

Abstract
Psalmopeotoxin I (PcFK1) is a 33-amino-acid residue peptide isolated from the venom of the tarantula Psalmopoeus cambridgei. It has been recently shown to possess strong antiplasmodial activity against the intra-erythrocyte stage of Plasmodium falciparum in vitro. Although the molecular target for PcFK1 is not yet determined, this peptide does not lyse erythrocytes, is not cytotoxic to nucleated mammalian cells, and does not inhibit neuromuscular function. We investigated the structural properties of PcFK1 to help understand the unique mechanism of action of this peptide and to enhance its utility as a lead compound for rational development of new antimalarial drugs. In this paper, we have determined the three-dimensional solution structure by 1H two-dimensional NMR means of recombinant PcFK1, which is shown to belong to the ICK structural superfamily with structural determinants common to several neurotoxins acting as ion channels effectors.
Keywords: Psalmopoeus cambridgei, PcFK1, spider toxin, structure determination, Plasmodium, malaria, NMR
Spider venoms have been shown to contain complex mixtures of a number of different types of molecules, such as neurotoxins (Escoubas et al. 2000) and necrotic and antimicrobial peptides (Corzo and Escoubas 2003). Also, spider toxins have recently emerged as interesting tools for exploring new antiparasitic targets (Silva et al. 2000; Choi et al. 2004).
Psalmopeotoxin I (PcFK1) (Choi et al. 2004), a peptide isolated from the venom of the tarantula Psalmopoeus cambridgei, has been recently shown to possess strong antiplasmodial activity against the intra-erythrocyte stage of Plasmodium falciparum in vitro. Malaria constitutes the most widespread infectious disease, affecting over 300 million people (Snow et al. 2001, 2005). Resistance of Plasmodium species to classical anti-malarial drugs is becoming a critical problem and new drug targets against Plasmodium are urgently needed. Searching for these targets requires a better understanding of the Plasmodium biology and interaction with its host. PcFK1 contains 33 amino acid residues, including six cysteine residues that form three disulfide bonds. Its sequence shows similarity with well-characterized spider toxins belonging to the Inhibitor Cystine Knot (ICK) superfamily (Ferrat and Darbon 2005), including HwTx-I (Zhou et al. 1997), HnTx-I (Li et al. 2003), and HaTx1 (Swartz and MacKinnon 1995) (identity, 27%, 21%, and 21%, respectively).
Although the molecular target for PcFK1 is not yet determined, this peptide does not lyse erythrocytes, is not cytotoxic to nucleated mammalian cells, and does not inhibit neuromuscular function (Choi et al. 2004). Besides ongoing pharmacological studies, investigation of structural properties of PcFK1 could be important to help understand the unique mechanism of action of this peptide and to enhance its utility as a lead compound for rational development of new antimalarial drugs. In this paper, we report the three-dimensional solution structure determined by 1H two-dimensional nuclear magnetic resonance (NMR) spectroscopy of recombinant PcFK1 expressed in Escherichia coli.
Results0
NMR resonance assignment and structure calculation
The sequential assignment procedure allowed us to determine the resonance frequency of almost all protons (data deposited in the Biological Magnetic Resonance Bank [BMRB] under accession no. 6636). Backbone proton frequencies of residues 9 and 30 were determined on a partially deuterium exchanged sample and the correlation between the amide and the α-proton of residue 10 was not assigned because of frequency degeneracy of amide protons of residues 10, 32, and 33.
The structure of recombinant PcFK1 was determined by using 625 NOE (nuclear Overhauser effect)-based distance restraints (including 293 intra-residue restraints, 230 sequential restraints, 30 medium range restraints, and 72 long-range restraints). In addition, 24 hydrogen bond restraints derived from hydrogen exchange data and 25 dihedral angle constraints derived from coupling constants were included, as well as nine distance restraints derived from the three disulfide bridges. Altogether, the final experimental set corresponds to 20.09 constraints per residue on the average. The best-fit superimposition of backbone atoms for 25 models is shown on Figure 1A, and the structural statistics are given in Table 1. Analysis of local root-mean-square deviation (RMSD) values shows that the precision of the calculation is fairly constant all along the sequence with the exception of N- and C-terminal ends. The poor resolution of N and C termini is due to the scarcity of NOE, most probably the result of higher mobility of these residues. All the solutions have good nonbonded contacts and good covalent geometry as evidenced by low values of CNS (Crystallography and NMR System) energy terms. The correlation with the experimental data shows no NOE-derived distance violation greater than 0.2 Å, and the analysis of the Ramachandran plot shows (in PROCHECK software nomenclature) 66.7% of the residues in the most favored regions, 32.9% in the additional regions, 0.4% in the generously allowed regions, and none in the disallowed regions.
Figure 1.
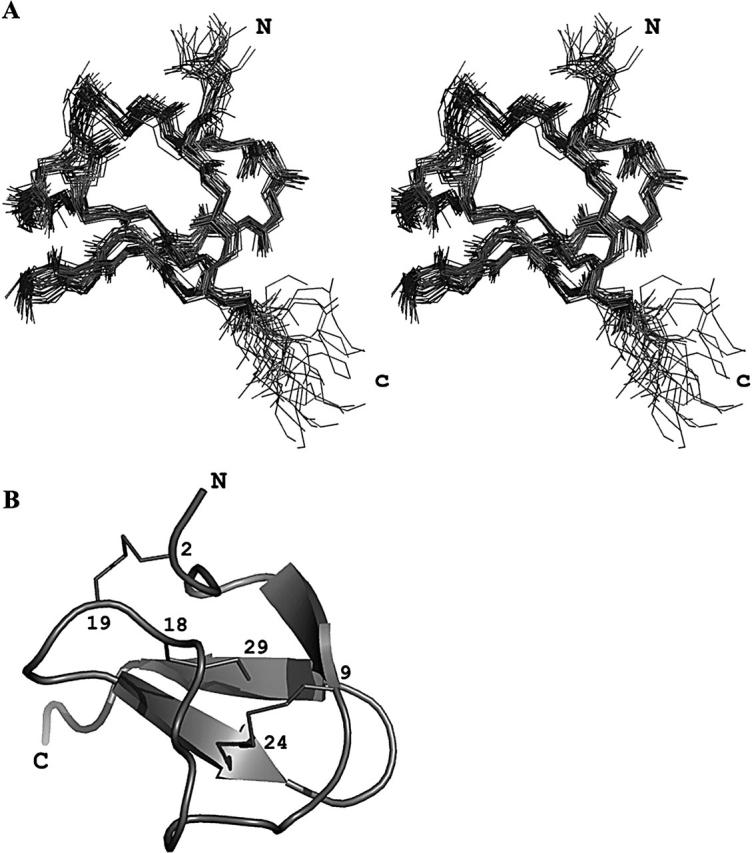
(A) Stereopair view of the best fit of 25 structures of PcFK1. Only the backbone atoms are shown. (B) Ribbon representation of PcFK1. The secondary structures are shown by arrows and the disulfide bridges are in sticks. The figure was done with the PyMOL software.
Table 1.
Structural statistics of the 25 best structures of PcFK1
| Residues | Residues | |
| RMSD (Å) | 1–34 | 2–33 |
| Backbone | 1.14 ± 0.32 | 0.80 ±0.19 |
| All heavy atoms | 2.18 ±0.32 | 1.99 ±0.26 |
| Energies (kcal/mol) | ||
| Total | −884.42 ±21.97 | |
| Bonds | 8.68 ±0.53 | |
| Angles | 139.29 ±3.74 | |
| Impropers | 28.21 ±5.83 | |
| Dihedrals | 159.81 ±3.24 | |
| van der Waals (repel) | −120.25 ±8.39 | |
| Elec | −1104.75 ±19.30 | |
| NOE | 4.36 ±1.24 | |
| Cdih | 0.19 ±0.19 | |
| RMSD | ||
| Bonds (Å) | 0.0041 ±0.0001 | |
| Angles (°) | 0.9910 ±0.0132 | |
| Impropers (°) | 1.726 ±0.200 | |
| Dihedral (°) | 42.35 ±0.58 | |
| NOE (Å) | 0.0132 ±0.0020 | |
| Cdih (°) | 0.3280 ±0.1762 | |
Structure description
The three-dimensional structure (PDB accession code 1X5V) of PcFK1 consists of a compact disulfide-bonded core, from which two loops and the C terminus emerge (Fig. 1B). The only element of regular secondary structure is a triple-stranded antiparallel β-sheet comprising residues 7–9, 22–25, and 28–31.
The first strand is connected with the second one by a large loop made of residues 10 to 21, while the second and third strands are connected by a type II-β turn. The β-sheet is confirmed by the analysis of the sequential connectivities (strong Hα/HN and weak HN/HN) and by NH-CO hydrogen bonds involving amide protons of residues 9, 23, 25, 28, 29, 30, and 32. Amide protons of residues 7 and 29 are highly overlapping in the spectra, preventing us from observing a correlation between these two residues and then to better describe the first strand. Other slowly exchanging amide protons, i.e., those belonging to residues 3, 7, 16, 19, and 22, are involved in hydrogen bonding with carbonyl oxygens of residues 17, 4, 13, 3, and 19, respectively.
The PcFK1 fold (Fig. 1B) can be classified as an ICK fold, already described in numerous toxic and inhibitory peptides (Bernard et al. 2004; Ferrat et al. 2005), as well as various protease inhibitors (Pallaghy et al. 1994). Of the 34 residues of PcFK1, only the side chain of Cys residues are buried. All others are solvent-exposed, and their conformation is constrained by steric interactions. The two N-terminal residues and the two C-terminal residues are poorly constrained. Finally, residue 34 is a homoserine lactone, which consists of a cyclization of the side chain.
Discussion
Most of the peptides classified as ICK folds have similar structural conformations despite their diverse functions. Analysis of the conformation therefore provides little insight into their mode of action; the cystine knot simply provides the structural framework onto which diverse functional motifs can be grafted (Wang et al. 2000; Vitt et al. 2001). The slight differences in the surface shape, which result from the orientation of the side chains of aromatic residues, and/or the distribution of the charged residues and/or the amino acid composition of the respective β-hairpin structure, may explain the difference in the binding affinity and selectivity with their receptors (Wang et al. 2000; Vitt et al. 2001). Deciphering the selectivity determinants of these peptides is a key issue and many studies have been done to get an insight into the structure–activity relationship of ion channel inhibitors with ICK fold and therefore to better understand the mechanism of interaction with their targets.
PcFK1 is classified as a member of the ICK superfamily and presents sequence similarities with two other toxins: HNTX-I (Li et al. 2003) and HWTX-I (Qu et al. 1997), effectors of Na+ and Ca+ channels, respectively. These three toxins present some surface similarities. Their molecular surfaces possess basic residues associated with hydrophobic ones. This cluster is made by the residues K3, K7, R25, K27, K30, F5, Y20, W28, for HNTX-I; K3, R20, K25, K27, K32, F6, H26, W28, W31 for HWTX-I; and R25, K28, Y11, Y26, H6 for PcFK1. The dipole moment, symbolizing the electrostatic anisotropy, emerges through the PcFK1 molecular surface by the R25 residue, close to two hydrophobic residues Y11 and Y26 (Fig. 2A) (DeLano 2002). An interesting feature is that it recalls the role of the basic-hydrophobic dyad demonstrated for scorpion toxins interacting with voltage-dependent Kv1 channels (Dauplais et al. 1997). Our group (Ferrat et al. 2001) and others (Yu et al. 2004; Huang et al. 2005) have previously proposed that the electrostatic anisotropy could be an orientating force within the electrostatic field of the membrane receptor and this led to the hypothesis that the molecular surface through which the dipole emerges could be construed as an indication of the interaction surface between a toxin and its target. Applied to HNTX-I and HWTX-I, the calculation shows that the dipole moments emerge through a basic/aromatic dyad, respectively, R25-W28 and K30-W28. A dyad is also present in κ-conotoxin PVIIA (Savarin et al. 1998) (Fig. 1B), which is active on K+ channel, and in almost all toxins active on ion channels, despite their three-dimensional fold and specificity. Additional basic residues are often associated with this dyad to form a basic/aromatic cluster.
Figure 2.
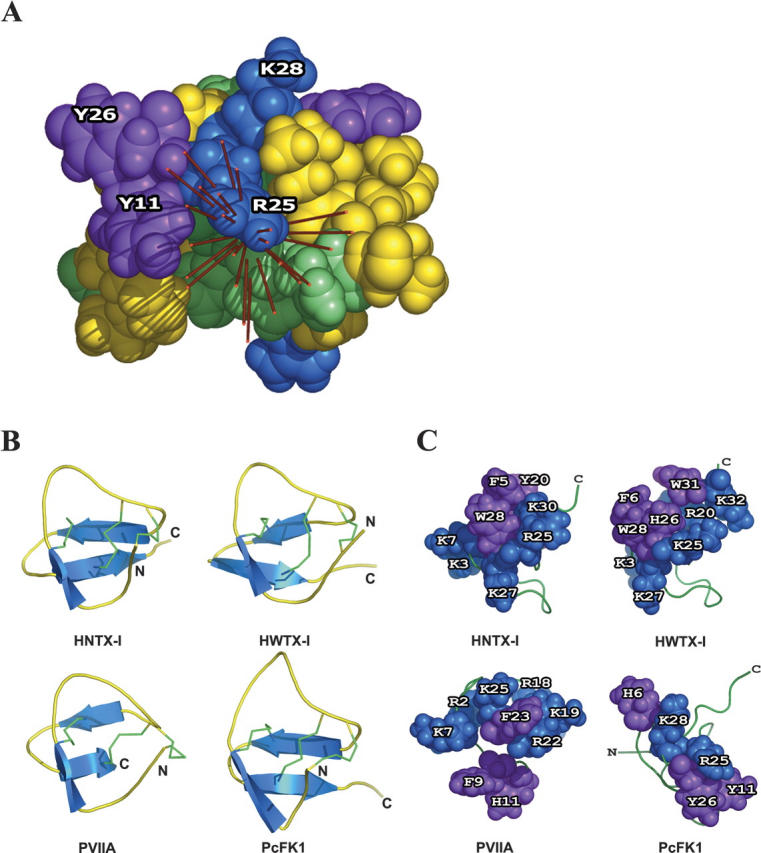
(A) Putative functional surface of PcFK1 and the dipole moment calculated on each individual NMR solution. The residues are colored green for polar uncharged residues, blue for basic residues, red for acidic residues, purple for aromatic residues, and yellow for aliphatic residues. (B) Secondary structure (blue) and disulfide bridges (green) of Hainantoxin-I (HNTX-I), Huwentoxin-I (HWTX-I), κ-conotoxin PVIIA, and PcFK1. (C) Representation of triplet of residues through which the dipole moment emerges. The residues are colored blue for basic residues and purple for aromatic residues. The figure was done with the PyMOL software.
The pharmacological target of PcFK1 remains currently unknown. However, it was demonstrated (Choi et al. 2004) that PcFK1 inhibits specifically the intra-erythrocyte stage of P. falciparum by a mechanism not yet elucidated. PcFK1 is neither hemolytic nor cytotoxic for nucleated eukaryotic cells and does not affect the main voltage-dependant ion channels involved in neuromuscular pre- and post-synaptic signal transduction. Furthermore, PcFK1 has neither antibacterial nor anti-fungal activity, contrary to other antimicrobial peptides (AMPs) with insect AMPs (Boman 2003; Bulet and Stocklin 2005), plant defensins (Thomma et al. 2002; Castro and Fontes 2005), β-defensins (Torres and Kuchel 2004), and cyclotide peptides (Tam et al. 1999), in which positively-charged residues may be important for the interaction with the microbial surface. Interestingly, our previous data have shown that PcFK1 could be adsorbed to red blood cells, probably through hydrophobic or electrostatic interactions. The best-characterized antiplasmodial peptides are dermaseptin S4 derivatives (Krugliak et al. 2000). Native dermaseptin S4, a 28-residue peptide isolated from the skin of a frog (Phyllomedusa genus), displayed high aggregation in solution, preventing the determination of its NMR structure. This led to the proposal that dermaseptin S4’s aggregation state in solution might be an important factor affecting selective cytotoxicity (Ghosh et al. 1997). In contrast, our determination of the three-dimensional solution structure of PcFK1 rules out such mechanism of action. It is, however, not sufficient to predict the target of this peptide in the infected red blood cells, owing to the unusual biology of P. falciparum and lack of completely elucidated metabolism and changes in the infected red blood cells (Bannister and Mitchell 2003). Our results also suggest that PcFK1 could be an ion channel effector, because of structural similarities with other neurotoxins. The basic/hydrophobic patch may be principally responsible for the binding of PcFK1 as do other ion channel effectors, to a still unknown ion channel. Therefore, binding studies must be performed against other ion channels and further studies on the pharmacology of the antiplasmodial activity will now be tested with mutants and/or modified peptides derived from PcFK1.
Materials and methods
Development of a recombinant expression system for PcFK1s
Annealing and ligation of the overlapping oligonucleotides constructing the synthetic gene for PcFK1 leads to a ladder of concatemers consisting mainly of the products of 100, 150, 200, and 300 bp easily separated on agarose gels (data not shown). We purified the 100-bp product for subcloning into the pET-31b(+) vector (Novagen). The KSI-toxin-(His)6Tag fusion protein was produced in E. coli strain BL21(DE3)plysE as inclusion bodies. The optimal temperature for production of the protein was 30°C. The fusion protein represented 10% of total proteins in the cell lysate.
The purified protein fusion was solubilized in 70% formic acid and cleaved by CNBr, which led to recombinant toxin with homoserine lactone (Hsl) at the C terminus. The mixture of cleavage products and (His)6Tag was then purified by RP-HPLC on C18 column with an appropriate elution a linear 0–60% gradient of acetonitrile in 0.1% aqueous trifluoroacetic acid (50 min at 4.0 mL/min) and these fractions were assessed by MALDI-TOF. The measured molecular mass of recombinant PcFK1 (3700.7 Da) showed 6-Da difference mass compared with the calculated molecular mass (3706.7 Da), suggesting spontaneous formation of three disulfide bonds. Mass spectrometry under Post Source Decay conditions (S.-J. Choi, C. Pimentel, M.-L. Celerier, C. Deregnaucourt, H.Darbon, and J.-M. Camadro, pers. comm.) allowed us to verify the spontaneous oxidation of the six cysteines and the formation of native disufide bonds (CI-CIV, CII-CV, and CIII-CVI connections), corresponding to classical ICK disulfide bond connections. The final yield of active recombinant PcFK1 was about 1 mg/L of starting BL21(DE3)plysE cells culture. The mass difference between natural PcFK1 and recombinant PcFK1 was 84 Da and corresponds to the mass of the homoserine lactone. Recombinant PcFK1 inhibited the development of P. falciparum in erythrocytes in vitro with similar IC50 value to that of natural PcFK1 (3–11 μM vs. 1–11 μM).
NMR spectroscopy
Sample preparation
A 1-mM sample of recombinant PcFK1 in 0.5 mL of H2O/D2O (90/10 by vol) at pH 3.0 was used for NMR spectra recordings. Amide proton exchange rate was determined after lyophilization of this sample and dissolution in 100% D2O.
NMR experiments
All 1H spectra were recorded on BRUKER DRX500 spectrometer equipped with an HCN probe and self-shielded triple axis gradients were used.
Two-dimensional NOESY (nuclear Overhauser effect spectroscopy) and TOCSY (total correlated spectroscopy) spectra were acquired at 290 and 300 K to solve assignment ambiguities. The spectra collected at 290 K provided the optimal resolution of overlapping NMR signals of PcFK1; therefore, this temperature was used for further studies of the protein.
Two-dimensional spectra were acquired using states-TPPI (time proportional phase incrementation) method (Marion et al. 1989) to achieve F1 quadrature detection (Marion and Wüthrich 1983). The spectral width in both dimensions was 6000 Hz. NOESY and TOCSY experiments were recorded with 2048 data points for t2 and 512 points for t1 increments, with 64 transients per experiment. DQF-COSY (double quantum filtered - correlation spectroscopy) experiment was recorded with 4096 data points in t2 and 1024 data points in t1. Water suppression was achieved using presaturation during the relaxation delay (1.5 sec), and during the mixing time in the case of NOESY experiments, or using a water gate 3-9-19 pulse train (Piotto et al. 1992) using a gradient at the magic angle obtained by applying simultaneous x-, y-, and z- gradients prior to detection. NOESY spectra were acquired using mixing time of 80 msec. TOCSY was performed with a spin-locking field strength of 8 kHz and spin lock time of 80 msec. The amide proton exchange experiments were recorded immediately after dissolution of the peptides in D2O. A series of NOESY spectra with a mixing time of 80 msec were recorded at 290K, the first one for 1 h, followed by spectra of 12 h each. Furthermore, the NOE build-up curves were obtained by successively recording NOESY spectra with 50-, 150-, 200-, and 300-msec mixing time to check for spin diffusion.
Data processing
Spectra were processed with XWIN-NMR version 2.1. The matrices were transformed to a final size of 2048 points in the acquisition dimension and to 1024 points in the other, except for coupling constant determination for which a 8192 · 1024 matrix was used in the COSY spectrum. The signal was multiplied by a shifted sine bell window in both dimensions prior to a Fourier transform, then a fifth-order polynomial baseline correction was applied.
Spectral analysis
Identification of amino acid spin systems and sequential assignment were done using the standard strategy described by Wüthrich (1986) applied with a graphical software, XEASY (Bartels et al. 1995). The comparative analysis of COSY and TOCSY spectra recorded in water gave the spin system signatures of the protein. The spin systems were then sequentially connected using the NOESY spectra.
Experimental restraints
The assignment and integration of NOE data using manual integration in the XEASY software allowed us to obtain a list of volumes, which were automatically translated into upper-limit distances by the calibration routine of the ARIA software (Linge et al. 2003). The Φ torsion angle constraints resulted from the 3JHN-Hα coupling constant measurements. They were estimated by the INFIT program (Szyperski et al. 1992). For a given residue, separated NOESY cross peaks with the backbone amide proton in the ω2 dimension were used. Several cross-sections through these cross peaks were selected that exhibited a good signal-to-noise ratio. They were added up, and only those data points of the peak region that were above the noise level were retained. The left and the right ends of the peak region were then brought to zero intensity by a linear baseline correction. After extending the baseline-corrected peak region with zeros on both sides, which is equivalent to oversampling in the time domain, an inverse Fourier transformation was performed. The value of the 3JHN-Hα coupling constant was obtained from the first local minimum. The Φ angles were restrained to −120 ± 40° for a 3JHN-Hα ≥ 8 Hz and to −65 ± 25° for a 3JHN-Hα ≤ 6 Hz. No angle constraint was assigned to a 3JHN-Hα = 7 Hz, a value considered as ambiguous.
Structure calculations
The automated assignment program ARIA was used for the assignment of ambiguous NOEs and to calibrate the NOE distance restraints. In later stages of the ARIA calculations, additional NOEs collected on the D2O NOESY spectrum and hydrogen bond restraints were added. Hydrogen bond acceptors were assigned based on the previous structures calculated by ARIA. Inter-strand hydrogen bonds were assigned when an amide proton and carbonyl oxygen were within 2.4 Å, as observed with TURBO software (Roussel and Cambillau 1989). The geometric quality of the obtained structures was assessed by PROCHECK 3.5 and PROCHECK-NMR software (Laskowski et al. 1993).
Electrostatic calculations
The electrostatic calculations together with the dipole moments and analysis were done by using the GRASP software (Nicholls et al. 1991). The potential maps were calculated with a simplified Poisson-Boltzmann solver (Nicholls and Honig 1991), on the basis of an AMBER-derived parameter file.
Acknowledgments
B.C. is the recipient of a doctoral fellowship from the Ministère de l’Education Nationale, de l’Enseignement Supérieur et de la Recherche. S.-J.C. is the recipient of a grant-in-aid from the French Government (BGF-French Embassy in Republic of Korea). We thank Christophe Jolivet and Nicole Buisson for their help in establishing the synthetic gene expression system and Dr. Dietmar Waidelich, from Applied Biosystems (Darmstadt, Germany), for high resolution MS/MS data handling.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051860606.
References
- Bannister, L. and Mitchell, G. 2003. The ins, outs and roundabouts of malaria. Trends Parasitol. 19: 209–213. [DOI] [PubMed] [Google Scholar]
- Bartels, C., Xia, T.-H., Billeter, M., Guntert, P., and Wüthrich, K. 1995. The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. J. Biomol. NMR 5: 1–10. [DOI] [PubMed] [Google Scholar]
- Bernard, C., Corzo, G., Adachi-Akahane, S., Foures, G., Kanemaru, K., Furukawa, Y., Nakajima, T., and Darbon, H. 2004. Solution structure of ADO1, a toxin extracted from the saliva of the assassin bug, Agriosphodrus dohrni. Proteins 54: 195–205. [DOI] [PubMed] [Google Scholar]
- Boman, H.G. 2003. Antibacterial peptides: Basic facts and emerging concepts. J. Intern. Med. 254: 197–215. [DOI] [PubMed] [Google Scholar]
- Bulet, P. and Stocklin, R. 2005. Insect antimicrobial peptides: Structures, properties and gene regulation. Protein Pept. Lett. 1: 3–11. [DOI] [PubMed] [Google Scholar]
- Castro, M.S. and Fontes, W. 2005. Plant defense and antimicrobial peptides. Protein Pept. Lett. 12: 13–18. [PubMed] [Google Scholar]
- Choi, S.J., Parent, R., Guillaume, C., Deregnaucourt, C., Delarbre, C., Ojcius, D.M., Montagne, J.J., Célérier, M.L., Phelipot, A., Amiche, M., et al. 2004. Isolation and characterization of Psalmopeotoxin I and II: Two novel antimalarial peptides from the venom of the tarantula Psalmopoeus cambridgei. FEBS Lett. 572: 109–117. [DOI] [PubMed] [Google Scholar]
- Corzo, G. and Escoubas, P. 2003. Pharmacologically active spider peptide toxins. Cell. Mol. Life Sci. 60: 2409–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauplais, M., Lecoq, A., Song, J., Cotton, J., Jamin, N., Gilquin, B., Roumestand, C., Vita, C., de Medeiros, C.L., Rowan, et al. 1997. On the convergent evolution of animal toxins. Conservation of a diad of functional residues in potassium channel-blocking toxins with unrelated structures. J. Biol. Chem. 272: 4302–4309. [DOI] [PubMed] [Google Scholar]
- DeLano, W.L. 2002. The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA.
- Escoubas, P., Diochot, S., and Corzo, G. 2000. Structure and pharmacology of spider venom neurotoxins. Biochimie 82: 893–907. [DOI] [PubMed] [Google Scholar]
- Ferrat, G. and Darbon, H. 2005. An overview of the three dimensional structures of short spider toxins. Toxin Rev. 24: 361–384. [Google Scholar]
- Ferrat, G., Bernard, C., Fremont, V., Mullmann, T.J., Giangiacomo, K.M., and Darbon, H. 2001. Structural basis for α-K toxin specificity for K+ channels revealed through the solution 1H NMR structures of two noxiustoxin-iberiotoxin chimeras. Biochemistry 40: 10998–11006. [DOI] [PubMed] [Google Scholar]
- Ferrat, G., Bosmans, F., Tytgat, J., Pimentel, C., Chagot, B., Gilles, N., Nakajima, T., Darbon, H., and Corzo, G. 2005. Solution structure of two insect-specific spider toxins and their pharmacological interaction with the insect voltage-gated Na(+) channel. Proteins 59: 368–379. [DOI] [PubMed] [Google Scholar]
- Ghosh,. J.K., Shaoo, D., Guillaud, P., Cicéron, L., Mazier, D., Kustanovich, I., Shai, Y., and Mor, A. 1997. Selective cytotoxicity of dermaseptin S3 toward intraerythrocytic Plasmodium falciparum and the underlying molecular basis. J. Biol. Chem. 272: 31609–31616. [DOI] [PubMed] [Google Scholar]
- Huang, X., Dong, F., and Zhou, H.X. 2005. Electrostatic recognition and induced fit in the j-PVIIA toxin binding to shaker potassium channel. J. Amer. Chem. Soc. 127: 6836–6849. [DOI] [PubMed] [Google Scholar]
- Krugliak, M., Feder, R., Zolotarev, V.Y., Gaidukov, L., Dagan, A., Ginsburg, H., and Mor, A. 2000. Antimalarial activities of dermaseptin S4 derivatives. Antimicrob. Agents Chemother. 44: 2442–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.M., and Thornton, J.M. 1993. A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26: 283–291. [Google Scholar]
- Li, D., Xiao, Y., Hu, W., Xie, J., Bosmans, F., Tytgat, J., and Liang, S. 2003. Function and solution structure of hainantoxin-I, a novel insect sodium channel inhibitor from the Chinese bird spider Selenocosmia hainana. FEBS Lett. 555: 616–622. [DOI] [PubMed] [Google Scholar]
- Linge, J.P., Habeck, M., Rieping, W., and Nilges, M. 2003. ARIA: Automated NOE assignment and NMR structure calculation. Bioinformatics 19: 315–316. [DOI] [PubMed] [Google Scholar]
- Marion, D. and Wüthrich, K. 1983. Application of phase sensitive two-dimensional correlated spectroscopy (COSY) for measurements of 1H-1H spin-spin coupling constants in proteins. Biochem. Biophys. Res. Commun. 113: 967–974. [DOI] [PubMed] [Google Scholar]
- Marion, D., Ikuto, M., Tschudin, R., and Bax, A. 1989. Rapid recording of 2D NMR spectra without phase cycling. Application to the study of hydrogen exchange in proteins. J. Mag. Res. 85: 393–399. [Google Scholar]
- Nicholls, A. and Honig, B. 1991. A rapid finite difference algorithm, utilizing successive over-relaxation to solve the Poisson-Boltzmann equation. J. Comp. Chem. 12: 435–445. [Google Scholar]
- Nicholls, A., Sharp, K.A., and Honig, B. 1991. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11: 281–296. [DOI] [PubMed] [Google Scholar]
- Pallaghy, P.K., Nielsen, K.J., Craik, D.J., and Norton, R.S. 1994. Common structural motif incorporating a cystine knot and a triple-stranded β-sheet in toxic and inhibitory polypeptides. Protein Sci. 3: 1833–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piotto, M., Saudek, V., and Sklenar, V. 1992. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 2: 661–665. [DOI] [PubMed] [Google Scholar]
- Qu, Y., Liang, S., Ding, J., Liu, X., Zhang, R., and Gu, X. 1997. Proton nuclear magnetic resonance studies on Huwentoxin-I from the venom of the spider Selenocosmia huwena:Three-dimensional structure in solution. J. Protein Chem. 16: 565–574. [DOI] [PubMed] [Google Scholar]
- Roussel, A. and Cambillau, C. 1989. TURBO-FRODO. In Silicon Graphics geometry partner directory (ed. Silicon Graphics), pp. 77–78. Silicon Graphics, Mountain View, CA.
- Savarin, P., Guenneugues, M., Gilquin, B., Lamthanh, H., Gasparini, S., Zinn-Justin, S., and Menez, A. 1998. Three-dimensional structure of κ-conotoxin PVIIA, a novel potassium channel-blocking toxin from cone snails. Biochemistry 37: 5407–5416. [DOI] [PubMed] [Google Scholar]
- Silva Jr., P.I., Daffre, S., and Bulet, P. 2000. Isolation and characterization of gomesin, an 18-residue cysteine-rich defense peptide from the spider Acanthoscurria gomesiana hemocytes with sequence similarities to horseshoe crab antimicrobial peptide of the tachyplesin family. J. Biol. Chem. 275: 33464–33470. [DOI] [PubMed] [Google Scholar]
- Snow, R.W., Trape, J.F., and Marsh, K. 2001. The past, present and future of childhood malaria mortality in Africa. Trends Parasitol. 17: 593–597. [DOI] [PubMed] [Google Scholar]
- Snow, R.W., Guerra, C.A., Noor, A.M., Myint, H.Y., and Hay, S.I. 2005. The global distribution of clinical episodes of Plasmodium falciparium malaria. Nature 434: 214–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz, K.J. and MacKinnon, R. 1995. An inhibitor of the Kv2.1 potassium channel isolated from the venom of a Chilean tarantula. Neuron 15: 941–949. [DOI] [PubMed] [Google Scholar]
- Szyperski, T., Guntert, P., Otting, G., and Wüthrich, K. 1992. Determination of scalar coupling constants by inverse Fourier transformation of in-phase multiples. J. Magn. Reson. Imaging 99: 552–560. [Google Scholar]
- Tam, J.P., Lu, Y.-A., Yang, J.-L., and Chiu, K.-W. 1999. An unusual structural motif of antimicrobial peptides containing end-to-end macrocycle and cystine-knot disulfides. Proc. Natl. Acad. Sci. 96: 8913–8918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomma, B.P., Cammue, B.P., and Thevissen, K. 2002. Plant defensins. Planta 216: 193–202. [DOI] [PubMed] [Google Scholar]
- Torres, A.M. and Kuchel, P.W. 2004. The β-defensin-fold family of polypeptides. Toxicon 44: 581–588. [DOI] [PubMed] [Google Scholar]
- Vitt, U.A., Hsu, S.Y., and Hsueh, A.J. 2001. Evolution and classification of cystine knot-containing hormones and related extracellular signaling molecules. Mol. Endocrinol. 15: 681–694. [DOI] [PubMed] [Google Scholar]
- Wang, X., Connor, M., Smith, R., Maciejewski, M.W., Howden, M.E., Nicholson, G.M., Christie, M.J., and King, G.F. 2000. Discovery and characterization of a family of insecticidal neurotoxins with a rare vicinal disulfide bridge. Nat. Struct. Biol. 7: 505–513. [DOI] [PubMed] [Google Scholar]
- Wüthrich, K. 1986. NMR of proteins and nucleic acids. Wiley, New York.
- Yu, K., Fu, W., Liu, H., Luo, X., Chen, K.X., Ding, J., and Shen, J. 2004. Computational simulations of interactions of scorpion toxins with the voltage-gated potassium ion channel. Biophysical J. 86: 3542–3555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, P.A., Xie, X.J., Li, M., Yang, D.M., Xie, Z.P., Zong, X., and Liang, S.P. 1997. Blockade of neuromuscular transmission by huwentoxin-I, purified from the venom of the Chinese bird spider Selenocosmia huwana. Toxicon 35: 39–45. [DOI] [PubMed] [Google Scholar]