

Abstract
We recently described a new apolipoprotein A1 variant presenting a Leu174Ser replacement mutation that is associated with a familial form of systemic amyloidosis displaying predominant heart involvement. We have now identified a second unrelated patient with very similar clinical presentation and carrying the identical apolipoprotein A1 mutation. In this new patient the main protein constituent of the amyloid fibrils is the polypeptide derived from the first 93 residues of the protein, the identical fragment to that found in the patient previously described to carry this mutation. The X-ray fiber diffraction pattern obtained from preparations of partially aligned fibrils displays the cross-β reflections characteristic of all amyloid fibrils. In addition to these cross-β reflections, other reflections suggest the presence of well-defined coiled-coil helical structure arranged with a defined orientation within the fibrils. In both cases the fibrils contain a trace amount of full-length apolipoprotein A1 with an apparent prevalence of the wild-type species over the variant protein. We have found a ratio of full-length wild-type to mutant protein in plasma HDL of three to one. The polypeptide 1–93 purified from natural fibrils can be solubilized in aqueous solutions containing denaturants, and after removal of denaturants it acquires a monomeric state that, based on CD and NMR studies, has a predominantly random coil structure. The addition of phospholipids to the monomeric form induces the formation of some helical structure, thought most likely to occur at the C-terminal end of the polypeptide.
Keywords: Amyloidosis, apolipoprotein A1, proteolysis, fibrillogenesis
Apolipoprotein A1 (apoA1) circulates in normal plasma as the predominant apolipoprotein of HDL particles, and variant forms of apoA1 are the amyloidogenic precursor proteins in some forms of familial non-neuropathic systemic amyloidosis (Bellotti et al. 1999). In all cases, an N-terminal fragment of apoA1, ranging in length from 83 to 94 residues, is incorporated into amyloid deposits. However, amyloid fibrils constituted by the N-terminal polypeptide of nonmutated apoA1 have been detected in aged dogs (Johnson et al. 1992) and in humans in the aortic intima when an atherosclerotic plaque is present (Westermark et al. 1995). ApoA1 is a 243-amino acid protein synthesised in the liver and small intestine; it binds and transports plasma lipid and increases cholesterol efflux from peripheral tissues in a process called ``reverse cholesterol transport'' (Glomset 1968). This exchangeable lipoprotein exhibits different structural conformations in the lipid-free and the lipid-bound states and the transition between the two could be extremely important for lipid binding, interaction with a putative receptor, protein catabolism, and pathological misassembly in amyloidosis. To date, nine different mutations in the apoA1 gene have been linked to various forms of hereditary amyloidosis. Four are missense mutations (Nichols et al. 1988; Soutar et al. 1992; Booth et al. 1995; Hamidi Asl L. et al. 1999), one is a deletion mutation (Persey et al. 1998), and one is a deletion/insertion mutation (Booth et al. 1996). All of these mutations are located within the first 90 residues of the protein.
Three other recently described missense mutations occur far from the N-terminal region and more precisely in position 174 (Obici et al. 1999 ), 173 (Hamidi Asl K. et al. 1999), and 178 (de Sousa et al. 2000). Despite the differences in location of the disease-inducing mutations, in all of the reported cases where the fibril constituents have been characterized in detail, it has been shown that the presence of the mutation causes the formation of amyloid fibrils composed primarily of an N-terminal-derived polypeptide of ∼90 amino acids. There are some reports of trace amounts (Soutar et al. 1992; Obici et al. 1999) of full-length apoA1 present in amyloid deposits composed of N-terminal fragments of the protein. In the most recently described report of apoA1 amyloidosis, which involved a Leu178His mutation, the deposits appear to contain both an N-terminal-derived fragment and substantial amounts of full-length apoA1, as well as transthyretin (de Sousa et al. 2000). In the cases described by our group (Obici et al. 1999) and by Hamidi Asl and colleagues (Hamidi Asl et al. 1999b), the site of mutation is distant in the sequence from the portion of the protein that becomes incorporated into amyloid fibrils and the apoA1 polypeptide isolated from fibrils does not include the mutated residue. These findings, in the context of the three-dimensional structure of a truncated form of human apoA1 (apo Δ[1–43]A1; Borhani et al. 1997), have suggested a particular mechanism of release of the amyloidogenic polypeptide in these cases. The site of mutation in one polypeptide chain is in close proximity to the site of cleavage in the adjacent chain (Obici et al. 1999). In this paper a new unrelated patient affected by the Leu174Ser mutation is presented, and clinical and biochemical findings fully overlap those of the previous observation, supporting the proposal of a specific, structurally dependent cleavage of the amyloidogenic N-terminal polypeptide in a process that involves the mutation site.
Results
Fibril morphology and structure
Amyloid fibrils were isolated from the transplanted heart of the patient (PER), which displayed heavy infiltration by amyloid deposits. The clinical phenotype was very similar to that previously observed in the unrelated patient (PIC) with the same Leu174Ser mutation in apoA1. In negatively stained electron micrographs, the fibrils exhibited classical amyloid morphology, with a diameter of approximately 7 nm and length in excess of 500 nm (Fig. 1 ▶). The X-ray fiber diffraction pattern obtained from partially aligned fibrils displayed the cross-β reflections characteristic of all amyloid fibrils (Fig. 2 ▶). These consist of an intense and sharp reflection at 4.81 (±0.03) Å on the meridian of the pattern, which arises from the interstrand spacing in the direction of the fibril axis, and weaker and more diffuse equatorial reflections at 9.35 (±0.11) and 11.82 (±0.11) Å which may reflect the intersheet spacing or may arise from close lateral packing of fibrils. In addition to these cross-β reflections, a sharp reflection was observed at 5.08 (±0.03) Å on the equator of all of the diffraction patterns obtained from preparations of these fibrils, as well as a similarly sharp but weaker reflection observed at 2.54 (±0.01) Å. The sharpness of these two reflections and their specific orientation on the equator of the patterns suggested the presence of a well-defined coiled-coil helical structure arranged specifically within the fibrils. Reflections with no preferential orientation were observed at 3.1 and 2.9 Å. The fibril samples were not specifically treated to remove lipid, and the intensity and appearance of these reflections suggest they could be derived from lipid associated with the fibrils.
Fig. 1.
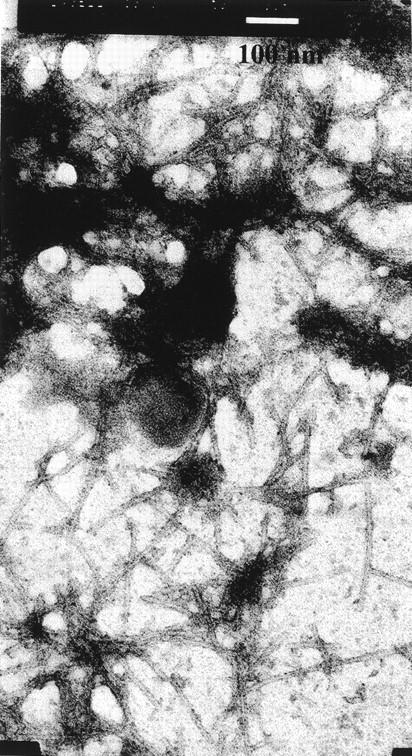
Electron micrograph of amyloid fibrils of patient PER. The bar corresponds to 100 nm.
Fig. 2.
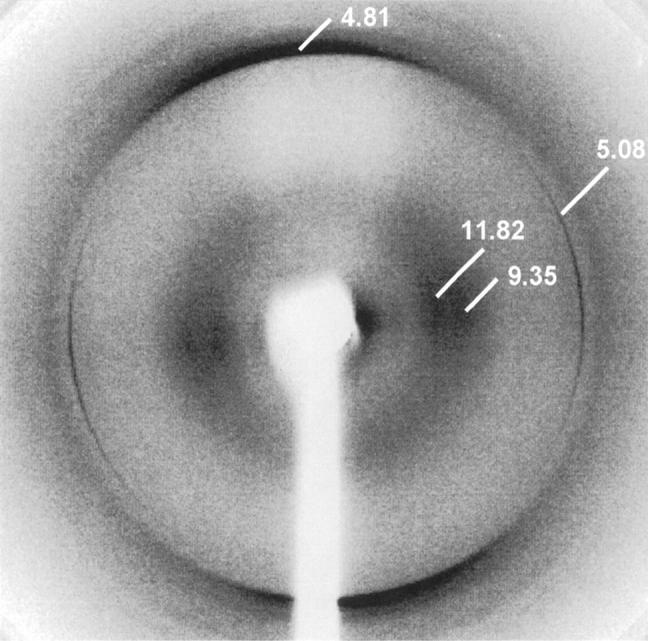
X-ray fiber diffraction image from partially aligned apoA1 fibrils. The cross-β reflections characteristic of amyloid fibrils are visible at 4.81 Å on the meridian and at 9.35 and 11.82 Å on the equator. The meridional reflection arises from the interstrand spacing between β-strands, lying at right angles to the fibril long axis. The two weaker and more diffuse equatorial reflections are a consequence of the intersheet spacing and close lateral packing of fibrils. In addition to these reflections, there are strong and sharp reflections at 5.08 and 2.54 Å on the equator, suggestive of the presence of well-defined coiled coil structure, preferentially aligned with the direction of hydrogen bonding at right angles to the fibril long axis.
The presence of lipid in samples usually manifests as concentric circles, and we (Sunde et al. 1997) and others (Damas et al. 1995) have previously observed reflections from lipid components at 4.1–4.2 Å and at other spacings that may be fitted to a 46-Å repeat, which is the thickness of a lipid layer in a gel phase (Damas et al. 1995).
Fibril constituents
The protein component of the fibrils was fractionated by gel filtration FPLC in 6 M guanidine and the low molecular weight component analyzed by SDS-PAGE, N-terminal sequencing, and mass spectrometry. The main protein component is a polypeptide corresponding to the N-terminal 93 residues of mature apoA1 (Fig. 3 ▶).
Fig. 3.

(A) Deconvoluted mass spectrum of the main constituent of patient PIC amyloid fibrils (predicted mass of polypeptide 1–93 = 10.720 Da). (B) Deconvoluted mass spectrum of the main constituent of patient PER amyloid fibrils.
The SDS-PAGE analysis of the fibrils of patients PER and PIC fractionated by gel filtration in denaturant as previously reported (Obici et al. 1999) shows three faint bands in the range of 25–28 kD immunostained with anti-apoA1 and one band immunostained by anti-SAP antibodies. The presence of SAP in these deposits was expected, given that this molecule is a universal constituent of amyloid fibrils. The pattern of apoA1 immunoreactivity in the western blot suggests that traces of intact or minimally proteolyzed apoA1 are present in these fibrils. The presence of an unusual 5.1-Å reflection in the X-ray diffraction image prompted us to investigate further the codeposition of full-length apoA1 with the 1–93 N-terminal fragment because the globular form of this protein has a predominantly helical structure, with some coiled-coil elements (Borhani et al. 1997). We have established the ratio of full-length apoA1 to peptide 1–93 within the fibrils through the analysis of proteolytic digests of nonfractionated fibrils and the identification of peptides as belonging to the N-terminal or C-terminal part of the molecule. The fibrillar material purified by water-extraction procedure was digested by V8 protease; the peptides generated in this way were separated by reversed-phase chromatography in HPLC and identified by electrospray mass spectrometry (Fig. 4 A–D ▶). The concentration of peptides derived from the region of apoA1 C-terminal to residue 93 was extremely low. Our analysis was mainly focused on the identification of peptide 170–179 that includes the site of the amyloidogenic mutation. Two peptides with the expected molecular weights for the wild-type (m/z 1225) and variant-derived (m/z 1199) species were identified at retention times around 11 and 16 min, but these did not correspond to well-identified chromatographic peaks, thus making peak integration and quantification difficult. However, according to the ion current, both forms were present and there was a slight prevalence of the wild-type species. Furthermore, a significant amount of the peptide 1–93 was not digested and was recovered as fibrillar, insoluble precipitate at the end of the 12-h incubation time. This finding was not surprising, given the well-known resistance of natural fibrils to proteases. Digestion of these fibrils with proteinase K, followed by analysis of proteolytic products by SDS-PAGE, shows that the full-length apoA1 component of the fibrils is rapidly degraded, whereas the 1–93 component is relatively resistant to digestion and that native, full-length apoA1 is also very susceptible to proteinase K digestion (M. Sunde, unpubl.). Although the experiments using proteolysis, followed by chromatographic separation and mass spectrometric characterization of fragments, do not allow determination of the precise ratio of full length to 1–93 fragment, they do confirm that the full-length protein associated with the amyloid material is a minor species. Not all of the peptides belonging to the 1–93 sequence were easily identified, because they did not all elute in pure forms or they were heterogeneous as a result of multiple cleavage sites, but, according to N-terminal sequencing and mass spectrometry, none of the main peaks included any sequence derived from the polypeptide C-terminal to residue 93.
Fig. 4.


(A) Reversed phase chromatography of V8 protease digestion of patient PER amyloid fibrils. (B) Reversed phase chromatography of V8 protease digestion of patient PIC amyloid fibrils. The polypeptide corresponding to the residues 170–179 with Ser in position 174 (marked with asterisk) elutes at 11.00 min and the same peptide but with Leu in position 174 is eluted at 16.00 min. In the chromatograms are also marked the peptide 63–72 (elution time 11.5 min) and the peptide 35–62 (elution time 33.6 min), which were isolated as pure peptides from the mixture. (C,D) Electrospray mass spectra of peptides 170–179* (with Ser in position 174) and 170–179 (with Leu in position 174), respectively. The labeled peaks indicate single and doubly charged ions.
ApoA1 species in plasma
To determine the ratio of the two species in the blood, we have purified apoA1 from the plasma of both patients. We failed in an attempt to separate the two species of full-length proteins by reversed-phase chromatography and therefore performed the same procedure of proteolysis with V8 protease, followed by separation and quantification of peptides. A chromatogram illustrating the separation of the peptides is presented in Figure 5 ▶, A and B. Peptides corresponding to residues 170–179 of the wild-type and variant chains are well separated, eluting at the same times as seen with protease digestion of the fibrillar peptide material. We recognized and quantified the wild-type and the mutant peptide by recognition of the characteristic (M + H)+ and double charged (M + 2H)2 + ion produced by the two peptides (Fig. 5C,D ▶). The integrated areas of the peaks indicate that the variant represents ∼25% of the total apoA1.
Fig. 5.




(A) Reversed phase chromatography of V8 protease digestion of PER apoA1. The peptide indicated as 170–179* has Ser in position 174 and peptide 170–179 has Leu in position 174. (B) Detail of V8 protease digest of apoA1 of patient PIC. Peptides 170–179* and 170–179 are indicated. (C) Mass spectrum of peptide 170–179* (eluted at 10.77 min) and peptide 170–179 (eluted at 15.82 min) from PER apoA1 shown in panel A. (D) Mass spectrum of peptide 170–179* (eluted at 10.50 min) and peptide 170–179 (eluted at 15.28 min) from PIC apoA1 shown in panel B. (Inset) SDS-PAGE analysis of plasma apoA1 stained by Coomassie blue. (1) apoA1 purified from plasma and used for V8 protease digestion; (2) molecular weight standards (phosphorylase b = 97.4 kD, bovine serum albumin = 66.2 kD, ovalbumin = 45.0 kD, carbonic anhydrase = 31.0 kD, soybean trypsin inhibitor = 21.5 kD, lysozyme = 14.4 kD).
Polypeptide 1–93 in aqueous solvent
The availability of abundant amyloid material made possible the purification of a few milligrams of highly purified 1–93 polypeptide (inset in Fig. 6A ▶). This polypeptide was solubilized in guanidine and refolded in phosphate buffer or in a solution containing phospholipids. Figure 6A ▶ shows the far-UV CD spectra of peptide 1–93 in 5 M guanidine hydrochloride, in phosphate buffer, and in the presence of phospholipids. Deconvolution of the far-UV CD spectra (Yang et al. 1986) suggests the presence of a high percentage of random coil structure (∼40%), about 30% β sheet, and a very limited amount of helical structure. It is worthy to note that the presence of phospholipids significantly increases the helical representation, whereas the percentage of β sheet and coil structure are not affected. The 93-residue polypeptide in the lipid-free state is eluted as a monomer from gel filtration performed in aqueous buffer (Fig. 6B ▶), and its apparent molecular weight is not modified by the presence of phospholipids. This suggests that phospholipids do not create a stable complex with the polypeptide chain, but it is likely that the more hydrophobic environment created by the aliphatic chains may favor the formation of the helical structure. The 1–93 polypeptide can therefore maintain a soluble state in physiological buffer, and it is likely that it could circulate in this state in plasma.
Fig. 6.
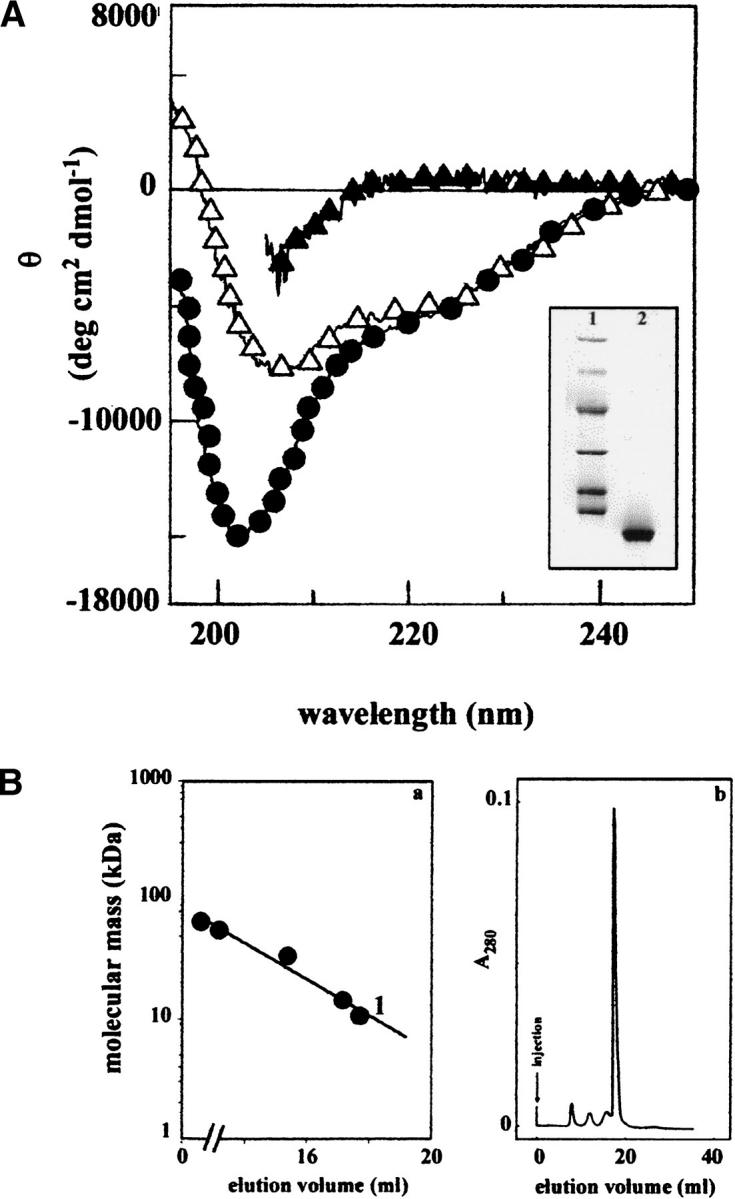
(A) CD spectra in the far-UV range of peptide 1–93 purified from PER natural fibrils. Sample in 5 M GdnHCl (filled triangle), in 0.1 M phosphate buffer (pH 7.5; filled circle), and in the presence of phospholipids (open triangle). (Inset) SDS-PAGE analysis of the polypeptide 1–93 used for all of the spectroscopic analysis (lane 2); in lane 1 are reported the molecular mass standards (phosphorylase b = 97.4 kD, bovine serum albumin = 66.2 kD, ovalbumin = 45.0 kD, carbonic anhydrase = 31.0 kD, soybean trypsin inhibitor = 21.5 kD, lysozyme = 14.4 kD). (B) (a) Calibration of gel filtration chromatography. Standard proteins: bovine serum albumin (66.2 kD); transthyretin (55.5 kD); A1 protein (34.2 kD); lysozyme (14.4 kD). 1, apoA1 1–93 (10.7 kD). (b) Gel filtration in 0.1 M phosphate buffer (pH 7.5) of polypeptide 1–93 solubilized from fibrils. The gel filtration was performed on a Superose 12 HR column (Pharmacia) having a bed dimension of 10 × 300 mm. Samples were injected in a volume of 0.5 mL. The apoA1 sample contained approximately 0.1 mg of the polypeptide 1–93.
Applying the algorithm of Chou and Fasman (1974) for the prediction of secondary structure, we would expect that the C-terminal part of the 93-residue polypeptide might be the region of the peptide more prone to acquire a helical conformation. The C-terminal part of the 1–93 apoA1 polypeptide contains a sequence that, according to the algorithm proposed by Lupas et al. (1991), should have a high tendency to assume a coiled-coiled conformation (Fig. 7 ▶). The NMR spectra of this apoA1 fragment, however, did not show any evidence of structure in aqueous solution.
Fig. 7.

Coils output for apoA1 sequence of 1–93. Prediction of coiled coil structure in apoA1 1–93 polypeptide according to Lupas et al. (1991). P (S) = probability of forming coiled coils. Windows of 14, 21, and 28 residues are reported.
The typical pattern of a disordered polypeptide was observed in 1D spectra, with all resonances clustered around the statistically random chemical shift values of aminoacyl residues (Wüthrich 1986). The 2D TOCSY and NOESY spectra confirmed this conclusion. The TOCSY fingerprint region did not show the frequency spreading of HN-Hα connectivities that could be expected if nearly 60% of the residues (Table 1) had adopted a secondary structure (at least), but rather an extended patch of cross-peaks collapsing into an unresolvable overlap (Fig. 8 ▶). The corresponding region in the NOESY spectrum exhibited only a few weak correlations, similar to the aromatic region where only side-chain primary amide cross-peaks from syn-anti exchange had remarkable intensity. Although the sequential amide NOESY pattern from the limited amount of expected helical structure (5%, according to Table 1) might have been lost because of unfavourable resonance overlap, the scarcity of NOESY connectivities even within the aromatic groups (at variance with the cross-peaks of the corresponding TOCSY region) matched with the poorness of dipolar connectivities obtained in the fingerprint and in the entire aliphatic region. Because the NMR spectra of a 93-residue peptide, albeit devoid of structure for 40% of the sequence, are expected to exhibit substantial NOE and chemical shift dispersion from the remaining structured portion of the molecule (especially considering the presence of ∼30% of β structure), the lack of either feature suggests that apoA1 1–93 is a flexible, unfolded polypeptide chain under the experimental conditions of the NMR measurements. The conflict of this finding with the results obtained from CD spectra (Table 1) may be ascribed to the limits of CD deconvolution because of the particular reference database of curve sets used for the fitting, the specific content of aromatic residues (9% for apoA1 1–93), and tertiary structure effects. The actual differences in the experimental conditions used for the CD and NMR experiments (i.e., temperature, pH, ionic strength, and concentration) should have affected their outcomes only marginally. The observed increase in helical structure within the 1–93 sample in the presence of phospholipids, detected by CD, seems convincing, but it was not possible to submit the NMR sample to the same refolding procedure as used for the CD sample with phospholipids because the presence of 10 mM Tris HCl, 20 mM POPC, and 27 mM sodium cholate would have hampered the observation of the 1H resonances from a peptide solution with a concentration below 1 mM. Performing NMR experiments in conditions analogous to those of the CD spectra would require 15–13C-labeled apoA1 samples.
Table 1.
Percentage of secondary structure according to the algorithm of Yang et al. (1986)
| Secondary structure | ApoA1 1-93 (5 M Guanidine, pH 7.5) | ApoA1 1-93 (100 mM Phosphate, pH 7.5) | ApoA1 1-93 (100 mM Phosphate, pH 7.5 and phospholipids) |
| Helix | 5% | 13% | |
| β sheet | 32% | 30% | |
| Turn | 19% | 18% | |
| Random | 100% | 44% | 39% |
| Total | 100% | 100% | 100% |
Fig. 8.

Fingerprint region of 2D TOCSY spectrum of apoA1 in aqueous phosphate showing extensive overlap of the HN-Hα connectivities.
Discussion
The mechanism of fibrillogenesis of apoA1 is a complex multistep process that probably involves a combination of different events, including protein misfolding, proteolytic remodelling, and fibrillogenesis of the N-terminal polypeptide. The ex vivo fibrils isolated from two unrelated patients affected by apoA1 amyloidosis and presenting the same Leu-to-Ser mutation in position 174 of apoA1 can shed light on the mechanism of fibrillogenesis of this protein. In both cases the fibrils were collected from the heart after it had been surgically removed under the transplantation procedure.
The tissue and the proteins were therefore not exposed to postmortem proteolytic activity. The perfect identity of the length of the polypeptide that constitutes the main fibrillar component in these two patients is very significant and may have been favored by the unique circumstances of sampling. In other proteins such as transthyretin (Hermansen et al. 1995), immunoglobulin light chains (Buxbaum 1992; Bellotti et al. 2000), and previously described apoA1 variants (Soutar et al. 1992; Booth et al. 1995; Booth et al. 1996) the polypeptide length from case to case is heterogeneous and many fragments coexist in the same samples. Our previous observation of a specific cleavage site between residues Val93 and Lys94 associated with the Leu174Ser replacement is therefore confirmed. Although a certain heterogeneity in the cleavage site of amyloidogenic apoA1 has been reported, the identification of the high susceptibility of the Val93-Lys94 bond or of the nearby peptide bonds to pathological cleavage is supported by the investigation of Pepys and colleagues, who have performed extensive chemical characterization of different ex vivo apoA1 amyloid fibrils (Soutar et al. 1992; Booth et al. 1995; Booth et al. 1996). The other recently reported case in which the amyloidogenic mutation in apoA1 occurs at position 173 and outside of the fibril-forming polypeptide, described by Benson and coworkers (Hamidi Asl K. et al. 1999), also supports this idea and has particular similarity to our reported cases with the Leu174Ser mutation because the effect of mutation is the release of an N-terminal polypeptide of ∼90 residues that accumulates as fibrillar material in the skin and heart. It is likely that the N-terminal polypeptide of this length is released physiologically at a low rate; in fact it is likely that it is a ubiquitous constituent of atherosclerotic plaques affecting blood vessels in the general population (Westermark et al. 1995).
We have previously hypothesized that the mutation in position 174 could favor the release of the N-terminal polypeptide 1–93, randomly from the wild-type or the mutant species. According to that hypothesis, in these heterozygous patients, we would expect a 50% distribution of the wild type and mutant in circulating HDL. On the contrary, we have found that in both our patients the mutant represents only ∼25% of all of the circulating apoA1 and therefore, in a stochastic distribution, we would expect a single pathogenic polypeptide in each apoA1 tetramer. In both patients, we observed an increase in the level of plasma apoA1 after heart transplantation; however, this did not reach a normal level. The ratio of the wild-type to mutant chain in the apoA1 purified from the plasma of one of the two patients did not change after the heart transplant (data not shown).
We have performed a similar analysis on a sample of apoA1 presenting the mutation Leu60Arg previously described by Soutar et al. (1992; material kindly provided by Prof. Mark Pepys), and we detected even less of the variant protein: The variant represented ∼10% of the total apoA1 (data not shown). An imbalance in the ratio of wild-type to variant protein was shown for the first time by Rader's group (Rader et al. 1992) on the basis of separation of plasma wild-type and Arg26Gly apoA1 isoforms by bidimensional electrophoresis and by metabolic studies. Isoelectric focusing separation performed in the cases of Leu60Arg (Soutar et al. 1992) and Arg26Gly apoA1-related amyloidosis (Nichols et al. 1988), as well as other metabolic studies (Genschel et al. 1998), confirms those findings, and in our study the quantification of the amyloidogenic and nonamyloidogenic species present within circulating apoA1 is reported. In addition to the increased extracellular metabolism demonstrated by Rader (1992) and Genschel et al. (1998), a disequilibrium between wild-type and variant proteins might be caused by reduction in the secretion of the pathogenic species brought about by the quality-control functions of the cell (Ellgaard et al. 1999). The higher percentage of Leu174Ser compared with the Leu60Arg variant would suggest that slightly different mechanisms are responsible for the abnormal protein metabolism in these two variants that for the following reason we consider to be two prototypic amyloidogenic variants of apoA1. The second carries the mutation inside the amyloidogenic polypeptide; the first has a mutation outside the fibrillogenic peptide but in a position able to affect the conformation of the antiparallel helical chain in the vicinity of the cleavage site. The peculiar stoichiometry of wild-type and mutant protein in circulating apoA1 should also be taken into account in future studies that investigate the abnormal structure and folding dynamics of amyloidogenic apoA1.
Proteins obtained from fibrillar deposits can be solubilized in chaotropic solvents, and the kinetics of their refolding pathways can yield information about their structural and functional properties (Booth et al. 1997; Bellotti et al. 1998). A similar approach was followed in this study, in which the Leu174Ser mutation has caused the massive production and deposition in the heart of a precisely cleaved 1–93 apoA1 fragment. Preliminary data regarding the structure and solubility of the polypeptide 1–93 purified from the natural amyloid fibrils are described here for the first time and have several physiopathological implications. This polypeptide can be solubilized from amyloid fibrils and analyzed by CD and NMR. Both techniques indicate that in phosphate buffer the polypeptide has a predominantly random coil conformation. On the contrary, full-length apoA1 purified from plasma and submitted to an identical procedure recovers a spectrum of predominantly helical structure (data not shown), as would be expected, given the helical nature of the native protein (Borhani et al. 1997). According to the CD analysis, the presence of phospholipids enhances the acquisition of a limited helical conformation, which most likely involves the C-terminal end of the peptide. The polypeptide is soluble in aqueous buffer and is eluted as a monomer in gel filtration chromatography. This suggests that the polypeptide could be produced and deposited in completely different compartments connected through the physiological fluids.
This is the first time that evidence from X-ray fiber diffraction studies has been found for residual ordered, oriented and repeating helical structure within ex vivo amyloid fibrils. The position of the 5.1- and 2.5-Å reflections on the equator of the patterns indicates that the helical structure extends at right angles to the fibril long axis.
Most amyloid fiber diffraction patterns are dominated by the meridional reflection at ∼4.7 Å because this arises from a very regular spacing that is repeated over long distances in the fibril. In the patterns from Leu174Ser apoA1 this is not the case, with the 5.1-Å equatorial reflection having a significant relative intensity.
It is possible that part of the helical contribution to the diffraction pattern could come from residual full-length, native apoA1 associated with the fibrils. If this is the case, then it can be inferred that the full-length material is associated in an integral, regular fashion with the cross-β core of the fibrils. However, given the fact that the full-length material represents only a minor component of this fibrillar material, and the observed relative intensities of the interstrand- and helical-derived reflections, it seems more likely that this helical component is contributed by a part of the 1–93 N-terminal fragment.
This suggests that the helical component of the fibrillogenic peptide may play an important role in the pathogenic structure. The morphology and staining characteristics of the amyloid fibrils from the two patients carrying the 174 mutation, as well as the main cross-β reflections, indicate that these fibrils do conform to the classical amyloid structure. However, for the first time, we have obtained an indication of the distinct nature of these apoA1 fibrils, which accommodate oriented, ordered helical components within their generic cross-β amyloid conformation. The study of the structural conversion of this polypeptide into the classical β-structure of amyloid fibrils should elucidate the role of the limited α-helical structure in the process of fibril formation and the maintenance of the stable fibrillar form.
Materials and methods
Case report
The patient (PER) presented at the age of 50 yr with exertional dyspnea and recurrent severe arrhythmia requiring positioning of a cardiac pacemaker. Diagnosis of amyloid cardiomyopathy was established on a heart biopsy. The patient's mother died at the age of 64 yr from congestive heart failure; a laryngeal biopsy performed at the age of 55 yr for progressive dysphony had revealed amyloid deposits that were not further characterized.
In a screening for putative mutations in genes related to hereditary systemic amyloidosis, we found a G-to-C transition at position 2069, producing a Leu-to-Ser substitution in position 174 of mature apoA1, previously identified in another patient (PIC). The patient underwent heart transplantation at the age of 53 yr and he is well 6 mo after surgical procedure. A low level of apoA1 was found in the plasma of both patients before the transplant procedure, 37 mg/dL in patient PIC and 80 mg/dL in patient PER, the reference range is 110–205 mg/dL. After the heart transplant, the apoA1 level increased to 60 mg/dL in PIC and 90 mg/dL in PER. Immunoelectron microscopy characterization of amyloid fibrils on heart tissue revealed strong reactivity with an anti-apoA1 antibody (rabbit anti-human apoA1 supplied by Genzyme). The immunostaining was negative by using antisera against immunoglobulin light-heavy chains, β2-microglobulin, lysozyme, transthyretin, fibrinogen, and amyloid A.
Fibril extraction
Fibrils were isolated from heart tissue by the classical water extraction procedure (Pras et al. 1968) in the presence of 1.5 mM phenylmethylsulfonyl fluoride (PhMeSO2F) after repeated homogenization in 2 mL of 10 mM Tris/EDTA 140 mM NaCl/ 0.1%NaN3 (pH 8) containing 1.5 mM PhMeSO2F/100 mg of tissue and centrifugation at 60,000g in Beckman L8–704 ultracentrifuge for 30 min. A pool of the IV and V water extraction fractions was used for further studies. All the material was birefringent at the polarized light when stained by Congo red.
X-ray fiber diffraction
The fibrils were kept at 4°C and X-ray was performed 1 wk after the extraction procedure.
Droplets of fibril-containing solutions were suspended between the ends of two wax-filled capillaries and allowed to dry slowly in air, at room temperature, yielding clumps of partially aligned fibrils (Serpell et al. 1999). These clumps were ∼0.25 mm3 and contained ∼20 μg of protein. X-Ray fiber diffraction images were collected in the Department of Biochemistry, University of Cambridge by using a Rigaku CuKα rotating anode source (wavelength 1.5418 Å) and a R-AXIS IV detector. Fibril clumps were aligned vertically in the beam.
Plasma apoA1 purification
ApoA1 was purified from plasma of the two patients, and the data reported in this study refer to samples obtained before the heart transplant. The purification was performed following the procedure of Sattler et al. (1994), and purified apoA1 migrates as a single band on a 12% SDS–polyacrylamide gel (inset in Fig. 5 ▶). The SDS-PAGE was performed in reducing conditions and stained by Coomassie brilliant blue.
Mass determination
Mass spectra were obtained on a Finnigan LCQ ion trap mass spectrometer (Finnigan) with electrospray ionization (ESI). The ESI spectra (positive ion mode) were collected from samples of ∼20 pmole/μL. The instrument was calibrated by caffeine (m/z = 195), the peptide MRFA (m/z = 525), and ultramark (m/z = 1022, 1122, 1222, 1322, 1422, 1522, 1622). Introduction of the solution into the ESI source was obtained via the syringe pump of the instrument at a flow rate of 5 μL/min for flow injection analyses. The same instrumentation coupled with a HPLC system (P4000, TSP Thermoquest) was used for HPLC-MS analysis.
The data were acquired in full-scan mode over the 300–2000 m/z range.
Multicharged ions resulting from mass spectra were processed through a deconvolution program to give a mass-range spectra.
The average masses of samples were calculated by using the Xcalibur, Biowork software.
Peptide preparation and separation
Amyloid fibrils or apoA1 purified from plasma were digested by Staphylococcus V8 protease (Pierce) that in bicarbonate buffer (50 mM at pH 8.0, 2mM EDTA) cleaves specifically at the C-terminal side of glutamate residues. An enzyme:protein ratio of 1:50 was used and the reaction was incubated at 37°C for 12 h. Peptides injected in a volume of 0.1 mL were separated by reversed phase HPLC through a C18 column (Vydac) at a flow of 1.5 mL/min. The mobile phases used were as follows: solvent A (0.05% TFA in water) and solvent B (0.05% TFA in acetonitrile). Peptides were eluted by a gradient of 5%–100% solvent B over 60 min. The elution of peptides was monitored by UV absorbance at 220 nm and by diversion of 20% of the column flow into the mass spectrometer. The same HPLC conditions were used for LC-MS analyses as well.
Solubilization of the polypeptide 1–93 and refolding procedure
The main protein constituent of amyloid fibrils of patients PER and PIC was purified as previously described (Obici et al. 1999) by gel filtration in 6 M GdnHCl.
GdnHCl was removed by extensive dialysis against H20 and the material separated into two aliquots. One sample was dissolved in 0.1 M phosphate buffer (pH 7.5), and another sample was submitted to the procedure of reconstitution of a protein–lipid complex following a modification of the method described previously by Sparks et al. for apoA1 (1992). In brief, POPC (supplied by SIGMA) was dissolved in CHCl3 in a conical glass tube and dried under nitrogen. Buffer containing 10 mM Tris-HCl (pH 8) with 1 mM EDTA, 1 mM NaN3, and 150 mM NaCl was added to obtain a concentration of POPC of 20 mM. Sodium cholate in Tris saline was added to give a molar ratio POPC:cholate of 1:1.35 and the mixture vortexed for 3 min. The dispersion was incubated at 37°C for 90 min before the addition of apoA1 polypeptide 1–93 (3 mg) and further incubation for 1 hr at 37°C with gentle shaking. A molar ratio POPC/polypeptide 1–93 of 100/L was used. The identical procedure, but without the addition of the apoA1 polypeptide was used to prepare a control buffer for the CD spectra.
Circular dichroism
Circular dichroism measurements were performed with a Jasco 710 spectropolarimeter. The instrument was calibrated by using a 0.06% (w/v) solution of ammonium d-10-camphorosulfonate. Measurements were performed at 20°C in cells with 0.1-cm path length in the far UV (195–250nm) at a protein concentration of 16 μM for all the samples with the exception of the sample containing 5 M GdnHCl, which required a 0.01-cm path-length cell and a protein concentration of 56 μM. Ellipticity was recorded every 1 sec with a response time of 1 sec and a bandwidth of 2 nm. Ellipticities in deg cm2 dmole-1 were expressed as (θ).
NMR spectroscopy
1H NMR experiments on the apoA1 fragment 1–93 were performed at 298 K with a Bruker Avance spectrometer operating at 500 MHz. The peptide concentration was ∼0.4 mM in 10 mM phosphate buffer (pH 7.2) with 10% D2O. 2D TOCSY (Braunschweiler & Ernst 1983) and NOESY (Jeener et al. 1979) spectra were acquired over 2048 points in t2 and 512 points in t1 with 8012.82 Hz spectral width in both dimensions. The mixing time intervals were 30 ms for TOCSY (DIPSI-2 spin-lock (Shaka et al. 1988), γB2/2π = 10.1 kHz) and 100 ms for NOESY. In all the experiments the solvent resonance was suppressed by excitation sculpting (Hwang and Shaka 1995) by using a repeated [hard- 180–soft-180] WATERGATE element (Piotto et al. 1992) with a water-selective Gaussian-shaped pulse of 4 ms. Data processing was performed by use of the software Felix (MSI).
Acknowledgments
We are grateful to Prof. Mark B. Pepys (Centre for Amyloidosis Acute Phase Proteins, Department of Medicine, Royal Free and University College Medical School, London) for kindly providing us with the plasma apoA1 with Leu60Arg mutation.
This work was supported by MURST (PRIN project: protein folding and misfolding), by Università di Pavia (progetto di Ateneo) IRCCS Policlinico S. Matteo, Pavia and CNR. M. Sunde holds a Royal Society University Research Fellowship.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ``advertisement'' in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
TIC, total ion current
ESI-MS, electrospray ionization mass spectrometry
apoA1, apolipoprotein A1
POPC, palmitoyl-oleoyl phosphatidylcholine
CD, circular dichroism
HDL, high density lipoprotein
NOE, nuclear Overhauser effect
TOCSY, total correlation spectroscopy
NOESY, NOE spectroscopy
1D, one dimensional
2D, two dimensional
SAP, serum amyloid-P component.
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/ps.29201.
References
- Bellotti, V., Stoppini, M., Mangione, P., Sunde, M., Robinson, C., Asti, L., Brancaccio, D., and Ferri, G. 1998. β2-Microglobulin can be refolded into a native state from ex vivo amyloid fibrils. Eur. J. Biochem. 258 61–67. [DOI] [PubMed] [Google Scholar]
- Bellotti, V., Mangione, P., and Stoppini, M. 1999. Biological activity and pathological implications of misfolded proteins. Cell. Mol. Life Sci. 55 977–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellotti, V., Mangione, P., and Merlini, G. 2000. Immunoglobulin light chain amyloidosis : The archetype of structural and pathogenic variability. J. Struct. Biol. 130 280–289. [DOI] [PubMed] [Google Scholar]
- Booth, D.R., Tan, S.Y., Booth, S.E., Hsuan, J.J., Totty, N.F., Nguyen, O., Hutton, T., Vigushin, D.M., Tennent, G.A., Hutchinson, W.L., et al. 1995. A new apolipoprotein AI variant, Trp50 Arg, causes hereditary amyloidosis. QJM 88 695–702. [PubMed] [Google Scholar]
- Booth, D.R., Tan, S.Y., Booth, S.E., Tennent, G.A., Hutchinson, W.L., Hsuan, J.J., Totty, N.F., Truong, O., Soutar, A.K., Hawkins, et al. 1996. Hereditary hepatic and systemic amyloidosis caused by a new deletion/insertion mutation in the apolipoprotein AI gene. J. Clin. Invest. 97 2714–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth, D.R., Sunde, M., Bellotti, V., Robinson, C.V., Hutchinson, W.L., Fraser, P.E., Hawkins, P.N., Dobson, C.M., Radford, S.E., Blake, C.C., et al. 1997. Instability, unfolding and aggregation of human lysozyme variants underlying amyloid fibrillogenesis. Nature 385 787–793. [DOI] [PubMed] [Google Scholar]
- Borhani, D.W., Rogers, D.P., Engler, J.A., and Brouillet, C.G. 1997. Crystal structure of truncated human apolipoprotein A-I suggests a lipid bound conformation. Proc. Natl. Acad. Sci. 94 12291–12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunschweiler, L. and Ernst, R.R. 1983. Coherence transfer by isotropic mixing: Application to proton correlation spectroscopy. J. Magn. Reson. 53 521–528. [Google Scholar]
- Buxbaum, J. 1992. Mechanisms of disease: Monoclonal immunoglobulin deposition. Amyloidosis, light chain deposition disease, and light and heavy chain deposition disease. Hematol. Oncol. Clin. North Am. 6 323–346. [PubMed] [Google Scholar]
- Chou, P.Y. and Fasman, G.D. 1974. Prediction of protein conformation. Biochemistry 13 222–245. [DOI] [PubMed] [Google Scholar]
- Damas, A., Sebastião, M.P., Domingues, F.S., Costa, P.P., and Saraiva, M.J. 1995. Structural studies on FAP fibrils: removal of contaminants is essential for the interpretation of X-ray data. Amyloid, Intl. J. Exp. Clin. Invest. 2 173–178. [Google Scholar]
- de Sousa, M.M., Vital, C., Ostler, D., Fernandes, R., Pouget-Abadie, J., Carles, D., and Saraiva, M.J. 2000. Apolipoprotein AI and transthyretin as components of amyloid fibrils in a kindred with apoA1 Leu178His amyloidosis. Am. J. Pathol. 156 1911–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellgaard, L., Molinari, M., and Helenius, A. 1999. Setting the standards: Quality control in the secretory pathway. Science 286 1882–1888. [DOI] [PubMed] [Google Scholar]
- Genschel, J., Haaas, R., Propsting, J.M., and Schmidt, H.H.J. 1998. Apolipoprotein A-I induced amyloidosis. FEBS Lett. 430 145–149. [DOI] [PubMed] [Google Scholar]
- Glomset, J.A. 1968. The plasma lecithins: Cholesterol acyltransferase reaction. J. Lipid Res. 9 155–167. [PubMed] [Google Scholar]
- Hamidi Asl, L., Liepnieks, J.J., Hamidi Asl, K., Uemichi, T., Moulin, G., Desjoyaux, E., Loire, R., Delpech, M., Grateau, G., and Benson, M.D. 1999. Hereditary amyloid cardiomyopathy caused by a variant apolipoprotein A1. Am. J. Pathol. 154 221–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamidi Asl, K., Liepnieks, J.J., Nakamura, M., Parker, F., and Benson, M. 1999. A novel apolipoprotein A1 variant, Arg173Pro, associated with cardiac and cutaneous amyloidosis. Biochem. Biophys. Res. Commun. 257 584–588. [DOI] [PubMed] [Google Scholar]
- Hermansen, L.F., Bergman, T., Jornval, H., Husby, G., Ranlov, I., and Sletten, K. 1995. Purification and characterization of amyloid related transthyretin associated with familial amyloidotic cardiomyopathy. Eur. J. Biochem. 227 772–779. [DOI] [PubMed] [Google Scholar]
- Hwang, T.L., and Shaka, A.J. 1995. Water suppression that works. Excitation sculpting using arbitrary waveforms and pulsed field gradients. J. Magn. Reson. A 112 76–80. [Google Scholar]
- Jeener, J., Meier, B.H., Bachmann, P., and Ernst, R.R. 1979. Investigation of exchange processes by two dimensional NMR spectroscopy. J. Chem. Phys. 71 286–292. [Google Scholar]
- Johnson, K.H., Sletten, K., Hayden, D.W., O'Brien, T.D., Roertgen, K., and Westermark, P. 1992. Pulmonary vascular amyloidosis in aged dogs: A new form of spontaneously occurring amyloidosis derived from apolipoprotein A1. Am. J. Pathol. 141 1013–1019. [PMC free article] [PubMed] [Google Scholar]
- Lupas, A., Van Dyke, M., and Stock, J. 1991. Predicting coiled coils from protein sequence. Science 252 1162–1164. [DOI] [PubMed] [Google Scholar]
- Nichols, W.C., Dwulet, F.E., Lipnieks, J., and Benson, M.D. 1988. Variant apolipoprotein A-I as a major constituent of a human hereditary amyloid. Biochem. Biophys. Res. Commun. 156 762– 768. [DOI] [PubMed] [Google Scholar]
- Obici, L., Bellotti, V., Mangione, P., Stoppini, M., Arbustini, E., Verga, L., Zorzoli, I., Anesi, E., Zanotti, G., Campana, C., et al. 1999. The new apolipoprotein A-I variant Leu(174) → Ser causes hereditary cardiac amyloidosis, and the amyloid fibrils are constituted by the 93-residue N-terminal polypeptide. Am. J. Pathol. 155 695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persey, M.R., Booth, D.R., Booth, S.E., van Zyl-Smit, R., Adams, B.K., Fattaar, A.B., Tennent, G.A., Hawkins, P.N., and Pepys, M.B. 1998. Hereditary nephropathic systemic amyloidosis caused by a novel variant apolipoprotein A-I. Kidney Int. 53 276–281. [DOI] [PubMed] [Google Scholar]
- Piotto, M., Saudek, V., and Sklenar, V. 1992. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 2 661–665. [DOI] [PubMed] [Google Scholar]
- Pras, M., Schubert, M., Zucker Franklin, D., Rimon, A., and Franklin, E.C. 1968. The characterization of soluble amyloid prepared in water. J. Clin. Invest. 47 924–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rader, D.J., Gregg, R.E., Meng, M.S., Schaefer, J.R., Zech, L.A., Benson, M.D., and Brewer, B.H. 1992. In vivo metabolism of a mutant apolipoprotein, ApoA-I Iowa, associated with hypoalphalipoproteinemia and hereditary systemic amyloidosis. J. Lipid Res. 33 755–763. [PubMed] [Google Scholar]
- Sattler, W., Mohr, D., and Stocker, R. 1994. Rapid isolation of lipoproteins and assessment of their peroxidation by high-performance liquid chromatography postcolumn chemiluminescence. Methods Enzymol. 233 469–489. [DOI] [PubMed] [Google Scholar]
- Serpell, L.C., Fraser, P.E., and Sunde, M. 1999. X-ray fiber diffraction of amyloid fibrils. Methods Enzymol. 309 526–536. [DOI] [PubMed] [Google Scholar]
- Shaka, A.J., Lee, C.J., and Pines, A. 1988. Iterative schemes for bilinear operators: Application to spin decoupling. J. Magn. Reson. 77 274–290. [Google Scholar]
- Soutar, A.K, Hawkins, P.N., Vigushin, D.M., Tennent, G.A., Booth, S.E., Hutton, T., Nguyen, O., Totty, N.F., Feest, T.G., Hsuan, J.J., et al. 1992. Apolipoprotein AI mutation Arg-60 causes autosomal dominant amyloidosis. Proc. Natl. Acad. Sci. 89 7389–7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks, D.L., Philips, M.C., and Lund-Katz, S. 1992. The conformation of Apolipoprotein A-I in discoidal and spherical recombinant high density lipoprotein particles. J. Biol. Chem. 267 25830–25838. [PubMed] [Google Scholar]
- Sunde, M., Serpell, L.C., Bartlam, M., Pepys, M.B., Fraser, P.E., and Blake, C.C.F. 1997. The common core structure of amyloid fibrils by synchrotron X-ray diffraction. J. Mol. Biol. 273 729–739. [DOI] [PubMed] [Google Scholar]
- Westermark, P., Mucchiano, G., Marthin, T., Johnson, K.H., and Sletten, K. 1995. Apolipoprotein AI derived amyloid in human aortic atherosclerotic plaques. Am. J. Pathol. 147 1186–1192. [PMC free article] [PubMed] [Google Scholar]
- Wüthrich, K. 1986. NMR spectroscopy of proteins and nucleic acids. Wiley & Sons, New York.
- Yang, J.T., Wu, C.C., and Martinez, H.M. 1986. Calculation of protein conformation from circular dichroism. In Methods in enzymology (eds. C.W. Hirs and S.N. Timasheff), Vol. 130, pp 208–269. Academic, San Diego. [DOI] [PubMed]