

Abstract
Based on the principle of dual prediction by segment hydrophobicity and nonpolar phase helicity, in concert with imposed threshold values of these two parameters, we developed the automated prediction program TM Finder that can successfully locate most transmembrane (TM) segments in proteins. The program uses the results of experiments on a series of host-guest TM segment mimic peptides of prototypic sequence KK AAAXAAAAAXAAWAAXAAAKKKK-amide (where X = each of the 20 commonly occurring amino acids) through which an HPLC-derived hydropathy scale, a hydrophobicity threshold for spontaneous membrane insertion, and a nonpolar phase helical propensity scale were determined. Using these scales, the optimized prediction algorithm of TM Finder defines TM segments by first searching for competent core segments using the combination of hydrophobicity and helicity scales, and then performs a gap-joining operation, which minimizes prediction bias caused by local hydrophilic residues and/or the choice of window size. In addition, the hydrophobicity threshold requirement enables TM Finder to distinguish reliably between membrane proteins and globular proteins, thereby adding an important dimension to the program. A full web version of the TM Finder program can be accessed at http://www.bioinformatics-canada.org/TM/.
Keywords: TM Finder, transmembrane segments, threshold hydrophobicity, nonpolar phase helicity, membrane protein prediction
Advances in DNA cloning and sequencing techniques have allowed the rapid derivation of many protein sequences. However, the extreme hydrophobic nature of most membrane proteins have made them difficult targets for detailed structural analysis by techniques such as nuclear magnetic resonance (NMR) or X-ray crystallography. Among the nearly 10,000 entries in the current PDB database (http://pdb. pdb.bnl.gov), only a handful are membrane proteins solved at atomic resolution (e.g., Henderson et al. 1990; Deisenhofer et al. 1984; Tsukihara et al. 1996; Doyle et al. 1998). Because of the lack of generally suitable routes to high resolution analysis, model construction and computer simulation have become necessary tools for understanding various detailed interactions within the membrane domain. Consequently, as a first step toward model building, the accurate delineation of sequences and their secondary structures within membranes becomes a prerequisite.
In the absence of high-resolution structural data, the approximate positions of the membrane-spanning segments within a TM domain can, in principle, be proposed from the amino acid sequence alone with the aid of hydropathy plots (Kyte and Doolittle 1982), which select TM segments based on a moving average of segment hydrophobicity. The development of such hydrophobicity scales traditionally has been based on a combination of statistical analysis (Munoz and Serrano 1994), and the partitioning properties of individual amino acids both as monomers and in globular proteins (Kyte and Doolittle 1982), and in short hydrophilic peptides (Hodges et al. 1994). We have previously reported the experimental determination of apparent hydrophobicity using a series of TM-mimetic model peptides of prototypic sequence KKAAAXAAAAAXAAWAAXAAAKKKK-amide (the underlined region represents the hydrophobic core of sufficient length to span a lipid bilayer, and the guest residues X = each of the 20 common amino acids) (Liu and Deber 1998a). This work led to a hydrophobicity scale for the 20 X-residues based on the individual high-pressure liquid chromatography (HPLC) retention time measurements of the model peptides. When corresponding peptide secondary structures were characterized in aqueous versus micellar environments, peptides exhibited random or partially helical structures in water, but adopted X-residue–dependent full helical structure only upon integration into the micellar membrane. These experiments revealed the existence of a threshold hydrophobicity approximately equivalent to that of a strand of poly-alanine, which, once met or exceeded, dictates the spontaneous insertion of the peptides into micelles. When hydropobicities of natural protein sequences in SWISS-PROT and TMbase were examined using the HPLC-based scale, and the threshold applied, it was found that close to 97% of protein TM segments have hydrophobicity above the threshold value, while nearly 80% of non-TM helices (>19 residues, derived from soluble proteins) fail to meet the same minimum requirement (Liu and Deber 1998a). Therefore, threshold hydrophobicity is an attribute that can be used to distinguish TM segments from helices in soluble proteins.
While transmembrane segments generally are α-helical, peptide helicity itself is influenced fundamentally by molecular environment, and accordingly, designations of particular residues as helix formers or breakers will differ significantly in membrane versus soluble protein domains. This circumstance is epitomized by the fact that β-branched residues (Val, Ile, Thr) and Gly residues predicted to disfavor α-helices by algorithms evolved from statistical analysis of residues in soluble proteins (Chou and Fasman 1978) among them can account for ∼40% of TM segment amino acid composition (Li and Deber 1994). Thus, a helicity scale developed from globular proteins cannot be applied to membrane proteins. We addressed this issue by using circular dichroism (CD) measurements to examine the series of TM-mimetic model peptides in a nonpolar organic phase (n-butanol), from which a scale of relative helical propensity for the 20 amino acids was constructed (Liu and Deber 1998b). When this scale was applied to protein databases, we observed a clear segregation between TM and non-TM helices; for example, a helicity value set at midscale selects correctly for 98% of the TM helices in a large database (Wang et al. 1999). These results suggested that in addition to the threshold hydrophobicity, there also exists a de facto minimum residue-dependent helicity requirement, which will promote and stabilize the folding of the nascent polypeptide chain within the membrane environment.
We have developed the program TM Finder, which automates and implements the concept of dual threshold hydrophobicity and nonpolar phase helicity requirements to locate protein TM segments from primary sequences alone. Here, we present the algorithm used for the TM Finder program, and discuss the refinements made to optimize the program for various protein sequences.
Construction of the TM Finder program
Development of the hydrophobicity scale
A hydrophobicity value for each amino acid was assigned according to the HPLC retention time of each peptide KKAAAXAAAAAXAAWAAXAAA-KKKK-amide (Liu and Deber 1998a), which in practice ranged from 22.58 min (Phe) to 18.88 min (Lys). To obtain a dimensionless relative scale between 5 and −5, retention times were converted to hydropathy values by the equation,
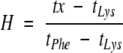 |
× 10 − 5,where H and Δt represent hydrophobicity and HPLC retention time, respectively. An exception was made for Cys, which was synthesized with only one Cys in the middle ``X,'' and two Leu residues at the other two guest positions, to avoid excessive disulfide formation. the hydropathy of Cys then was calculated according to
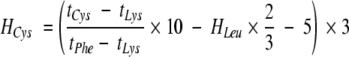 |
which corresponds to the hydropathy of a peptide with three Cys substitutions. The Liu-Deber hydrophobicity scale, with relative residue rankings, is presented in Table 1, column 1. Comparative residue rankings for three established hydropathy scales, those developed by Kyte and DooLittle (1982), Engelman et al. (1986), and Eisenberg et al. (1984), are presented in columns 3, 4, and 5.
Table 1.
Hydrophobicitya and helicityb scales, determined experimentally from the properties of KKAAAXAAAAAXAAWAAXAAAKKKK-amide peptides, and used in development of TM finder
| AA | Hydrophobicitya | Helicityb | KDc | GESd | Eisenberge |
| F | 5.00 (1) | 1.26 (4) | 2.8 (4) | 3.7 (1) | 1.19 (2) |
| W | 4.88 (2) | 1.07 (10) | −0.9 (11) | 1.9 (7) | 0.81 (5) |
| L | 4.76 (3) | 1.28 (2) | 3.8 (3) | 2.8 (4) | 1.06 (4) |
| I | 4.41 (4) | 1.29 (1) | 4.5 (1) | 3.1 (3) | 1.38 (1) |
| M | 3.23 (5) | 1.22 (6) | 1.9 (6) | 3.4 (2) | 0.64 (6) |
| V | 3.02 (6) | 1.27 (3) | 4.2 (2) | 2.6 (5) | 1.08 (3) |
| C | 2.50 (7) | 0.79 (19) | 2.5 (5) | 2 (6) | 0.29 (9) |
| Y | 2.00 (8) | 1.11 (8) | −1.3 (12) | −0.7 (13) | 0.26 (10) |
| A | 0.16 (9) | 1.24 (5) | 1.8 (7) | 1.6 (8) | 0.62 (7) |
| T | −1.08 (10) | 1.09 (9) | −0.7 (9) | 1.2 (9) | −0.05 (12) |
| E | −1.50 (11) | 0.85 (18) | −3.5 (18) | −8.2 (17) | −0.74 (15) |
| D | −2.49 (12) | 0.89 (16) | −3.5 (17) | −9.2 (19) | −0.9 (18) |
| Q | −2.76 (13) | 0.96 (13) | −3.5 (15) | −4.1 (15) | −0.85 (17) |
| R | −2.77 (14) | 0.95 (14) | −4.5 (20) | −12.3 (20) | −2.53 (20) |
| S | −2.85 (15) | 1.00 (11) | −0.8 (10) | 0.6 (11) | −0.18 (13) |
| G | −3.31 (16) | 1.15 (7) | −0.4 (8) | 1.0 (10) | 0.48 (8) |
| N | −3.79 (17) | 0.94 (15) | −3.5 (16) | −4.8 (16) | −0.78 (16) |
| H | −4.63 (18) | 0.97 (12) | −3.2 (14) | −3 (14) | −0.4 (14) |
| P | −4.92 (19) | 0.57 (20) | −1.6 (13) | −0.2 (12) | 0.12 (11) |
| K | −5.00 (20) | 0.88 (17) | −3.9 (19) | −8.8 (18) | −1.5 (19) |
a Hydrophobicity of each guest ``X'' residue, scaled from HPLC retention times (Liu and Deber 1998a).
b Nonpolar phase helical propensity of each guest ``X'' residue, scaled from circular dichroism measurements of peptides in n-butanol (Liu and Deber 1998b; Wang et al. 1999).
c Kyte and Doolittle (1982).
d Engelman et al. (1986).
e Eisenberg et al. (1984).
Development of the helicity scale
Helix propensity was assigned according to CD measurements in the nonpolar solvent n-butanol (Liu and Deber 1998b; Wang et al., 1999). In nonpolar solution, helic propensity was calculated by the equation,
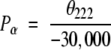 |
so that Pα = 1 corresponds to −30,000 deg cm2/dmol, a typical helicity value for a protein to adopt ∼50% α-helical conformation in a nonpolar environment (Liu and Deber 1998b; Parker et al. 1992). This helicity scale, with relative residue rankings is given in Table 1, column 2.
TM Finder parameters
The original TM Finder program was prototyped using Microsoft Visual Basic and Excel. A web-enabled version of the program written in C and perl is available free to all users at http://www.bioinformatics-canada.org/TM/. Figure 1 ▶ illustrates the operation of the web version of the TM Finder program. The program accepts protein sequences in SWISS-PROT format, and then outputs TM segments based on the combined prediction from hydrophobicity and helicity. Hydrophobicity and helicity values are assigned to each amino acid, and then a sliding window is applied to calculate the moving average. TM segments are selected on the basis of residues, which exceed both the imposed threshold hydrophobicity and helicity levels. A gap joining operation (vide infra) was implemented to ligate segments within short breaks from the main segments. Users may adjust five parameters depending on the size and nature of the protein to produce the best prediction. These five parameters are: N-terminal window size, C-terminal window size, minimum core length, closed-gap length, and minimum segment length. The default settings of these parameters are values obtained from optimizing the five parameters against a collection of solved membrane protein structures (training sets) which are collected on the TM Finder website. It is emphasized that the program is designed to evaluate proteins that contain transmembrane α-helices; membrane proteins such as the porins, which contain transmembrane β-strands as components of β-barrel structures, should be treated through algorithms developed specifically from this protein subset (Liu and Deber 1998a).
Fig. 1.

TM Finder output taken directly from the website (http://www.bioinformatics-canada.org/TM/). The program accepts the primary sequence in FASTA format as input, and generates a dual hydropathy/helicity plot as a function of protein primary sequence. Hydrophobicity values (red line) and helicity values (blue line) are based on input from the scales given in columns 1 and 2 of Table 1. TM segments (yellow bars) are predicted based upon considerations of segment length and gap size, in conjunction with the combined requirements of threshold hydrophobicity (>0.4) and threshold helicity (>1.1); the dual threshold is shown as a horizontal line drawn through the prediction profile. The example presented is chain III of cytochrome C oxidase (SWISSPROT accession number: P00415). The sequence used to generate this plot is 1MTHQTHAYHM VNPSPWPLTG ALSALLMTSG LTMWFHFNSM TLLMIGLTTN 51MLTMYQWWRD VIRESTFQGH HTPAVQKGLR YGMILFIISE VLFFTGFFWA 101FYHS SLAPTP ELGGCWPPTG IHPLNPLEVP LLNTSVLLAS GVSITWAH HS 151LMEGDRKHML QALFITITLG VYFTLLQASE YYEAPFTISD GVYGSTFFVA 201TGFHGLHVII GSTFLIVCFF RQLKFHFTSN HHFGFEAGAW YWHFVDVVWL 251FLYVSIYWWGS261. The plot using default parameters is shown for illustration.
Results and discussion
Principles of operation: Combination of hydrophobicity and helicity
The fundamental principle of the TM Finder prediction is that a candidate TM segment must satisfy both hydrophobicity and helicity thresholds (Liu and Deber 1999). As well, the use of peptide TM mimics offers an experimental alternative to guide formulation of hypotheses that explain/predict polypeptide conformation as a function of peptide primary sequence in the membrane environment. Based on peptide studies, the experimentally derived hydrophobicity and helicity scales are the two key elements of the TM Finder program. The hydrophobicity scale (Liu and Deber 1998a) used is distinct from conventional scales in several aspects. For example, this scale established the poor hydrophobicity of Gly, which ranks 16th out of 20 (Table 1), while KD, GES, and Eisenberg scales consider Gly as less hydrophilic (8th to 10th). In another instance, Trp is measured as highly hydrophobic (2nd), presumably due, in part, to its avid water-to-membrane transfer potential (Wimley and White 1996), but it ranks 5th to 11th in the other scales.
To a significant extent, hydrophobicity rankings of the 20 residues (Table 1, column 1) parallel helicity rankings (column 2). Inspection of the rankings reveals that five of the top six helix-promoting residues (F, L, I, M, and V) also are among the top six hydrophobes in the Liu-Deber scale. Yet a few residues do move drastically in their relative rankings, for example, the rankings of Gly and Trp in hydrophobicity (16th and 2nd, respectively) interchange versus their nonpolar phase helicity rankings (7th and 10th, respectively). As well, Ala is relatively less hydrophobic than it is a helix former in nonpolar phases. These circumstances suggest that hydrophobicity and helicity should be considered as uncoupled entities, and therefore the outcome of the prediction should improve when the two factors work in concert.
Gap-joining operation: Elimination of window size bias
Strict conformity to threshold hydrophobicity and helicity values can produce a lengthy list of predicted segments including many spikes. With the application of a small window size, we noted that the peaks tended to become narrow and break into several regions within one TM segment; such apparent shorter segments may arise in regions of local hydrophilicity (common to membrane transport proteins). In light of these observations, we modified TM Finder in the following manner. First, a variable m (usually set at 10) was set up, which allows the elimination of small segments with length m AA; and second, to resolve the problem of fragmentation when smaller window sizes are used, the program allows the fusion of neighboring smaller segments to a larger hydrophobic nucleus.
The importance of the gap-joining operation is demonstrated in Table 2. Cytochrome C oxidase had been chosen as the example because its crystal structure (Tsukihara et al. 1996) provides a good reference to determine the accuracy of the TM Finder prediction. As seen from Table 2, without the gap-joining operation (gap size = 0), two to three segments will be missed from the output. In addition, the prediction accuracy is highly dependent on the size of the sliding window chosen, i.e., the prediction accuracy is raised 7% simply by increasing the window size from 11 AA to 19 AA. With the application of gap size = 2 AA, all 12 segments of the protein are identified by the program, the prediction accuracy is improved by 6%–12%, and essentially the same level of accuracy is achieved regardless of the choice of window size. However, it must be noted that overuse of gap-joining operations can produce fused segments and result in reduced accuracy, as demonstrated in the case of gap size = 4 AA, and window size of 11 AA. The corrective action for fused segments is to reduce the gap size while at the same time increase the window size; when a larger window size is chosen, the resulting segments are more concentrated and have better separation. The eventual optimized choice (3 AA gap, 15 AA window) between gap size and window size that would allow the prediction of well-separated and reasonably sized segments is the choice that produces the best accuracy (81%).
Table 2.
Effect of gap-joining operations of TM finder
| Trial conditions | Optimized | ||||||
| Gap = 0AA | Gap = 2AA | Gap = 4AA | Gap = 3AA | ||||
| Solved Structure TM # | 11AA | 19AA | 11AA | 19AA | 11AA | 15AA | |
| 1 | 12–41 | 17–38 | 20–34 | 17–38 | 10–34 | 17–38 | 13–36 |
| 2 | 51–87 | 58–84 | 55–85 | 58–84 | 55–85 | 52–84 | 53–82 |
| 3 | 95–116 | XXX | XXX | 107–117 | 105–120 | 107–117 | 106–117 |
| 4 | 141–170 | 144–155 | 149–164 | 144–167 | 149–167 | 144–167 | 143–166 |
| 5 | 183–212 | 185–208 | 184–208 | 185–211 | 184–212 | 185–211 | 182–214 |
| 6 | 228–262 | XXX | XXX | 247–256 | 238–254 | 242–256 | 230–254 |
| 7 | 270–285 | 271–292 | 273–296 | 271–292 | 273–296 | 271–292 | 273–293 |
| 8 | 299–327 | XXX | 311–320 | 306–325 | 311–320 | 306–325 | 304–327 |
| 9 | 336–359 | 339–355 | 336–359 | 339–363 | 336–366 | 339–398 | 337–354 |
| 10 | 371–401 | 381–398 | 370–398 | 367–398 | 370–401 | '' | 359–399 |
| 11 | 407–433 | 412–423 | 408–427 | 412–423 | 408–427 | 412–428 | 410–426 |
| 12 | 445–478 | 460–475 | 454–481 | 455–475 | 454–481 | 451–475 | 453–475 |
| Accuracy | 64% | 71% | 76% | 77% | 79% | 81% | |
Prediction results on chain I of the solved structure cytochrome C oxidase (Tsukihara et al. 1996) with and without the gap-joining operation, using window sizes of 11 AA and 19 AA. The residual accuracy of prediction is calculated as P = 1 − (No + Nu) /Nt, where Nt represents the total number of amino acids in the protein, while No and Nu represent the number of overpredicted and underpredicted amino acids, respectively.
These protocols also act to improve the prediction for lower molecular weight proteins. For example, M13 coat protein has only 50 AA's; with the application of a window size = 19, its single TM segment is predicted to be at residues 19–38. With the modified protocol using window size = 7, the position of the TM segment is predicted to be 23–42 (Fig. 2 ▶), significantly closer to the result of residues 25–45 deduced experimentally from NMR studies (Papavoine et al. 1998).
Fig. 2.
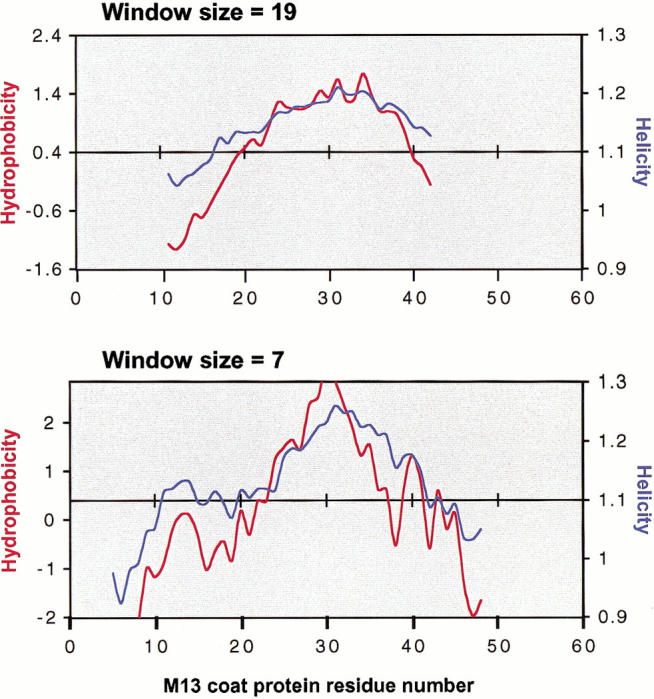
TM Finder prediction for M13 major coat protein, showing hydrophobicity (red trace) and helicity (blue trace). The sequence of the protein is 1AEGDDPAKAA 11FNSLQASATE 21YIGYAWAMVV 31VIVGATIGIK 41LFKKFTSKAS50. M13 major coat protein contains a single transmembrane segment that may extend maximally from 25–45 as deduced from nuclear magnetic resonance studies (Papavoine et al. 1998). Application of window size = 19 AA predicts the TM segment to occur at residues 19–38, while using a smaller window size = 7, the TM segment is identified at 23–42, closer to its experimentally determined location.
While TM segment prediction programs should, as indicated above, strive for the highest percent accuracy in identifying the actual TM segment residues, the notion of accuracy eventually will require an operational definition of membrane entry/exit points of protein segments. Given that a water/membrane interface is a broad cross-section of lipid substituents through which a full turn of α-helix could readily traverse at both N- and C-termini, it remains problematical to designate the precise residue membrane entry/exit points of a given TM segment. Although the helicity requirement used in the current work helps to refine prediction of entry/exit points (Liu and Deber 1999), it may ultimately become useful to distinguish between a core TM helix in the hydrocarbon phase, and a full TM helix, with the latter defined in terms of inclusion of the adjacent (usually hydrophilic) N- and C-capping helical regions.
Globular protein database used to refine the prediction
In order to further codify the power of TM Finder to distinguish between membrane and nonmembrane proteins, we constructed a database composed of helices from globular proteins and from aqueous-based domains of membrane proteins, restricting the analysis to segments of comparable length to TM segments. Application of the threshold hydrophobicity criterion to this latter database of non-TM helices produced a few outliers containing 3–4 charged residues, but for which hydropathy is raised to just above the threshold value because of (1) an increased occurrence of aromatic residues (Tyr, Phe, Trp); and (2) an increased amount of Leu residues as compared to the majority of globular protein helices (i.e., those below the threshold). We have termed such helices δ-regions (Wang et al. 2000), which we speculate may occur as helices that function to bridge core-to-surface regions in globular protein structures. To eliminate false positives from this source, TM Finder has been programmed to flag potential TM segments, which contain three or more charged residues per 19 AA's.
Application of TM Finder to databases of membrane and nonmembrane proteins
To assess the generic ability of TM Finder to locate TM segments of membrane proteins, we constructed two databases. We first applied the program to a database of 21 protein subunits (containing 75 reported TM segments) in a set of membrane proteins considered to be solved to high resolution (Table 3) (see references herein and the TM Finder website). Recognizing that segment entry/exit points will vary from the specific positions reported from crystallography (data not shown), TM Finder correctly locates 67 of 75 TM segments, while mispredicting three segments, for a predictive value of a positive test of 67/70 = 96%. The segments underpredicted by TM Finder likely result from its stringency in that both hydrophobicity and helicity profiles must exceed the threshold for a positive prediction; thus, the program will occasionally bypass a Gly-rich and/or hydrophilic residue-rich (e.g., His) TM segment for which it computes insufficient hydrophobicity (example from Table 3: TM 3 of photosystem I [PsaA], residues 197–221: LNHHLAGLLGLGSLAWAGHQIHVSL). Comparison with three other hydropathy scales (Table 3) indicates roughly comparable performance for TM segment prediction in membrane proteins, viz., the KD scale correctly locates 70 segments while mispredicting eight (predictive value = 90%); the GES scale found 66 while mispredicting five (93%), and the Eisenberg scale found 73 while mispredicting 15 (83%).
Table 3.
Transmembrane segment predictions in membrane proteins (min. segment length = 14)
| Protein | Accession No. | Reported No. TMs | TM Finder | Kyte Doolittle | GES | Eisenberg |
| Bacteriorhodopsin | 3659944 | 7 | 7 (0)* | 7 (0)* | 5 (0)* | 7 (0)* |
| Photoreaction Center (L chain) | P11846 | 5 | 5 (0) | 5 (0) | 5 (0) | 5 (0)* |
| Photoreaction Center (M chain) | P02953 | 5 | 5 (0) | 5 (0) | 5 (0) | 5 (0)* |
| Photoreaction Center (H chain) | P11846 | 1 | 1 (0) | 1 (1) | 1 (0) | 1 (3) |
| Light harvesting complexes (A, D, G, J) | P26789 | 1 | 1 (0) | 1 (0) | 1 (0) | 1 (0) |
| Light harvesting complexes (B, E, H, K) | P26790 | 1 | 1 (0) | 1 (0) | 1 (0) | 1 (0) |
| Photosystem I (PsaA) | P25896 | 11 | 8 (1) | 10 (2) | 11 (1)* | 10 (3) |
| Photosystem I (PsaB) | P25897 | 11 | 10 (0) | 10 (1) | 10 (1) | 11 (2)* |
| Photosystem I (PsaI) | P25900 | 1 | 1 (0) | 1 (0) | 1 (0) | 1 (0) |
| Photosystem I (PsaJ) | P25901 | 1 | 1 (0) | 1 (0) | 1 (0) | 1 (0) |
| Photosystem I (PsaK) | P20453 | 2 | 1 (0) | 2 (0) | 2 (0) | 2 (0) |
| Photosystem I (PsaL) | P25902 | 2 | 2 (0) | 2 (1) | 2 (0) | 2 (2) |
| Cytochrome C Oxidase (A chain, I) | P00396 | 12 | 12 (0)* | 11 (0)* | 12 (0)* | 12 (1)* |
| Cytochrome C Oxidase (B chain, II) | P00404 | 2 | 2 (0) | 2 (1) | 2 (0) | 3 (2)* |
| Cytochrome C Oxidase (C chain, III) | P00415 | 7 | 7 (0) | 7 (0)* | 5 (0)* | 7 (0)* |
| Cytochrome C Oxidase (D chain, IV) | P00423 | 1 | 1 (0) | 1 (0) | 0 (1) | 1 (0) |
| Cytochrome C Oxidase (G chain, VI-A) | P07471 | 1 | 1 (0) | 1 (0) | 1 (0) | 1 (0) |
| Cytochrome C Oxidase (J chain, VII-A) | P07470 | 1 | 0 (0) | 1 (0) | 0 (0) | 1 (0) |
| Cytochrome C Oxidase (K chain, VII-B) | P13183 | 1 | 0 (1) | 0 (1) | 0 (1) | 0 (1) |
| Cytochrome C Oxidase (L chain, VII-C) | P00430 | 1 | 1 (0) | 1 (0) | 1 (0) | 1 (0) |
| Cytochrome C Oxidase (M chain, VIII) | P10175 | 1 | 0 (1) | 0 (1) | 0 (1) | 0 (1) |
Transmembrane segment prediction results for TM Finder of 75 TM segments reported for a training set of membrane protein structures solved to high resolution (see references herein and the TM Finder web site (http://www.bioinformatics-canada.org/TM/). Accession numbers are given. Runs were performed using the TM Finder default parameters (N-terminal window = 5; C-terminal window = 5; minimum core length = 6; minimum gap length = 4), along with a minimum segment length = 14. Predictions of hydrophobic segments of 14 or more consecutive residues as found in this database are also shown for the KD scale (window size = 9), the GES scale (window size = 20), and the Eisenberg scale (window size = 9). The KD and Eisenberg scales were accessed throug the ProtScale program on the Expasy server (http://www.expasy.ch/cgi-bin/protscale.pl). The GES scale was accessed through the pepplot program in the Genetics Computer Group (GCG) software package distributed through Oxford Molecular. Columns for the predictive methods indicate the number of TM segments correctly located for each protein/subunit; numbers in parenthesis indicate the number, if any, of mispredicted segments for each entry. Asterisks (*) indicate one or more TM segments merged in the prediction because of short intervening loops. In all cases, segments with 10 or more residues of overlap with the training set sequences are scored as ``correctly predicted''. See text for a further discussion.
To illustrate the capacity of TM Finder to distinguish between membrane and non-membrane proteins, we next constructed a database of 21 globular proteins, with entries chosen randomly from Genbank but with attention to inclusion of examples of representative folds/motifs (i.e., leucine zipper). As seen in Table 4, TM Finder correctly identifies the entire collection as nonmembrane proteins, with the sole exception of one unusual Leu-rich 17-residue N-terminal segment in rat ribonuclease 4 (residues 6–22: TQSLLL LLLLTLLGLGL). While hydrophobic segments detected by the other hydrophobicity scales can give useful information concerning buried versus exposed residues, it is clear from Table 4 that these scales tend to over-predict putative TM segments (KD = 20, GES = 9, Eisenberg = 68) in globular proteins. While further refinements can be made, the overall results from the two databases suggest that if one wishes to determine whether it is a membrane protein or not, TM Finder is a reliable indicator.
Table 4.
Predicted transmembrane segments (min. segment length = 14) in nonmembrane proteins
| Protein | Accession | TM Finder | Kyte Doolittle | GES | Eisenberg |
| Drosophila Bicoid | P09081 | 0 | 1 | 1 | 4 |
| Mouse GLI 1 zinc finger | P47806 | 0 | 0 | 1 | 9 |
| Chicken p53 | P10360 | 0 | 0 | 0 | 4 |
| E. Coli homoserine kinase | AAG14779 | 0 | 2 | 1 | 5 |
| Yeast RNA pol II | T29959 | 0 | 5 | 5 | 14 |
| Human Rab2 | NP_002856 | 0 | 0 | 0 | 2 |
| Yeast Yap7p bZIP | NP_014614 | 0 | 0 | 0 | 1 |
| Arabidopsis alcohol dehydrogenase | BAB10198 | 0 | 4 | 0 | 6 |
| Rat ribonuclease 4 | NP_064467 | 1 | 1 | 0 | 1 |
| Mouse calmodulin | P41040 | 0 | 0 | 0 | 0 |
| Rabbit β actin | P29751 | 0 | 1 | 1 | 6 |
| Mouse ribosomal protein S14 | NP_065625 | 0 | 1 | 0 | 1 |
| Mouse gastrin precursor | S68861 | 0 | 1 | 0 | 1 |
| Human histone H4 | CAC0427 | 0 | 0 | 0 | 0 |
| HIV GAG | AAG15248 | 0 | 0 | 0 | 0 |
| Mouse p23 telomerase binding protein | NP_062740 | 0 | 0 | 0 | 0 |
| Human albumin | CAA35749 | 0 | 0 | 0 | 0 |
| Human IgG light chain | CAB93577 | 0 | 0 | 0 | 1 |
| Human plasminogen | P00747 | 0 | 2 | 0 | 8 |
| Mouse cyclin-dependent kinase 5 | NP_031694 | 0 | 2 | 0 | 4 |
| Human RB associated protein | AAG13723 | 0 | 0 | 0 | 3 |
Transmembrane segment prediction results for a database of nonmembrane (globular) protein structures selected from Genbank. Accession numbers are indicated. Predictions of hydrophobic segments of 14 or more consecutive residues as found in this database are given for TM Finder, the KD scale, the GES scale, and the Eisenberg scale. Runs were performed with input parameters as given in Table 3. See text for a further discussion.
The nonmembrane protein elastin (Fig. 3 ▶) provides a further example of the use of TM Finder. Elastin is a fibrous protein rich in Ala, Gly, and Pro residues. A typical section of the sequence (residues 281–430) from human elastin is as follows: GVPGVPGAIPGIGGIAGVGTPAAAAAAAAAA KAAKYGAAAGLVPGGPGFGPGVVGVPGAGVPGVG VPGAGIPVVPGAGIPGAAVPGVVSPEAAAKAAAKAA KYGARPGVGVGGIPTYGVGAGGFPGFGVGVGGIPG VAGVPSVGGVPGVG. Upon initial inspection, some relatively hydrophobic regions appear to be compatible with a membrane environment. In fact, as shown for the full sequence of elastin in Figure 3b-d, hydropathy profiles produced from three hydrophobicity scales (KD, GES, and Eisenberg) (refer to Table 1) contain several occurrences of broad positive regions that might suggest the existence of TM segments in elastin. However, despite the apparently compatible nonpolar phase helicity of elastin, the threshold hydrophobicity requirement imposed by TM Finder (Fig. 3a ▶) virtually eliminates the possibility of a false positive. In fact, elastin hydrophobicity and helicity predictions (Fig. 3a ▶) coincide above the threshold only for a 10-residue stretch (469–478: AQFALLNLAG), nominally too short for a typical TM segment. One reason that elastin has apparent low hydrophobicity is its high Gly content; as discussed above, the Liu-Deber hydrophobicity scale regards Gly as more hydrophilic than do the other three scales.
Fig. 3.
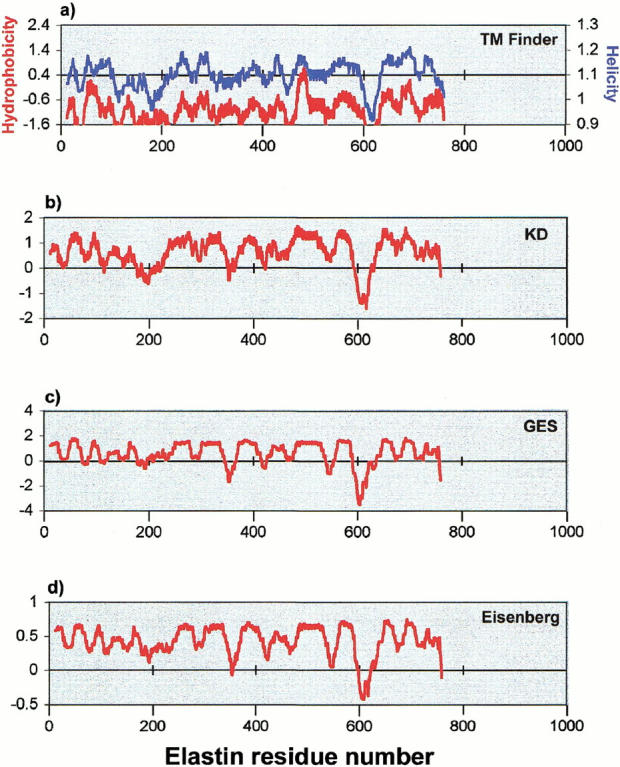
Transmembrane segment prediction profiles of human elastin (SWISSPROT accession number: P15502). (a) TM Finder profiles of hydrophobicity (red) and helicity (blue) (Liu and Deber 1998a,b), with the dual threshold shown as a horizontal line drawn through the prediction profiles. Hydrophobicity profiles for the same sequence are also shown for (b) the KD scale (Kyte and Doolittle 1982); (c) the GES scale (Engelman et al. 1986); and (d) the Eisenberg scale (Eisenberg et al. 1984). See text for a further discussion.
Conclusion
Using a combination of hydrophobicity and helicity scales derived from membrane-interactive TM mimic peptides, with experimentally determined thresholds imposed in each case, TM Finder locates ∼90% of TM segments from the training set of crystallized membrane proteins containing transmembrane α-helices, with a predictive value of 96%. Applied to membrane proteins not yet solved to high resolution, TM Finder constitutes a useful screening operation for identifying their membrane-based segments. The program also has the power to distinguish essentially unequivocally between membrane and nonmembrane proteins, e.g., in a representative database of soluble proteins, no protein (with one minor exception) is mispredicted as a membrane protein. This latter feature of TM Finder would be of particular value, for example, in functional genomics research, where open reading frames present may code for as-yet-uncharacterized proteins.
Acknowledgments
This work was supported, in part, by grants to C.M.D. from the Natural Sciences and Engineering Research Council of Canada, and the Canadian Institutes of Health Research. L.-P.L and C.W. acknowledge support from the Research Training Committee at the Hospital for Sick Children. We thank Dr. Raisa B. Deber for helpful discussions.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ``advertisement'' in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/ps.30301.
References
- Chou, P.Y. and Fasman, G.D. 1978. Empirical predictions of protein conformation. Annu. Rev. Biochem. 47 251–276. [DOI] [PubMed] [Google Scholar]
- Deisenhofer, J., Epp, O., Miki, K., Huber, R., and Michel, H. 1984. X-ray structure analysis of a membrane protein complex. Electron density map at 3 Å resolution and a model of the chromophores of the photosynthetic reaction center from R. viridis. J. Mol. Biol. 180 385–398. [DOI] [PubMed] [Google Scholar]
- Doyle, D.A., Cabral, J.M., Pfuetzner, R.A., Kuo, A., Gulbis, J.M., Cohen, S.L., Chait, B.T., and MacKinnon, R. 1998. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science 280 69–77. [DOI] [PubMed] [Google Scholar]
- Eisenberg, D., Schwarz, E., Komaromy, M., and Wall, R. 1984. Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J. Mol. Biol. 179 125–142. [DOI] [PubMed] [Google Scholar]
- Engelman, D.M., Steitz, T.A., and Goldman, A. 1986. Identifying nonpolar transbilayer helices in amino acid sequences of membrane proteins. Annu. Rev. Biophys. Biophys. Chem. 15 321–353. [DOI] [PubMed] [Google Scholar]
- Fazio, M.J., Olsen, D.R., Kauh, E.A., Baldwin, C.T., Indik, Z., Ornstein-Goldstein, N., Yeh, H., Rosenbloom, J., and Uitto, J. 1988. Cloning of full-length elastin cDNAs from a human skin fibroblast recombinant cDNA library: further elucidation of alternative splicing utilizing exon-specific oligonucleotides. J. Invest. Dermatol. 91 458–464. [DOI] [PubMed] [Google Scholar]
- Henderson, R., Baldwin, J.M., Ceska, T.A., Zemlin, F., Beckmann, E., and Downing, K.H. 1990. Model for the structure of bacteriorhodopsin based on high-resolution electron cryo-microscopy. J. Mol. Biol. 213 899–929. [DOI] [PubMed] [Google Scholar]
- Hodges, R.S., Zhu, B.Y., Zhou, N.E., and Mant, C.T. 1994. Reversed-phase liquid chromatography as a useful probe of hydrophobic interactions involved in protein folding and protein stability. J. Chromatography 676 3–15. [DOI] [PubMed] [Google Scholar]
- Kyte, J. and Doolittle, R.F. 1982. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157 105–132. [DOI] [PubMed] [Google Scholar]
- Li, S.C. and Deber, C.M. 1994. A measure of helical propensity for amino acids in membrane environments. Nat. Struct. Biol. 1 368–373. [DOI] [PubMed] [Google Scholar]
- Liu, L.P. and Deber, C.M. 1998a. Guidelines for membrane protein engineering derived from de novo designed model peptides. Biopolymers 47 41–62. [DOI] [PubMed] [Google Scholar]
- Liu, L.P. and Deber, C.M. 1998b. Uncoupling hydrophobicity and helicity in transmembrane segments: α-helical propensities of the amino acids in non-polar environments. J. Biol. Chem. 273 23645–23648. [DOI] [PubMed] [Google Scholar]
- Liu, L.P. and Deber, C.M. 1999. Combining hydrophobicity and helicity: A novel approach to membrane protein structure prediction. Bioorg. Med. Chem. 7 1–7. [DOI] [PubMed] [Google Scholar]
- Munoz, V. and Serrano, L. 1994. Intrinsic secondary structure propensities of the amino acids, using statistical phi-psi matrices: Comparison with experimental scales. Proteins 20 301–311. [DOI] [PubMed] [Google Scholar]
- Papavoine, C.H., Christiaans, B.E., Folmer, R.H., Konigs, R.N., and Hilbers, C.W. (1998). Solution structure of the M13 major coat protein in detergent micelles: A basis for a model of phage assembly involving specific residues. J. Mol. Biol. 282 401–419. [DOI] [PubMed] [Google Scholar]
- Park, K., Perczel, A., and Fasman, G.D. 1992. Differentiation between transmembrane helices and peripheral helices by the deconvolution of circular dichroism spectra of membrane proteins. Protein Sci. 1 1032–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowell, M.H., McPhillips, T.M., Rees, D.C., Soltis, S.M., Abresch, E., and Feher, G. 1997. Light-induced structural changes in photosynthetic reaction center: Implications for mechanism of electron-proton transfer. Science 276 812–816. [DOI] [PubMed] [Google Scholar]
- Tsukihara, T., Aoyama, H., Yamashita, E., Tomizaki, T., Yamaguchi, H., Shinzawa-Itoh, K., Nakashima, R., Yaono, R., and Yoshikawa, S. 1996. The whole structure of the 13-subunit oxidized cytochrome C oxidase at 2.8 Å. Science 272 1136–1144. [DOI] [PubMed] [Google Scholar]
- Wang, C., Liu, L.P., and Deber, C.M. 1999. Helicity of hydrophobic peptides in polar vs. non-polar environments. Phys. Chem. Chem. Phys. 1 1539–1542. [Google Scholar]
- Wang, C., Liu, L.P., and Deber, C.M. 2000. δ-Regions in proteins: Helices mispredicted as transmembrane segments by the threshold hydrophobicity requirement. Proc. 16th Amer. Peptide Symp. (Fields, G.B., Tam, J.P., and Barany, G., eds.), pp. 367–369.
- Wimley, W.C . and White, S.M. 1996. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 3 842–848. [DOI] [PubMed] [Google Scholar]