pH 5 (Fig. 3, right panels). The net result from these analyses is that domains lacking or containing a mutant carboxylate clamp position have similar conformations and cannot account for the structural differences observed when comparing full-length wtHop and mutant Hop forms, particularly at pH 5.0 (Fig. 1). We conclude that point mutations are affecting global Hop conformation apart from changes in domain conformations. A reasonable explanation would be that TPR ligand-binding pockets are also involved in domain:domain interactions that influence global Hop conformation.
Figure 3.
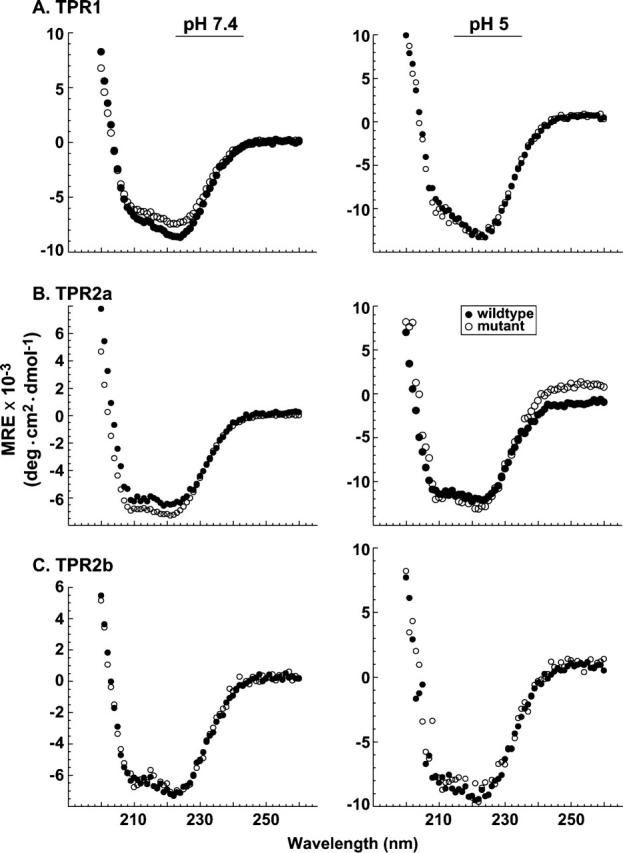
The structure of isolated TPR domains is unaffected by point mutation. Recombinant domain fragments were individually generated and purified for wtHop, the TPR1 point mutant K73E (A), the TPR2a mutant R305E (B), and the TPR2b mutant K429E (C). In each set of plots, Far UV-CD spectra were compared for the wild-type (solid circle) or the mutant domain (open circle). Measurements were made at pH 7.4 (left-hand panels) and at pH 5 (right-hand panels). Protein concentrations ranged from 0.3 μM to 0.8 μM for measurements at pH 7.4 and from 1.5 μM to 4 μM for measurements conducted at pH 5.
If carboxylate clamp residues within the ligand-binding pockets participate in intramolecular interactions, then one would predict that competitive binding of peptides corresponding to the C-termini of Hsp70 and Hsp90 would disrupt these intramolecular interactions. To test this prediction, wtHop was mixed with the following heptapeptides: SRMAAVD as a negative control peptide, SRMEEVD that corresponds to the Hsp90 C terminus and has been co-crystallized with TPR2a (Scheufler et al. 2000), and PTIEEVD that corresponds to the Hsp70 C terminus and has been co-crystallized with TPR1 (Scheufler et al. 2000). Far UV-CD spectra were generated for individual components and for the wtHop + peptide mixtures (Fig. 4); also, the individual wtHop and peptide spectra were added to generate a theoretical additive spectrum for a mixture in which wtHop and peptide do not interact. Separate spectral sets were generated at pH 7.4 and at pH 5.0 to increase the neutral acidic residues in the peptide without affecting protein structure to a large extent. At either pH, the measured spectrum for wtHop plus control peptide overlapped with the theoretical additive spectrum (Fig. 4A); thus, the control peptide has no effect on wtHop conformation. On the other hand, the pH 7.4 spectrum for either wtHop plus Hsp90 peptide (Fig. 4B, left) or wtHop plus Hsp70 peptide (Fig. 4C, left) differed from the corresponding theoretical spectra and showed an enhancement in helical structure for the spectra corresponding to wtHop plus peptide. Thus, peptide binding induces a conformational change in wtHop. In contrast to the altered wtHop conformation observed at pH 7.4, no apparent change in wtHop conformation is observed at pH 5 (right), as would be expected if protonated peptide fails to bind wtHop. These Far UV-CD results complement crystallographic and calorimetric findings that have characterized the binding of Hop to EEVD peptides (Scheufler et al. 2000; Brinker et al. 2002).
Figure 4.
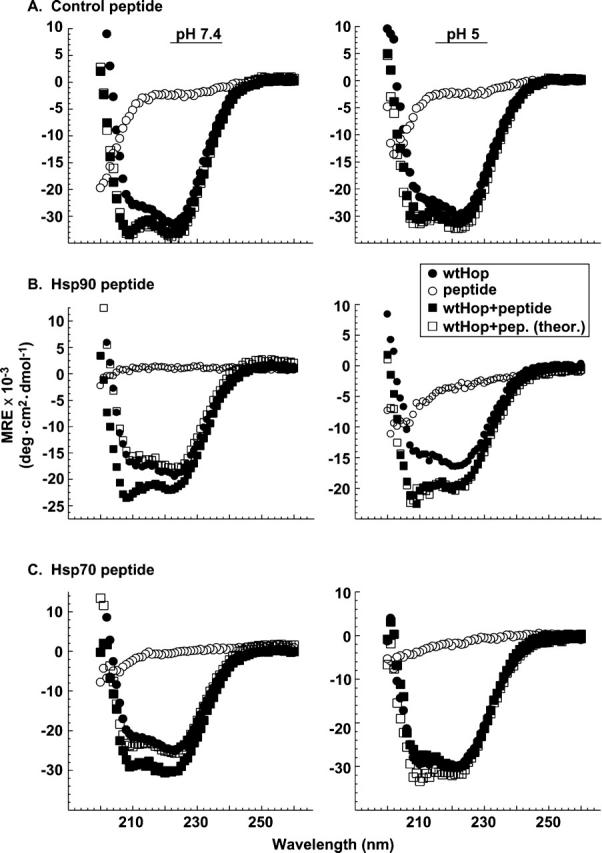
Effects of peptide ligands on wtHop secondary structure. Far UV-CD measurements were obtained for purified recombinant wtHop (1 μM) in the presence or the absence of synthetic peptide (100 μM). The peptides used were a control SRMAAVD (A), the Hsp90-related peptide SRMEEVD (B), or the Hsp70-related peptide PTIEEVD (C). Spectra were obtained for wtHop alone (solid circles), peptide alone (open circles), and the wtHop+ peptide mixture (solid squares). Also, theoretical spectra were calculated by adding the spectra for wtHop alone and peptide alone (open squares). Measurements were made in triplicate at either pH 7.4 (left-hand panels) or pH 5 (right-hand panels); representative data from one of three replicate experiments are shown.
Since peptide binding alters wtHop conformation, we addressed whether this conformational change could be attributed to a corresponding conformational change in the isolated peptide-binding domain. Each of the three TPR domains was generated and purified as described in Materials and Methods. The domains were combined or not with PTIEEVD or SRMEEVD and CD spectra were obtained, as shown in Figure 5. In contrast to the changes observed in the CD spectra for full-length protein (Fig. 4), no changes are observed with isolated domains. The global conformational change induced by peptide binding to full-length wtHop could reflect a change in domain:-domain interactions involving the peptide-binding sites.
Figure 5.
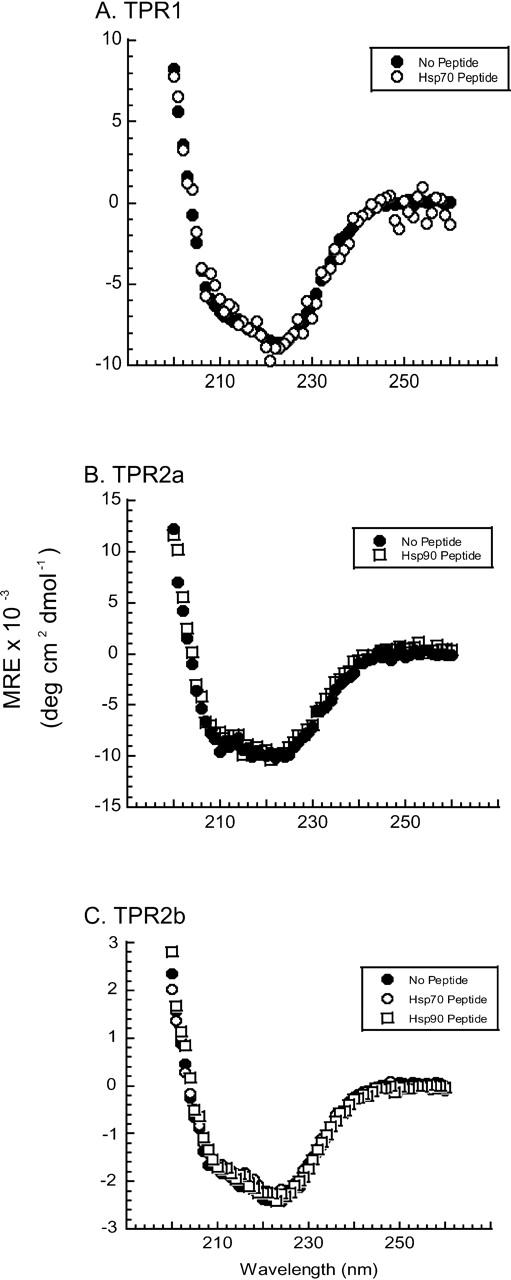
Peptide ligands do not affect individual Hop TPR domain structures. Individual TPR domains were incubated overnight with or without a 100-fold molar excess of peptide corresponding to Hsp70 or Hsp90, and CD spectra were obtained. (A) TPR1 was incubated in the absence or the presence of the Hsp70-related peptide PTIEEVD, which is known to bind specifically to TPR1. The MRE values for TPR1 with PTIEEVD were divided by 2 to correct for instrument differences. (B) TPR2a was incubated in the absence or the presence of the Hsp90-related peptide SRMEEVD, which is known to bind specifically to TPR2a. (C) Since a ligand for TPR2b is unknown, this domain was incubated with either Hsp70 or Hsp90 peptide. In no instance did peptide alter the secondary structure of TPR domains.
We next examined whether peptide binding alters conformation of full-length Hop point mutants K73E, R305E, or K429E (Fig. 6). As observed with wtHop, control peptide did not alter the conformation of any of the mutants (Fig. 6, left panels). In contrast to wtHop, the Hsp90 peptide fails to alter conformation of K73E or R305E (Fig. 6A,B, middle panels), but peptide does alter conformation of K429E (Fig. 6C, middle panel). Since R305 in TPR2a is directly involved in Hsp90 binding and EEVD interaction, it is likely that SRMEEVD fails to bind this mutant and thus fails to alter conformation, as expected. TPR1 is not necessary for Hsp90 binding and K73E retains binding to full-length Hsp90 (Carrigan et al. 2004), thus one might have expected K73E conformational change in the presence of SRMEEVD. Several reasons could explain the absence of K73E conformational change in the presence of Hsp90 SRMEEVD peptide. One possible reason could be that the peptide negative charges create a local microenvironment of lower pH that could affect K73E structure in a way that the binding pocket is no longer present. Figure 1, B and C, shows that low pH (pH 5 and pH 3) affects K73E structure and thermal stability much more than the other mutants. Another reason could be that K73E has affected the global architecture of Hop, either in the presence or the absence of bound SRMEEVD, and it appears to be a conformation similar to wtHop bound to Hsp90 peptide. In other words, peptide binding to TPR2a might release a domain:domain interaction involving the ligand-binding pocket of TPR1, an interaction that has been already disrupted with the mutation K73E.
Figure 6.

Peptide ligands differentially affect structures of Hop TPR mutants. Far UV-CD spectra were obtained for the TPR1 point mutant K73E(A), the TPR2 a point mutant R305E(B), and the TPR2b point mutant K429E(C) (in each case, 1μM purified, full-length recombinant protein) in the presence or the absence of synthetic peptide ligand (100 μM). The peptides used were control SRMAAVD (left-hand panels), Hsp90-related peptide SRMEEVD (middle panels), or Hsp70-related peptide PTIEEVD (right-hand panels). Spectra were obtained for mutant Hop (mutHop) forms alone (solid circles), peptide alone (open circles), and the mutHop +peptide mixtures (solid squares). Also, theoretical spectra were calculated by adding the spectra for mutHop alone and peptide alone (open squares). All measurements were made at pH 7.4, and the data shown are representative of three replicate experiments.
Hsp70 peptide stimulates a similar conformational change in K73E and R305E, while it has some modest conformation enhancement with K429E (Fig. 6, right panels). This contrasts with our earlier finding that binding to full-length Hsp70 is disrupted with all of these mutants (Carrigan et al. 2004). One could argue that TPR2a and TPR2b mutations do not directly alter PTIEEVD binding to TPR1, and thus R305E and K429E mutants should retain the conformational change induced by PTIEEVD binding to TPR1 even if full-length Hsp70 is hindered from binding these mutants. Nonetheless, co-crystallographic studies argue strongly that K73E should greatly disfavor EEVD binding (Scheufler et al. 2000); therefore, one must still account for how PTIEEVD induces a conformational change in K73E. One possibility is that PTIEEVD can bind a unique site in Hop that does not involve carboxylate clamp interactions in TPR1 or other TPR domains. If such a novel site exists, then this could explain why each of the point mutants and wtHop are all affected similarly by PTIEEVD interaction. Another possibility is that, as we mentioned before, K73E could affect the overall architecture of Hop and may help the protein adopt a conformation it adopts when Hsp90 is present. This conformational change may help K73E to bind to PTIEEVD.
Discussion
Hop plays a key role in coordinating the actions of Hsp70 and Hsp90, and Hop TPR domains are clearly important for Hsp binding. Binding of Hsp90 to the Hop TPR2a domain appears straightforward and requires the MEEVD terminus of Hsp90, as supported by mutagenic and co-crystallization studies. Binding of Hsp70 to Hop is a more complex matter. Previous studies have shown that multiple TPR domains and DP2 all affect Hsp70 binding and that the PTIEEVD terminus of Hsp70 is not required for binding, despite suggestive evidence from co-crystallization results. We and others have suggested (Nelson et al. 2003; Odunuga et al. 2003, 2004; Carrigan et al. 2004) that Hop domain:domain interactions play a role in Hsp70 binding, and it was our goal here to provide additional structural evidence for such interactions.
TPR ligand binding pockets influence Hop global conformation
We have established by CD spectroscopy that carboxylate clamp residues in the ligand-binding pocket of each of three TPR domains influence the global conformation and stability of Hop. Under mild conditions at pH 7.4 and 4°C, Hop point mutants K73E (TPR1) and R305E (TPR2a) have α-helical content similar to wtHop (Fig. 1A), but K429E (TPR2b) has an apparent reduction in α-helical content. At pH 5, all of the mutants have dramatically reduced α-helical content compared to wtHop (Fig. 1B). We determined that the apparent reduction in secondary structure is not due to local destabilization of TPR domains since the isolated mutant and wild-type domains have similar CD spectra at both pH 7.4 and pH 5 (Fig. 3). Therefore, TPR ligand-binding pockets appear to influence Hop global conformation in the absence of bound Hsp70 or Hsp90. These findings extend to all TPR domains an earlier observation (Odunuga et al. 2003) that point mutations in TPR1 can alter Hop conformation beyond any change in the isolated domain conformation.
Further support for the involvement of TPR ligand-binding sites in domain:domain interactions is provided by our observation that the SRMEEVD or PTIEEVD peptides, but not a control peptide, can alter the wtHop CD spectrum at pH 7.4 (Fig. 4). When CD measurements are made at pH 5, which should promote more protonation of the peptide carboxylates, no conformational difference is noted with addition of SRMEEVD (Fig. 4B). On the other hand, PTIEEVD induced a conformational change at pH 5, although different from the change noted at pH 7.4 (Fig. 4C). Importantly, peptides did not induce a corresponding conformational change in the individual domains (Fig. 5). These observations implicate TPR ligand-binding pockets in domain:domain interactions and raise the possibility that Hsp binding at one TPR site could influence Hop’s global conformation and perhaps impact Hsp binding at an alternative TPR site.
SRMEEVD interactions with carboxylate clamp mutants
When the effect of peptide binding was examined for carboxylate clamp point mutants, neither the control peptide nor Hsp90 peptide (SRMEEVD) had an effect on K73E conformation. This was unexpected for SRMEEVD since its binding site has been localized to TPR2a (Scheufler et al. 2000) and K73E is known to retain binding to Hsp90 (Carrigan et al. 2004). Our interpretation is that SRMEEVD likely binds K73E but does not induce a conformational change because that change has already resulted from K73 mutation. In other words, we think this result provides evidence that TPR1 interaction with another Hop domain is relieved by SRMEEVD binding to TPR2a.
As expected, SRMEEVD did not induce a conformational change in R305E (Fig. 6B), the TPR2a mutant that lacks Hsp90-binding ability (Carrigan et al. 2004). Furthermore, SRMEEVD induces a conformational change in K429E (Fig. 6C), which is consistent with retention of Hsp90-binding ability by this TPR2b carboxylate clamp mutant.
We have observed that while Hsp90 binds Hop readily and stoichiometrically in a purified system, Hsp70-binding levels are much reduced compared to the level observed in Hop complexes purified from cell extracts (Chen et al. 1996), although Hop–Hsp70 binding may be enhanced by the Hsp70 co-chaperone Hsp40 (Hernandez et al. 2002). Our observation that SRMEEVD induces a conformational change in wtHop but not in K73E suggested the possibility that SRMEEVD binding at TPR2a might release TPR1 from intramolecular interactions and enhance TPR1 interaction with Hsp70. However, when we added SRMEEVD to a mixture of wtHop and Hsp70, we did not observe an increase in Hsp70 binding (results not shown). Apparently, there is some factor or condition in cells and crude cell extracts that further facilitates Hsp70–Hop binding; whether MEEVD binding at TPR2a will assist in this interaction awaits identification of other factors required for Hsp70 binding.
PTIEEVD interactions with carboxylate clamp mutants
The Hsp70 peptide (PTIEEVD) induced conformational changes in all three carboxylate clamp point mutants (Fig. 6) despite the fact that none of these mutants retains Hsp70-binding ability (Carrigan et al. 2004). This is especially surprising for K73E since the TPR1-PTIEEVD co-crystal showed that K73 carboxylate clamp position participates directly in peptide binding, and a switch to glutamic acid at this position should greatly disfavor peptide binding. An intriguing possibility suggested by these data is that PTIEEVD binds a unique site in Hop that is separate from the TPR ligand-binding pockets. Thus, even though PTIEEVD is not required for Hsp70 to bind Hop (Carrigan et al. 2004), an exchange of PTIEEVD interactions between the putative unique site and TPR1, which is suggested by co-crystallographic results, could influence Hsp70 activity in the context of client protein complexes.
Conclusions
This study demonstrates that the TPR ligand-binding sites are involved in domain:domain interactions as well as the previously described roles in Hsp binding. There are independent indications for interaction between TPR1 and TPR2b (Odunuga et al. 2003; Carrigan et al. 2004; Flom et al. 2005), between TPR2a and TPR2b (Chen et al. 1998; Song and Masison 2005), and between TPR1 and DP2 (Nelson et al. 2003; Carrigan et al. 2004; Flom et al. 2005). As of yet, however, there is no definitive biochemical evidence for the exact domain pairings that form intramolecular interactions in Hop. As supported by our findings here, carboxylate clamp residues appear to participate both in intramolecular and Hsp-binding interactions. Since these distinct interactions involving the same TPR domain are likely to be mutually exclusive, there is the intriguing possibility that Hsp binding stimulates structural changes in Hop that could translate into changes in Hop interactions with the alternative Hsp. TPR binding site exchange might be an important aspect of Hop’s ability to coordinate Hsp70 and Hsp90 interactions and to promote progressive assembly and refolding of client protein complexes.
Materials and methods
Generation of Hop recombinant proteins
Bacterial expression plasmids for wild-type and mutant Hop forms were generated as previously described (Carrigan et al. 2004). All forms of Hop were subcloned into pET28a for expression of untagged proteins in bacteria. Bacterial cell lysates were then purified by three-step chromatography (AKTA FPLC, Amersham Biosciences). Briefly, bacterial cell lysates were fractionated by HiPrep 16/10 Heparin-Sepharose chromatography followed by Resource Q-FPLC and 16/60 Superdex 200 FPLC. Final peak fractions for Hop were pooled and concentrated using Amicon Centricon-10 filters (Millipore). The purity of all Hop forms was judged to be >95%. The following full-length mutants were generated: K73E and K73A (TPR1 mutants), R305E and R305A (TPR2a mutants), and K429E and K429A (TPR2b mutants).
Purification of TPR domains
Hop truncation mutants that separately encode each TPR domain were first expressed as C-terminal fusions with GST using a bacterial expression plasmid (pGex-5X). The TPR1 construct encoded amino acids 1–122, the TPR2a construct encoded amino acids 214–362, and the TPR2b construct encoded amino acids 349–481. All cDNA sequences were verified by automated sequencing. Bacterial extracts containing GST-fusion proteins were separately loaded onto a 5-mL glutathione-Sepharose 4B resin column (Amersham Biosciences). Columns were washed with 15 mL of ice-cold phosphate-buffered saline (PBS) to remove unbound proteins, and the GST-TPR fusion was eluted with 10 mM reduced glutathione in 50 mM Tris-HCl. Glutathione was removed from the eluate by dialysis against PBS containing 0.02 mg/mL complete protease inhibitors (Roche Diagnostics). The TPR domain was cleaved from the purified GST-fusion by digestion at room temperature for 16 h with 10 units of Factor Xa protease per milligram of GST-fusion. After protease digestion, the samples were reapplied to an equilibrated 1-mL glutathione-Sepharose column to capture free GST and undigested GST-TPR. Removal of the Factor Xa protease was accomplished by applying the flowthrough of the glutathione-Sepharose column onto a 1-mL HiTrap Benzamidine Sepharose 4 fast flow column (Amersham). Proteins were analyzed on a precast 4%–20% gradient gel (BioRad) and quantitated by measuring protein absorbance at 280 nm.
Peptide synthesis
PTIEEVD and SRMEEVD, corresponding respectively to the Hsp70 or Hsp90 C terminus, and a negative control peptide (SRMAAVD) were synthesized using an automated Advanced ChemTech MPS-396 peptide synthesizer by the Peptide Core Facility at Mayo Clinic Rochester. Peptides were cleaved from a solid support resin, and side-chain protecting groups were removed by acid hydrolysis. Crude peptides were purified by a Beckman Integrated Analytical High Performance Liquid Chromatography. Peptide integrity was assessed by amino acid analysis, mass spectrometry, and sequence analysis.
Secondary structure analysis
Proteins were equilibrated overnight in experimental buffers (10 mM sodium acetate, 10 mM boric acid, 10 mM sodium citrate for pH 3 and pH 5; 10 mM Tris-HCl at pH 7.4). CD spectra were recorded on an AVIV 215 Circular Dichroism spectrometer (AVIV Biomedical Inc.). Protein secondary structure was measured through Far UV-CD spectra (260–200 nm), in the continuous mode, taking measurements every 1 nm with an averaging time of 5 sec at 4°C. Readings were taken with protein solutions (up to 4 μM concentration) in 0.2-cm path-length cells. Far-UV CD spectra were analyzed in triplicate.
Thermal denaturation
The maximum α-helical signal (ellipticity at 222 nm) for each particular protein was chosen and used to analyze protein unfolding. The ellipticity of the maximum α-helix signal (~222 nm) was monitored every 2°C, from 4°C to 90°C, with an equilibration time of 1 min between each temperature point and an averaging time of 60 sec. Refolding curves from 90°C to 4°C were collected immediately after the unfolding curves using the same parameters. All proteins achieved at least 60% refolding, and aggregation was not evident by light scattering measurements.
The thermal denaturation curves were analyzed according to a two-state transition model. Linear extrapolation of the folded and the unfolded baselines based on a minimum of 10 points was performed. The fraction folded (FF) at each temperature was calculated by using the equation FF = (ellipticity observed − ellipticity of the folded state) / (ellipticity of the folded state − ellipticity of the unfolded state).
The ellipticities of the folded and unfolded states were derived from the extrapolated baselines. Thermal denaturation experiments were performed in triplicate. Each data point averages 30–60 scans per temperature. Errors calculated for the spectral data points range from 0.27 to 0.3 (pH 7.4), from 0.35 to 0.5 (pH 5), and from 0.36 to 0.55 (pH 3).
Acknowledgments
We thank members of the Ramirez-Alvarado laboratory for technical assistance. This work was supported by NIH R01-DK44923 (to D.F.S.) and the Mayo Foundation.
Abbreviations
CD, circular dichroism
HOP, Hsp70/Hsp90 organizing protein
HSP, heat shock protein
TPR, tetratricopeptide repeat
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051810106.
References
- Brinker, A., Scheufler, C., Von Der Mulbe, F., Fleckenstein, B., Herrmann, C., Jung, G., Moarefi, I., and Hartl, F.U. 2002. Ligand discrimination by TPR domains. Relevance and selectivity of EEVD-recognition in Hsp70 × Hop × Hsp90 complexes. J. Biol. Chem. 277: 19265–19275. [DOI] [PubMed] [Google Scholar]
- Carrigan, P.E., Nelson, G.M., Roberts, P.J., Stoffer, J., Riggs, D.L., and Smith, D.F. 2004. Multiple domains of the co-chaperone Hop are important for Hsp70 binding. J. Biol. Chem. 279: 16185–16193. [DOI] [PubMed] [Google Scholar]
- Carrigan, P.E., Riggs, D.L., Chinkers, M., and Smith, D.F. 2005. Functional comparison of human and Drosophila Hop reveals novel role in steroid receptor maturation. J. Biol. Chem. 280: 8906–8911. [DOI] [PubMed] [Google Scholar]
- Chen, S. and Smith, D.F. 1998. Hop as an adaptor in the heat shock protein 70 (Hsp70) and hsp90 chaperone machinery. J. Biol. Chem. 273: 35194–35200. [DOI] [PubMed] [Google Scholar]
- Chen, S., Prapapanich, V., Rimerman, R.A., Honore, B., and Smith, D.F. 1996. Interactions of p60, a mediator of progesterone receptor assembly, with heat shock proteins hsp90 and hsp70. Mol. Endocrinol. 10: 682–693. [DOI] [PubMed] [Google Scholar]
- Chen, S., Sullivan,W.P., Toft, D.O., and Smith, D.F. 1998. Differential interactions of p23 and the TPR-containing proteins Hop, Cyp40, FKBP52 and FKBP51 with Hsp90 mutants. Cell Stress Chaperones 3: 118–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung-Flynn, J., Roberts, P.J., Riggs, D.L., and Smith, D.F. 2003. C-terminal sequences outside the tetratricopeptide repeat domain of FKBP51 and FKBP52 cause differential binding to Hsp90. J. Biol. Chem. 278: 17388–17394. [DOI] [PubMed] [Google Scholar]
- Demand, J., Luders, J., and Hohfeld, J. 1998. The carboxy-terminal domain of Hsc70 provides binding sites for a distinct set of chaperone cofactors. Mol. Cell. Biol. 18: 2023–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flom, G., Weekes, J., Williams, J.J., and Johnson, J.L.2005. Effect of mutation of the TPR and DP2 domains of Sti1 on Hsp90 signaling and interaction in Saccharomyces cerevisiae. Genetics http://www.genetics.org. [DOI] [PMC free article] [PubMed]
- Gebauer, M., Zeiner, M., and Gehring, U. 1997. Proteins interacting with the molecular chaperone hsp70/hsc70: Physical associations and effects on refolding activity. FEBS Lett. 417: 109–113. [DOI] [PubMed] [Google Scholar]
- Hernandez, M.P., Sullivan, W.P., and Toft, D.O. 2002. The assembly and intermolecular properties of the hsp70–Hop–hsp90 molecular chaperone complex. J. Biol. Chem. 277: 38294–38304. [DOI] [PubMed] [Google Scholar]
- Lassle, M., Blatch, G.L., Kundra, V., Takatori, T., and Zetter, B.R. 1997. Stress-inducible, murine protein mSTI1. Characterization of binding domains for heat shock proteins and in vitro phosphorylation by different kinases. J. Biol. Chem. 272: 1876–1884. [DOI] [PubMed] [Google Scholar]
- Nelson, G.M., Huffman, H., and Smith, D.F. 2003. Comparison of the carboxy-terminal DP-repeat region in the co-chaperones Hop and Hip. Cell Stress Chaperones 8: 125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odunuga, O.O., Hornby, J.A., Bies, C., Zimmermann, R., Pugh, D.J., and Blatch, G.L. 2003. Tetratricopeptide repeat motif-mediated Hsc70- mSTI1 interaction. Molecular characterization of the critical contacts for successful binding and specificity. J. Biol. Chem. 278: 6896–6904. [DOI] [PubMed] [Google Scholar]
- Odunuga, O.O., Longshaw, V.M., and Blatch, G.L. 2004. Hop: More than an Hsp70/Hsp90 adaptor protein. Bioessays 26: 1058–1068. [DOI] [PubMed] [Google Scholar]
- Prapapanich, V., Chen, S., and Smith, D.F. 1998. Mutation of Hip’s carboxy-terminal region inhibits a transitional stage of progesterone receptor assembly. Mol. Cell. Biol. 18: 944–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell, L.C., Whitt, S.R., Chen, M.S., and Chinkers, M. 1999. Identification of conserved residues required for the binding of a tetratricopeptide repeat domain to heat shock protein 90. J. Biol. Chem. 274: 20060–20063. [DOI] [PubMed] [Google Scholar]
- Scherrer, L.C., Dalman, F.C., Massa, E., Meshinchi, S., and Pratt, W.B. 1990. Structural and functional reconstitution of the glucocorticoid receptor–hsp90 complex. J. Biol. Chem. 265: 21397–21400. [PubMed] [Google Scholar]
- Scheufler, C., Brinker, A., Bourenkov, G., Pegoraro, S., Moroder, L., Bartunik, H., Hartl, F.U., and Moarefi, I. 2000. Structure of TPR domain–peptide complexes: Critical elements in the assembly of the Hsp70–Hsp90 multichaperone machine. Cell 101: 199–210. [DOI] [PubMed] [Google Scholar]
- Smith, D.F. 1993. Dynamics of heat shock protein 90–progesterone receptor binding and the disactivation loop model for steroid receptor complexes. Mol. Endocrinol. 7: 1418–1429. [DOI] [PubMed] [Google Scholar]
- ———. 2004. Tetratricopeptide repeat cochaperones in steroid receptor complexes. Cell Stress Chaperones 9: 109–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, Y. and Masison, D.C. 2005. Independent regulation of Hsp70 and Hsp90 chaperones by Hsp70/Hsp90-organizing protein Sti1 (Hop1). J. Biol. Chem. 280: 34178–34185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Spuy, J., Kana, B.D., Dirr, H.W., and Blatch, G.L. 2000. Heat shock cognate protein 70 chaperone-binding site in the co-chaperone murine stress-inducible protein 1 maps to within three consecutive tetratricopeptide repeat motifs. Biochem. J. 345 (Pt. 3): 645– 651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward, B.K., Allan, R.K., Mok, D., Temple, S.E., Taylor, P., Dornan, J., Mark, P.J., Shaw, D.J., Kumar, P., Walkinshaw, M.D., et al. 2002. A structure-based mutational analysis of cyclophilin 40 identifies key residues in the core tetratricopeptide repeat domain that mediate binding to Hsp90. J. Biol. Chem. 277: 40799–40809. [DOI] [PubMed] [Google Scholar]
- Wegele, H., Muller, L., and Buchner, J. 2004. Hsp70 and Hsp90—A relay team for protein folding. Rev. Physiol. Biochem. Pharmacol. 151: 1–44. [DOI] [PubMed] [Google Scholar]