

Abstract
This is a personal account of the early history of ubiquitin research, by one of its protagonists. The field of ubiquitin and regulated protein degradation was created in the 1980s, largely through the complementary discoveries by the laboratory of A. Hershko (Technion, Haifa, Israel) and by my laboratory, then at MIT (Cambridge, MA). I describe the elegant insights by Hershko and his colleagues that yielded the initial understanding of ubiquitin conjugation and ubiquitin-mediated proteolysis in cell extracts, including the identification of E1, E2, and E3 enzymes. These advances were followed by a set of interconnected discoveries in my laboratory that revealed the biology of the ubiquitin system, i.e., its necessity for the protein degradation in vivo, its specific physiological functions (in the cell cycle, DNA repair, protein synthesis, transcriptional regulation, and stress responses), the source of its selectivity (specific degradation signals in short-lived proteins), and its key mechanistic attributes, such as the polyubiquitin chain and the subunit selectivity of protein degradation. The above biological (function-based) insights produced the main discovery of the physiological regulation by intracellular protein degradation. These advances caused the enormous expansion of the ubiquitin field in the 1990s. Together with the initial discovery of ubiquitin-mediated proteolysis by Hershko and coworkers, our biological discoveries in the 1980s led to a radically changed understanding of the logic of intracellular circuits, as it became clear that the control through regulated protein degradation rivals, and often surpasses in significance, the classical regulation through transcription and translation.
Keywords: ubiquitin, proteasome, proteolysis, N-end rule, nitric oxide
This article, on the occasion of the 2005 Stein and Moore Award to Avram Hershko and me, is a personal account of the early history of ubiquitin research. Proteolysis (protein degradation) is mediated by proteases, which vary from small proteins such as extracellular trypsin and the intracellular caspases to large, ATP-dependent, multifunctional proteases called proteasomes. For a very long time, and despite some evidence to the contrary (Schoenheimer 1942), most intracellular proteins were believed to be long-lived. This assumption survived nearly intact until the 1980s, when two complementary sets of discoveries were made, largely by two groups of researchers, Hershko’s laboratory at the Technion (Haifa, Israel) and my laboratory, then at the Massachusetts Institute of Technology (MIT; Cambridge, MA). Through the elegant use of biochemical fractionation and enzymology, Hershko, his student A. Ciechanover, his collaborator I. Rose, and their colleagues discovered in 1978–1983 that some proteins added to a reticulocyte extract became covalently conjugated to a protein called ubiquitin, and that ubiquitylated proteins were processively destroyed by an ATP-dependent protease in the extract (Ciechanover et al. 1978; Hershko et al. 1980). Hershko and colleagues went on to identify and characterize the enzymes, termed E1, E2, and E3, that carry out ubiquitin–protein conjugation (Ciechanover et al. 1982; Hershko et al. 1983, 2000). The ATP-dependent protease that mediates the destruction of ubiquitin–protein conjugates (Hershko et al. 1984) was characterized by several laboratories much later, in the 1990s, and is now called the 26S proteasome (Baumeister et al. 1998; Lee and Goldberg 1998; Rechsteiner 1998; Groll and Huber 2004; Pickart and Cohen 2004; Wolf and Hilt 2004; Rechsteiner and Hill 2005).
In 1984–1990, my colleagues and I discovered the first biological functions of the ubiquitin system; deciphered the source of its specificity, i.e., the primary degradation signals in short-lived proteins; and identified some of the system’s fundamental attributes, such as the polyubiquitin chain and the subunit selectivity of protein degradation. Through genetic, biochemical, and cell biological studies with mammalian cells and the yeast Saccharomyces cerevisiae, we discovered that the ubiquitin system (until then defined in cell extracts) was essential for the bulk of protein degradation in living cells, was required for cell viability, and played major roles in the cell cycle, DNA repair, protein synthesis, transcriptional regulation, and stress responses (Ciechanover et al. 1984; Finley et al. 1984, 1987, 1989; Özkaynak et al. 1984, 1987; Bachmair et al. 1986; Jentsch et al. 1987; Goebl et al. 1988; Bachmair and Varshavsky 1989; Chau et al. 1989; Gonda et al. 1989; Bartel et al. 1990; Hochstrasser and Varshavsky 1990; Johnson et al. 1990).
What follows is an account of the discoveries in the 1980s that cofounded the ubiquitin field as we know it today. Later advances, including our own ubiquitin work after 1990, are mentioned only to an extent required to explain the meaning of early discoveries. While the narrative encompasses most of the major ubiquitin-linked developments in the 1980s, the review’s relative brevity does not allow a comprehensive and detailed account of all significant advances in the early years, a task for other scholars and later times.
Discovering the biological functions and degradation signals of the ubiquitin system
Through preparation, help from friends, and a lot of luck, I was able to leave the former Soviet Union in the fall of 1977, and ended up in Boston, Massachusetts. A month later I was a faculty member at the Biology Department of MIT, before I knew what exactly grants were (and before the colleagues who hired me became aware of that fact). In Moscow, I studied chromosome structure and regulation of gene expression, and looked forward to continuing this work.
There were few similarities between my Moscow milieu and the astonishing new life. The libraries were one of them. They were just as quiet and pleasant in Cambridge as in Moscow, and a library at MIT soon became my second home. Reading there in late 1977, I came across a curious paper, of the same year, by I. Goldknopf and H. Busch (Goldknopf and Busch 1977). They found a DNA-associated protein that had one C terminus but two N termini, an unprecedented structure. The short arm of that Y-shaped protein was joined, through its C terminus, to an internal lysine of histone H2A. The short arm was soon identified, by L. Hunt and M. Dayhoff (Hunt and Dayhoff 1977), as ubiquitin (Ub), a universally present protein of unknown function that had been previously described (as a free protein) by G. Goldstein and colleagues (Goldstein et al. 1975).
I became interested in that first ubiquitin conjugate, Ub–H2A. Back in Russia, I had begun to develop a method for high-resolution analysis of nucleosomes, based on the electrophoresis of DNA–protein complexes in a low-ionic-strength polyacrylamide gel, a forerunner of the gel shift assay (Varshavsky et al. 1976). At MIT, my first post-doc L. Levinger and I developed this method further in 1978–1982, by adding the second-dimension electrophoresis of either DNA or proteins and mapping the spots of fractionated DNA by southern hybridization. We located Ub–H2A in a subset of nucleosomes, succeeded in separating these nucleosomes from those lacking Ub–H2A (Levinger and Varshavsky 1980), and showed that ubiquitin-containing nucleosomes were enriched on transcribed genes and absent from transcriptionally inactive regions such as the centromeric hetero-chromatin (Levinger and Varshavsky 1982).
In the meantime Hershko, his graduate student Ciechanover, and their colleagues in the Hershko laboratory at the Technion were studying the ATP-dependent protein degradation in extracts from rabbit reticulocytes. In 1978, they discovered that a small protein, termed APF-1 (ATP-dependent proteolytic factor 1), was covalently conjugated to proteins before their degradation in the extract (Ciechanover et al. 1978). In 1980, they suggested that a protein-linked APF-1 served as a signal for a downstream protease (Hershko et al. 1980) and began dissecting the enzymology of APF-1 conjugation. In 1981–1983, Hershko and coworkers identified a set of three enzymes involved, termed E1 (ubiquitin-activating enzyme), E2 (ubiquitin carrier protein or ubiquitin-conjugating enzyme), and E3 (an accessory component that appeared to confer specificity on E2) (Ciechanover et al. 1982; Hershko et al. 1983). Although our studies of ubiquitin in chromosomes began in 1978, I did not know about the 1978 APF-1 paper by Hershko and coworkers, since the identity of APF-1 and ubiquitin was unknown to them as well. The disposition changed in 1980, when APF-1 and ubiquitin were shown to be the same protein (Wilkinson et al. 1980), by K. Wilkinson, M. Urban and A. Haas, who worked in the laboratory of I. Rose, a collaborator of Hershko during his sojourns at the Philadelphia’s Fox Chase Cancer Center.
When I read the 1980 papers of Hershko et al. (1980), which described the APF-1 conjugation, and of Wilkinson et al. (1980), which described the identity of APF-1 and ubiquitin, two previously independent realms—protein degradation and chromatin-associated ubiquitin—came together for me, suggesting a regulatory system of great complexity and broad, still-to-be-discovered biological functions. I decided to find genetic approaches to the entire problem because a system of such complexity was unlikely to be understood through biochemistry alone. In 1980, reverse genetic techniques were about to become feasible with the yeast S. cerevisiae but were still a decade away in mammalian genetics. I kept reading, as widely as I could. Near the end of 1980, I came across a paper by M. Yamada and colleagues (Marunouchi et al. 1980) that described a conditionally lethal, temperature-sensitive mouse cell line called ts85. The investigators showed that a specific nuclear protein disappeared at elevated temperatures from ts85 cells, and suggested that this protein may be Ub–H2A. Glancing at their data, I had to calm down to continue reading, being virtually certain that the protein was Ub–H2A: In the preceding two years we had learned much about electrophoretic properties of this ubiquitin conjugate. On the hunch that mouse ts85 cells might be a mutant in a component of the ubiquitin system, I wrote to Yamada and received from him, in 1981, both ts85 and the parental (“wild-type”) cell line.
D. Finley, then a graduate student, joined my lab at that time to study regulation of gene expression. He didn’t need much convincing to switch to ts85 cells. A few months into the project, Finley and I made the critical observation that ubiquitin conjugation in an extract from ts85 cells was temperature-sensitive, in contrast to an extract from parental cells. While this was going on, I met Ciechanover, who came from the Hershko laboratory in Israel for a post-doctoral stint in the MIT lab of H. Lodish and was studying growth factor receptors. Presuming that Ciechanover was still interested in ubiquitin (very few people were), I told him about our results with ts85 cells and invited him to join, part-time, with Finley and me to complete the ts85 study. Ciechanover did, the work continued, and in 1984 we submitted two papers that described, primarily, the following discoveries: (1) Mouse ts85 cells have a temperature-sensitive ubiquitin-activating (E1) enzyme, and (2) these cells, in contrast to their wild-type counterpart, stop degrading the bulk of their short-lived proteins at nonpermissive temperature (Ciechanover et al. 1984; Finley et al. 1984).
This was the first evidence that ubiquitin conjugation was required for protein degradation in vivo. (The earlier studies by Hershko and coworkers were carried out with cell-free systems.) The results with ts85 cells also indicated that ubiquitin conjugation was essential for cell viability, the first hint of the enormous, many-sided biological importance of the ubiquitin system. In addition, ts85 cells were preferentially arrested in the G2 phase of the cell cycle, and the synthesis of heat-stress proteins was strongly induced in these cells at the non-permissive temperature, suggesting that ubiquitin conjugation was involved in the cell cycle progression and stress responses (Ciechanover et al. 1984; Finley et al. 1984). In 1983, T. Hunt and colleagues discovered unusual proteins in sea urchin and clam embryos. These proteins, which they called cyclins, were degraded at the end of mitosis (Evans et al. 1983). We suggested in 1984 that cyclins were destroyed by the ubiquitin system (Ciechanover et al. 1984; Finley et al. 1984), a hypothesis shown to be correct in 1991 by Glotzer, Murray, and Kirschner (Glotzer et al. 1991), and independently by Hershko and coworkers (Hershko et al. 1991).
It may be helpful to place the above advance in historical context. Despite some evidence to the contrary, until the 1980s and the two 1984 Cell papers (Ciechanover et al. 1984; Finley et al. 1984), the prevailing view was that intracellular protein degradation was a simple and even mundane process, serving largely to dispose of “aged” or otherwise damaged proteins. Cellular regulation was believed to be a separate affair, mediated primarily by repressors and activators of gene expression, which were assumed, often tacitly, to be long-lived. Among the reasons for this lopsided perspective was the difficulty of connecting the long-recognized proteolytic system in the lysosomes to specific pathways of intracellular regulation. Thus, most people studying gene expression in the 1960s and 1970s assumed that the regulatory circuits they cared about did not involve short-lived proteins. As we now know, just the opposite proved true, especially in eukaryotes, where most regulators of transcription are conditionally short-lived proteins whose levels in a cell are determined at least as much by the rates of their ubiquitin-dependent destruction as by the rates of their synthesis. Ironically, the first physiological (as distinguished from engineered) substrate of the ubiquitin system was a transcriptional regulator, Matα2, which M. Hochstrasser (then a post-doc) and I demonstrated in 1990 to be short-lived in vivo and delineated its degradation signal (Hochstrasser and Varshavsky 1990). As mentioned above, a mitotic cyclin was the second such substrate, identified in 1991 (Glotzer et al. 1991; Hershko et al. 1991).
In addition to having been a breakthrough that indicated the requirement of the ubiquitin system for intra-cellular proteolysis, cell viability, and cell cycle progression, the ts85 papers were also the first to address the in vivo workings of this system. In 2004, this pair of papers (Ciechanover et al. 1984; Finley et al. 1984) was selected for republication by the editors of Cell as being among the most important papers that have been published in Cell’s 30-year history. In a review accompanying republication, C. Pickart, one of the early pioneers in the ubiquitin field, summed up the papers’ contribution: “The two papers…led to a new world-view; not only was the ubiquitin/proteasome pathway a major proteolytic mechanism in the average mammalian cell, but it was also likely to regulate cell cycle progression. These conclusions are so well accepted today that it is difficult to appreciate the magnitude of their impact at the time the two papers appeared” (Pickart 2004).
Although the ts85 discoveries left little doubt, among the optimists, about the importance of the ubiquitin system in cellular physiology, it was difficult to extend these findings in the same system, owing to limitations of mammalian somatic cell genetics, which was hampered at that time by the impossibility of altering genes at will. In addition, the advances with ts85 cells produced little more than hints about specific physiological functions of the ubiquitin system and also did not address another fundamental problem: the source of selectivity of ubiquitin conjugation, i.e., the existence and structure of degradation signals in short-lived proteins that make them the targets for ubiquitylation.
Therefore in 1983, even before the completion of ts85 work, Finley and I, together with other colleagues in the lab, began a systematic analysis of the ubiquitin system in the genetically tractable yeast S. cerevisiae (Fig. 1), a project that soon expanded to occupy the entire laboratory. Between 1983 and 1990, this work revealed the first specific biological functions of ubiquitin conjugation. Briefly mentioned below are key advances of those early years that established the physiological fundamentals of the ubiquitin field.
Figure 1.
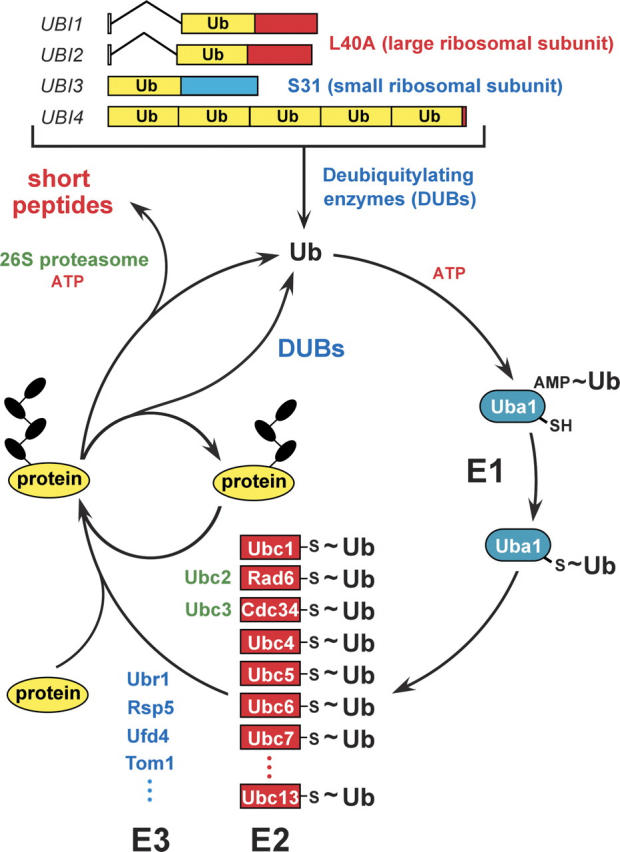
The ubiquitin system of the yeast S. cerevisiae (Varshavsky 1997, 2005). The fundamental design of this system is conserved among eukaryotes. The yeast ubiquitin genes UBI1–UBI4, two of which contain introns, encode fusions of ubiquitin either to itself or to one of two ribosomal proteins. These fusions are cleaved by deubiquitylating enzymes (DUBs), yielding mature ubiquitin. Thioester bonds between ubiquitin and the active-site Cys residues of ubiquitin-specific enzymes are denoted by the “~” sign. The conjugation of ubiquitin to other proteins involves a preliminary ATP-dependent step, in which the last (Gly-76) residue of ubiquitin is joined, via a thioester bond, to a Cys residue of the ubiquitin-activating (E1) enzyme, encoded by UBA1. The activated ubiquitin is transferred to a Cys residue in one of several ubiquitin-conjugating (E2) enzymes, encoded by the UBC-family genes, and from there to a Lys residue of an ultimate acceptor protein. This last step, and the formation of a substrate-linked polyubiquitin chain (black ovals) require participation of another component, called E3, whose mechanistic functions include the recognition of a substrate’s degradation signal (degron). The names of some of the currently known yeast E3s are indicated as well. The term “ubiquitin ligase” denotes either an E2–E3 holoenzyme or its E3 component. A targeted, ubiquitylated protein is processively degraded to short peptides by the ATP-dependent 26S proteasome. (For reviews, see Varshavsky 1997; Hershko et al. 2000; Pickart 2004.)
In 1984, E. Özkaynak, Finley, and I cloned the first ubiquitin gene and found it to encode a polyubiquitin precursor protein (Özkaynak et al. 1984). By 1987, we showed that this gene, UBI4, was strongly induced by a variety of stresses, and that a deletion of UBI4 resulted in stress-hypersensitive cells (Finley et al. 1987). These genetically based results validated and deepened the earlier indirect evidence with mouse ts85 cells (Finley et al. 1984), thereby establishing one broad and essential function of the ubiquitin system.
In 1986, A. Bachmair, Finley, and I discovered, through the invention of the ubiquitin fusion technique, the first degradation signals (degrons) that target proteins for ubiquitin conjugation and proteolysis (Bachmair et al. 1986). By revealing the basis of specificity of intracellular protein degradation, this critical advance has spawned the field of degradation signals, a major arena of current research. The term “degron,” proposed later (Varshavsky 1991), has since become a standard acronym for “degradation signal,” “destruction box,” and related terms. One set of degrons discovered in 1986 gives rise to the N-end rule, a relation between the in vivo half-life of a protein and the identity of its N-terminal residue (Bachmair et al. 1986). The seemingly simple N-end rule is “implemented” by the remarkably complex N-end rule pathway (Fig. 2), the first complete pathway of the ubiquitin system to be discovered. Other specific pathways of the ubiquitin system, including those that underlie the cell cycle oscillator, were identified nearly a decade later, in the mid-1990s. The N-end rule pathway is still a focus of our work, surprising us by what it has up its sleeve, including its functions, which continue to emerge (Turner et al. 2000; Rao et al. 2001; Kwon et al. 2002, 2003; Yoshida et al. 2002; Ditzel et al. 2003; Varshavsky 2003; Hu et al. 2005).
Figure 2.

(A) The N-end rule pathway in mammals (Kwon et al. 2002, Hu et al. 2005). This proteolytic pathway was the first specific pathway of the ubiquitin system to be discovered, initially in yeast (Bachmair et al. 1986; Varshavsky 1996). It is present in all eukaryotes examined, from fungi to animals and plants. Although prokaryotes lack ubiquitin conjugation and ubiquitin itself, they, too, contain the N-end rule pathway, a ubiquitin-independent version of it (Tobias et al. 1991; Shrader et al. 1993). Studies of this pathway, its mechanisms and functions, have become a major focus of my laboratory. N-terminal residues are indicated by single-letter abbreviations for amino acids. The ovals denote the rest of a protein substrate. MetAPs, methionine aminopeptidases. The “cysteine” (Cys) sector, in the upper left corner, describes the recent discovery of a nitric oxide (NO)-mediated oxidation of N-terminal Cys, with subsequent arginylation of oxidized Cys by the ATE1-encoded isoforms of Arg-tRNA-protein transferase (R-transferase) (Hu et al. 2005). This advance identified the N-end rule pathway as a new kind of NO sensor. C* denotes oxidized Cys, either Cys-sulfinic acid (CysO2[H]) or Cys-sulfonic acid (CysO3[H]). Type 1 and type 2 primary destabilizing N-terminal residues are recognized by multiple E3 ubiquitin ligases of the N-end rule pathway, including UBR1 and UBR2. Through their other substrate-binding sites, these E3 enzymes also recognize internal (non-N-terminal) degrons in other substrates of the N-end rule pathway, denoted by a larger oval. (B) MetAPs remove Met from the N terminus of a polypeptide if the residue at position 2 belongs to the set of residues shown.
In 1987, S. Jentsch, J. McGrath, and I discovered that RAD6, a protein known to yeast geneticists as an essential component of DNA repair pathways, was a ubiquitin-conjugating (E2) enzyme, the first ubiquitin-conjugating enzyme with a specific physiological function (Jentsch et al. 1987). We noticed that the sequence of RAD6 was weakly similar to that of CDC34, an essential cell cycle regulator (of unknown enzymatic activity) that had been defined genetically by L. Hartwell (1974).
In 1988, a collaboration between B. Byer’s and my laboratories demonstrated that CDC34 was a ubiquitin-conjugating enzyme (Goebl et al. 1988). This discovery was the first definitive evidence for a function of the ubiquitin system in the cell cycle control, the role suggested but not proved by our earlier ts85 studies.
In 1989, Finley, B. Bartel, and I discovered the functions of UBI1–UBI3, the other (non-polyubiquitin) ubiquitin genes, which were shown to encode fusions of ubiquitin to one protein of the large ribosomal subunit and one protein of the small ribosomal subunit (Finley et al. 1989), an arrangement conserved from yeast to mammals. (K. Redman and M. Rechsteiner independently identified mammalian counterparts of the yeast UBI1–UBI3 proteins as ubiquitin fusions to ribosomal proteins [Redman and Rechsteiner 1989].) In vivo experiments with mutationally altered yeast UBI proteins indicated that the presence of ubiquitin in front of a ribosomal protein moiety, despite being transient in vivo (the ubiquitin moiety was rapidly removed by deubiquitylating enzymes) was required for the efficient biogenesis of ribosomes (Finley et al. 1989). Remarkably, ubiquitin acts, in these settings, not as a degradation signal but as a cotranslational chaperone. This first nonproteolytic function of ubiquitin, mediated by its fusions to ribosomal proteins (Finley et al. 1989), appeared to be an exceptional case until years later, when L. Hicke and H. Riezman demonstrated that ubiquitylation of a plasma membrane–embedded receptor signals its endocytosis (Hicke and Riezman 1996). Ubiquitin is now recognized to have numerous nonproteolytic functions.
In 1985, the in vitro experiments by Hershko and H. Heller with a chemically modified ubiquitin suggested that substrate-linked ubiquitin moieties might be in the form of a polyubiquitin chain (Hershko and Heller 1985). In 1989, using new approaches that included two-dimensional protein mapping assays, V. Chau and other colleagues in my laboratory proved that the in vivo ubiquitin conjugation produces a polyubiquitin chain, and discovered its unique topology, with the isopeptide bonds between adjacent ubiquitin moieties through a specific (Lys-48) residue of ubiquitin. In addition, this study demonstrated that a substrate-linked polyubiquitin chain was essential for the substrate’s degradation (Chau et al. 1989). We proposed that a major function of the polyubiquitin chain is to bind the targeted substrate to the proteasome, a hypothesis subsequently confirmed by others, in part through the identification of polyubiquitin-binding proteins as components of the 26S proteasome. The 1989 insights about polyubiquitin chains (Chau et al. 1989) were yet another beginning of a major arena of ubiquitin studies.
In 1990, Bartel, I. Wünning, and I employed genetic and biochemical approaches to clone and analyze the first specific E3 ubiquitin ligase, UBR1, the E3 of the S. cerevisiae N-end rule pathway (Bartel et al. 1990). Many more E3 enzymes, whose functions include the recognition of specific degradation signals in targeted proteins, were identified in the 1990s and later—a process of discovery that continues as I write—in part because the number of distinct E3 ubiquitin ligases in a mammal is estimated, at present, to exceed a thousand.
A key feature of the ubiquitin-dependent protein degradation is its subunit selectivity, that is, the ability of the ubiquitin system to eliminate one subunit of an oligomeric protein or multiprotein complex, leaving intact the rest of it and thereby making possible protein remodeling. This fundamental property was discovered and dissected in 1990 by E. Johnson, D. Gonda, and myself, in the context of the N-end rule pathway (Johnson et al. 1990). Also in 1990, Hochstrasser and I detected subunit selectivity in the degradation of Matα2 (see above), the first physiological substrate of the ubiquitin system (Hochstrasser and Varshavsky 1990). Subunit-selective proteolysis is a biologically crucial property of the ubiquitin system, a feature both powerful and flexible, in that it enables protein degradation to be wielded as an instrument of protein remodeling for either positive or negative regulation, including the control of the eukaryotic cell cycle, the regulation of transcription, and a number of other processes. Among many specific examples are the activation of a major transcription factor NF-κB via the subunit-selective degradation of its inhibitory subunit IκB, and the inactivation of cyclin-dependent kinases (which drive the cell cycle oscillator) via the subunit-selective degradation of their cyclin subunits.
In summary, the complementary discoveries in the 1980s by Hershko’s and my laboratories revealed three sets of previously unknown facts:
That ATP-dependent protein degradation involves a new protein modification, ubiquitin conjugation, which is mediated by specific enzymes, termed E1, E2, and E3 (Ciechanover et al. 1978, 1982; Hershko et al. 1980, 1983, 2000).
That the selectivity of ubiquitin conjugation is determined by specific degradation signals (degrons) in short-lived proteins, including the degrons that give rise to the N-end rule (Bachmair et al. 1986; Bachmair and Varshavsky 1989; Gonda et al. 1989).
That ubiquitin-dependent processes play a strikingly broad, previously unsuspected part in cellular physiology, primarily by controlling the in vivo levels of specific proteins. Ubiquitin conjugation was demonstrated by us to be required for protein degradation in vivo (Ciechanover et al. 1984; Finley et al. 1984), for cell viability, and also, specifically, for the cell cycle (Goebl et al. 1988), DNA repair (Jentsch et al. 1987), protein synthesis (Finley et al. 1989), transcriptional regulation (Hochstrasser and Varshavsky 1990), and stress responses (Özkaynak et al. 1984; Finley et al. 1987). In addition, ubiquitin-dependent proteolysis was discovered to involve a substrate-linked polyubiquitin chain of unique topology that is required for protein degradation (Chau et al. 1989). The ubiquitin system was also discovered to possess the critically important property of subunit selectivity, i.e., the ability to destroy a specific subunit of oligomeric protein, leaving intact the rest of it and thereby making possible protein remodeling (Johnson et al. 1990).
The Hershko laboratory produced the first of these fundamental advances (item 1), and my laboratory produced the other two (items 2 and 3). Over the last 15 years, these complementary “chemical” and “biological” discoveries of the 1980s led to the enormous expansion of the ubiquitin field, which became one of the largest arenas in biomedical science, the point of convergence of many disparate disciplines. Our biological discoveries (Ciechanover et al. 1984; Finley et al. 1984, 1987, 1989; Bachmair et al. 1986; Jentsch et al. 1987; Özkaynak et al. 1987; Goebl et al. 1988; Bachmair and Varshavsky 1989; Chau et al. 1989; Gonda et al. 1989; Bartel et al. 1990; Hochstrasser and Varshavsky 1990; Johnson et al. 1990), together with later studies by many excellent laboratories that entered the field in the 1990s and afterward, yielded the modern paradigm of the central importance of regulated proteolysis for the control of the levels of specific proteins in vivo, as distinguished from their control by transcription and protein synthesis. In other words, these advances revealed that the control through regulated protein degradation rivals, and often surpasses in significance, the classical regulation through transcription and translation. This radically changed understanding of the logic of biological circuits will have (in fact, is already having) a major impact on medicine, given the astounding functional range of the ubiquitin system and the multitude of ways in which ubiquitin-dependent processes can malfunction in disease or in the course of aging, from cancer and neurodegenerative syndromes to perturbations of immunity and many other illnesses, including birth defects. A number of pharmaceutical companies are developing compounds that target specific components of the ubiquitin system. The fruits of their labors have already become, or will soon become, clinically useful drugs. Efforts in this area may yield not only “conventional” inhibitors or activators of enzymes but also more sophisticated drugs that will direct the ubiquitin system to target, destroy, and thereby inhibit functionally any specific protein.
I feel privileged having been able to contribute to the birth of this field, and to partake in its later development. The dynamism and surprises of this endeavor remain undiminished even today, two decades after the 1980s.
Acknowledgments
Our studies are supported by grants from the NIH and the Ellison Medical Foundation. I am most grateful to the past and the present colleagues in my laboratory for their contributions, some of which are mentioned above. I also thank Christopher Brower for his helpful comments on the article.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.052012306.
References
- Bachmair, A. and Varshavsky, A. 1989. The degradation signal in a short-lived protein. Cell 56: 1019–1032. [DOI] [PubMed] [Google Scholar]
- Bachmair, A., Finley, D., and Varshavsky, A. 1986. In vivo half-life of a protein is a function of its amino-terminal residue. Science 234: 179–186. [DOI] [PubMed] [Google Scholar]
- Bartel, B., Wünning, I., and Varshavsky, A. 1990. The recognition component of the N-end rule pathway. EMBO J. 9: 3179–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumeister, W., Walz, J., Zühl, F., and Seemüller, E. 1998. The proteasome: Paradigm of a self-compartmentalizing protease. Cell 92: 367–380. [DOI] [PubMed] [Google Scholar]
- Chau, V., Tobias, J.W., Bachmair, A., Marriott, D., Ecker, D.J., Gonda, D.K., and Varshavsky, A. 1989. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science 243: 1576–1583. [DOI] [PubMed] [Google Scholar]
- Ciechanover, A., Hod, Y., and Hershko, A. 1978. A heat-stable polypeptide component of an ATP-dependent proteolytic system from reticulocytes. Biochem. Biophys. Res. Commun. 81: 1100–1105. [DOI] [PubMed] [Google Scholar]
- Ciechanover, A., Elias, S., Heller, H., and Hershko, A. 1982. “Covalent affinity” purification of ubiquitin-activating enzyme. J. Biol. Chem. 257: 2537–2542. [PubMed] [Google Scholar]
- Ciechanover, A., Finley, D., and Varshavsky, A. 1984. Ubiquitin dependence of selective protein degradation demonstrated in the mammalian cell cycle mutant ts85. Cell 37: 57–66. [DOI] [PubMed] [Google Scholar]
- Ditzel, M., Wilson, R., Tenev, T., Zachariou, A., Paul, A., Deas, E., and Meier, P. 2003. Degradation of DIAP1 by the N-end rule pathway is essential for regulating apoptosis. Nat. Cell Biol. 5: 467–473. [DOI] [PubMed] [Google Scholar]
- Evans, T., Rosenthal, E.T., Youngblom, J., Distel, D., and Hunt, T. 1983. Cyclin: A protein specified by maternal mRNA in sea urchin eggs that is destroyed at each cleavage division. Cell 33: 389–396. [DOI] [PubMed] [Google Scholar]
- Finley, D., Ciechanover, A., and Varshavsky, A. 1984. Thermolability of ubiquitin-activating enzyme from the mammalian cell cycle mutant ts85. Cell 37: 43–55. [DOI] [PubMed] [Google Scholar]
- Finley, D., Özkaynak, E., and Varshavsky, A. 1987. The yeast polyubiquitin gene is essential for resistance to high temperatures, starvation, and other stresses. Cell 48: 1035–1046. [DOI] [PubMed] [Google Scholar]
- Finley, D., Bartel, B., and Varshavsky, A. 1989. The tails of ubiquitin precursors are ribosomal proteins whose fusion to ubiquitin facilitates ribosome biogenesis. Nature 338: 394–401. [DOI] [PubMed] [Google Scholar]
- Glotzer, M., Murray, A.W., and Kirschner, M. 1991. Cyclin is degraded by the ubiquitin pathway. Nature 349: 132–138. [DOI] [PubMed] [Google Scholar]
- Goebl, M.G., Yochem, J., Jentsch, S., McGrath, J.P., Varshavsky, A., and Byers, B. 1988. The yeast cell cycle gene CDC34 encodes a ubiquitin-conjugating enzyme. Science 241: 1331–1335. [DOI] [PubMed] [Google Scholar]
- Goldknopf, I.A. and Busch, H. 1977. Isopeptide linkage between nonhistone and histone 2A polypeptides of chromosomal conjugate-protein A24. Proc. Natl. Acad. Sci. 74: 864–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein, G., Scheid, M., Hammerling, U., Boyse, E.A., Schlesinger, D.H., and Niall, H.D. 1975. Isolation of a polypeptide that has lymphocyte-differentiating properties and is probably represented universally in living cells. Proc. Natl. Acad. Sci. 72: 11–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonda, D.K., Bachmair, A., Wünning, I., Tobias, J.W., Lane, W.S., and Varshavsky, A. 1989. Universality and structure of the N-end rule. J. Biol. Chem. 264: 16700–16712. [PubMed] [Google Scholar]
- Groll, M. and Huber, R. 2004. Inhibitors of the eukaryotic 20S proteasome core particle: A structural approach. Biochim. Biophys. Acta 1695: 33–44. [DOI] [PubMed] [Google Scholar]
- Hartwell, L.H. 1974. Saccharomyces cerevisiae cell cycle. Bacteriol. Rev. 38: 164–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko, A. and Heller, H. 1985. Occurrence of a polyubiquitin structure in ubiquitin–protein conjugates. Biochem. Biophys. Res. Commun. 128: 1079–1086. [DOI] [PubMed] [Google Scholar]
- Hershko, A., Ciechanover, A., Heller, H., Haas, A.L., and Rose, I.A. 1980. Proposed role of ATP in protein breakdown: Conjugation of protein with multiple chains of the polypeptide of ATP-dependent proteolysis. Proc. Natl. Acad. Sci. 77: 1783–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko, A., Heller, H., Elias, S., and Ciechanover, A. 1983. Components of ubiquitin–protein ligase system. Resolution, affinity purification, and role in protein breakdown. J. Biol. Chem. 258: 8206–8214. [PubMed] [Google Scholar]
- Hershko, A., Leshinsky, E., Ganoth, D., and Heller, H. 1984. ATP-dependent degradation of ubiquitin–protein conjugates. Proc. Natl. Acad. Sci. 81: 1619–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko, A., Ganoth, D., Pehrson, J., Palazzo, R.E., and Cohen, L.H. 1991. Methylated ubiquitin inhibits cyclin degradation in clam embryo extracts. J. Biol. Chem. 266: 16376–16379. [PubMed] [Google Scholar]
- Hershko, A., Ciechanover, A., and Varshavsky, A. 2000. The ubiquitin system. Nat. Med. 10: 1073–1081. [DOI] [PubMed] [Google Scholar]
- Hicke, L. and Riezman, H. 1996. Ubiquitination of a yeast plasma membrane receptor signals its ligand-stimulated endocytosis. Cell 84: 277–287. [DOI] [PubMed] [Google Scholar]
- Hochstrasser, M., and Varshavsky, A. 1990. In vivo degradation of a transcriptional regulator: The yeast Matα2 repressor. Cell 61: 697–708. [DOI] [PubMed] [Google Scholar]
- Hu, R.G., Sheng, J., Xin, Q., Xu, Z., Takahashi, T.T., and Varshavsky, A. 2005. The N-end rule pathway as a nitric oxide sensor controlling the levels of multiple regulators. Nature 437: 981–986. [DOI] [PubMed] [Google Scholar]
- Hunt, L.T. and Dayhoff, M.O. 1977. Amino-terminal sequence identity of ubiquitin and the nonhistone component of nuclear protein A24. Biochem. Biophys. Res. Commun. 74: 650–655. [DOI] [PubMed] [Google Scholar]
- Jentsch, S., McGrath, J.P., and Varshavsky, A. 1987. The yeast DNA repair gene RAD6 encodes a ubiquitin-conjugating enzyme. Nature 329: 131–134. [DOI] [PubMed] [Google Scholar]
- Johnson, E.S., Gonda, D.K., and Varshavsky, A. 1990. Cis–trans recognition and subunit-specific degradation of short-lived proteins. Nature 346: 287–291. [DOI] [PubMed] [Google Scholar]
- Kwon, Y.T., Kashina, A.S., Davydov, I.V., Hu, R.-G., An, J.Y., Seo, J.W., Du, F., and Varshavsky, A. 2002. An essential role of N-terminal arginylation in cardiovascular development. Science 297: 96–99. [DOI] [PubMed] [Google Scholar]
- Kwon, Y.T., Xia, Z.X., An, J.Y., Davydov, I.V., Seo, J.W., Xie, Y., and Varshavsky, A. 2003. Female lethality and apoptosis of spermatocytes in mice lacking the UBR2 ubiquitin ligase of the N-end rule pathway. Mol. Cell. Biol. 23: 8255–8271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, D.H. and Goldberg, A.L. 1998. Proteasome inhibitors: Valuable new tools for cell biologists. Trends Cell Biol. 8: 397–403. [DOI] [PubMed] [Google Scholar]
- Levinger, L. and Varshavsky, A. 1980. Separation of nucleosomes containing and lacking ubiquitin-H2A semihistone. Proc. Natl. Acad. Sci. 77: 3244–3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1982. Selective arrangement of ubiquitinated and D1 protein-containing nucleosomes within the Drosophila genome. Cell 28: 375–385. [DOI] [PubMed] [Google Scholar]
- Marunouchi, T., Mita, S., Matsumoto, Y., and Yamada, M. 1980. Disappearance of a chromosomal basic protein from cells of a mouse temperature-sensitive mutant defective in histone phosphorylation. Biochem. Biophys. Res. Commun. 95: 126–131. [DOI] [PubMed] [Google Scholar]
- Özkaynak, E., Finley, D., and Varshavsky, A. 1984. The yeast ubiquitin gene: Head-to-tail repeats encoding a polyubiquitin precursor protein. Nature 312: 663–666. [DOI] [PubMed] [Google Scholar]
- Özkaynak, E., Finley, D., Solomon, M.J., and Varshavsky, A. 1987. The yeast ubiquitin genes: A family of natural gene fusions. EMBO J. 6: 1429–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart, C. 2004. Back to the future with ubiquitin. Cell 116: 181–190. [DOI] [PubMed] [Google Scholar]
- Pickart, C. and Cohen, R.E. 2004. Proteasomes and their kin: Proteases in the machine age. Nat. Rev. Mol. Cell Biol. 5: 177–187. [DOI] [PubMed] [Google Scholar]
- Rao, H., Uhlmann, F., Nasmyth, K., and Varshavsky, A. 2001. Degradation of a cohesin subunit by the N-end rule pathway is essential for chromosome stability. Nature 410: 955–960. [DOI] [PubMed] [Google Scholar]
- Rechsteiner, M.1998. The 26S proteasome. In Ubiquitin and the biology of the cell (eds. J.M. Peters et al.), pp. 147–189. Plenum Press, New York.
- Rechsteiner, M. and Hill, C.P. 2005. Mobilizing the proteolytic machine: Cell biological roles of proteasome activators and inhibitors. Trends Cell Biol. 15: 27–33. [DOI] [PubMed] [Google Scholar]
- Redman, K.L. and Rechsteiner, M. 1989. Identification of the long ubiquitin extension as ribosomal protein S27a. Nature 338: 438–440. [DOI] [PubMed] [Google Scholar]
- Schoenheimer, R.1942. The dynamic state of body constituents. Harvard University Press, Cambridge, MA.
- Shrader, T.E., Tobias, J.W., and Varshavsky, A. 1993. The N-end rule in Escherichia coli: Cloning and analysis of the leucyl, phenylalanyl-tRNA-protein transferase gene aat. J. Bacteriol. 175: 4364–4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobias, J.W., Shrader, T.E., Rocap, G., and Varshavsky, A. 1991. The N-end rule in bacteria. Science 254: 1374–1377. [DOI] [PubMed] [Google Scholar]
- Turner, G.C., Du, F., and Varshavsky, A. 2000. Peptides accelerate their uptake by activating a ubiquitin-dependent proteolytic pathway. Nature 405: 579–583. [DOI] [PubMed] [Google Scholar]
- Varshavsky, A. 1991. Naming a targeting signal. Cell 64: 13–15. [DOI] [PubMed] [Google Scholar]
- ———. 1996. The N-end rule: Functions, mysteries, uses. Proc. Natl. Acad. Sci. 93: 12142–12149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1997. The ubiquitin system. Trends Biochem. Sci. 22: 383–387. [DOI] [PubMed] [Google Scholar]
- ———. 2003. The N-end rule and regulation of apoptosis. Nat. Cell Biol. 5: 373–376. [DOI] [PubMed] [Google Scholar]
- ———. 2005. Regulated protein degradation. Trends Biochem. Sci. 30: 283–286. [DOI] [PubMed] [Google Scholar]
- Varshavsky, A., Bakayev, V.V., and Georgiev, G.P. 1976. Heterogeneity of chromatin subunits in vitro and location of histone H1. Nucleic Acids Res. 3: 477–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson, K.D., Urban, M.K., and Haas, A.L. 1980. Ubiquitin is the ATP-dependent proteolytic factor of rabbit reticulocytes. J. Biol. Chem. 255: 7529–7532. [PubMed] [Google Scholar]
- Wolf, D.H. and Hilt, W. 2004. The proteasome: A proteolytic nanomachine of cell regulation and waste disposal. Biochim. Biophys. Acta 1695: 19–31. [DOI] [PubMed] [Google Scholar]
- Yoshida, S., Ito, M., Gallis, J., Nishida, I., and Watanabe, A. 2002. A delayed leaf senescence mutant is defective in arginyl–tRNA–protein arginyl-transferase, a component of the N-end rule pathway in Arabidopsis. Plant J. 32: 129–137. [DOI] [PubMed] [Google Scholar]