

Abstract
The structure of Herpes simplex virus type 1 thymidine kinase (TKHSV1) is known at high resolution in complex with a series of ligands and exhibits important structural similarities to the nucleoside monophosphate (NMP) kinase family, which are known to show large conformational changes upon binding of substrates. The effect of substrate binding on the conformation and structural stability of TKHSV1, measured by thermal denaturation experiments, far-UV circular dichroism (CD) and fluorescence is described, and the results indicate that the conformation of the ligand-free TKHSV1 is less ordered and less stable compared to the ligated enzyme. Furthermore, two crystal structures of TKHSV1 in complex with two new ligands, HPT and HMTT, refined to 2.2 Å are presented. Although TKHSV1:HPT does not exhibit any significant deviations from the model of TKHSV1:dT, the TKHSV1:HMTT complex displays a unique conformationally altered active site resulting in a lowered thermal stability of this complex. Moreover, we show that binding affinity and binding mode of the ligand correlate with thermal stability of the complex. We use this correlation to propose a method to estimate binding constants for new TKHSV1substrates using thermal denaturation measurements monitored by CD spectroscopy. The kinetic and structural results of both test substrates HPT and HMTT show that the CD thermal denaturation system is very sensitive to conformational changes caused by unusual binding of a substrate analog.
Keywords: Herpes simplex virus type 1 thymidine kinase, structural stability, conformation, circular dichroism, thermal denaturation, crystal structure
Thymidine kinases (TK, EC 2.7.1.21) are key enzymes in the pyrimidine salvage pathway catalyzing the γ-phosphate transfer from ATP to thymidine (dT) in the presence of Mg2+ and thus, yielding thymidine monophosphate (dTMP) and ADP. Herpesviridae, such as Herpes simplex virus type 1, encode for their own, multifunctional TK. Unlike the very specific human cytosolic TK (TK1), it is able to phosphorylate pyrimidine as well as purine analogs and demands less stereochemical restrictions concerning the sugar moiety also accepting acyclic side chains as phosphate acceptors (e.g., aciclovir; Elion et al. 1977; Keller et al. 1981). Therefore, the difference in substrate specificity of human TK 1 and TKHSV1 is a crucial point in establishing a molecular basis for selective antiviral therapy, featuring TKHSV1 as the center of activation of antiviral drugs such as aciclovir (ACV), penciclovir, and ganciclovir (GCV). First being activated by phosphorylation by viral encoded TK, these nucleoside analogs in their triphosphate form block the viral replication by subsequently terminating DNA elongation at the viral DNA polymerase. In combination with GCV TKHSV1 is an established tool used as a prodrug-activating enzyme, so-called suicide enzyme, in gene therapy of cancer (Culver et al. 1992, 1994; Moolten 1994; Morgan et al. 1997; Sacco et al. 1995; Tong et al. 1997) and AIDS (Caruso and Bank 1997), and in controlling graft-versus-host disease by allogenic bone marrow transplant (allo BMT) (Bonini et al. 1997; Verzeletti et al. 1998).
To improve the efficiency of gene therapy based on TKHSV1, mutations have been successfully introduced within the active site and at positions near the active site but not directly involved in ligand binding (Black et al. 1996; Kussmann-Gerber et al., 1998; Christians et al. 1999; Drake et al. 1999; Kokoris et al. 1999; Prota et al. 2000). To explain the effect of distant mutations conformational rearrangement has been postulated (Kussmann-Gerber et al. 1998), based on the structural homology with adenylate kinase (ADK) shown by high resolution structures of TKHSV1 in complex with a series of ligands (Brown et al. 1995; Wild et al. 1995, 1997; Champness et al. 1998; Bennett et al. 1999; Prota et al. 2000; Vogt et al. 2000). ADK, a member of the nucleoside monophosphate (NMP) kinases, is known to undergo major conformational changes upon binding of substrates (Vonrhein et al. 1995). These motions of internal chain segments relative to the CORE domain ranged between a ``closed'' conformation caused by ligand binding, and a less well-defined ``open'' conformation of the ligand-free enzymes. The hypothesis that TKHSV1 shows a similar behavior is substantiated by a recent isothermal calorimetry study on nucleoside (dT) and cofactor (ATP)-binding, suggesting that the formation of the TK:dT:ATP complex is accompanied by significant structural rearrangements of the enzyme (Perozzo et al. 2000).
Here, we report the effects of substrate binding on the conformation and structural stability of TKHSV1. We describe thermal denaturation measurements, far-UV circular dichroism (CD) and fluorescence spectroscopy experiments and the crystal structure analysis of two novel TKHSV1 substrates in complex with the enzyme at 2.2 Å resolution. On the basis of these studies, we propose a new and easy-to-perform method to determine binding constants for new TKHSV1 substrates using thermal denaturation measurements monitored by CD spectroscopy.
Results
Far-UV CD spectra
The first CD spectroscopy analyses for TKHSV1 are reported. Figure 1 ▶ presents the far-UV CD spectra of TKHSV1 without ligands, TKHSV1 incubated with either dT or ATP, and the enzyme incubated with both substrate dT and cosubstrate ATP, which account for background signal produced by the buffer and ligands. All four CD spectra show the characteristic shape of a α-helix-rich protein with a typical double minimum at 222 and 209 nm and a maximum at 193 nm. The solved crystal structures of the ligated TKHSV1 reveals an α-helix content of ∼46% (Brown et al. 1995; Wild et al. 1995, 1997; Champness et al. 1998; Bennett et al. 1999; Prota et al. 2000; Vogt et al. 2000). Addition of ligands resulted in significant changes in both shape and intensity of the CD spectra, most strikingly in an increase of the CD signal around 222 nm, and a shift of the maximum at 193 nm toward 196 nm, the latter only in presence of dT.
Fig. 1.

Conformational changes of TKHSV1 induced by ligand binding, monitored by far-UV CD spectroscopy. The different spectra resulting from the subtraction of dialysis buffer and ligand spectra from protein spectra are shown. Spectra of TKHSV1 (—), TK with 100 μM ATP (-.-.-), TK with 100 μM dT (. . . . .), and TKHSV1 with 100 μM ATP and dT (-..-).
Thermal denaturation studies monitored by CD spectroscopy
A series of thermal denaturation scans of TKHSV1 in the absence and presence of the natural substrate dT and cosubstrate ATP, monitored by changes in the CD signal at 223 nm, is shown in Figure 2A ▶. All samples showed symmetrical, sigmoid, and heating rate-independent decrease in ellipticity when the temperature was increased. This behavior is usually interpreted in terms of a simple two-state denaturation process. Because all denaturation curves could be perfectly fitted to the two-state thermal denaturation function described below (Fig. 3 ▶) and TKHSV1 shows a dimeric quaternary structure in the crystals (Brown et al. 1995; Wild et al. 1995, 1997; Bennett et al. 1999; Champness et al. 1998; Prota et al. 2000; Vogt et al. 2000), the curves probably describe a process where dissociation and unfolding of the protein are occurring simultaneously.
Fig. 2.

Thermal denaturation curves of ligand-free and ligated TKHSV1 (0.4 mg/mL) in TBSE. CD signal was recorded at 223 nm in a temperature range from 20° to 70°C. (A) Influence of the natural TKHSV1 substrates ATP and thymidine on the melting temperature. The melting curves of TKHSV1 without substrates (♦), TKHSV1 with 1 mM ATP (□), TKHSV1 with 1 mM dT (X), TKHSV1 with 1 mM ATP and 1mM dT (○) are displayed. (B) Influence of 1 mM ligand of TKHSV1 in the presence of 1 mM ATP. The melting profiles of TKHSV1 with ATP (♦), TKHSV1 with HMTT:ATP (▵), TKHSV1 with ACV:ATP (▴), TKHSV1 with GCV:ATP (□), TKHSV1 with HPT:ATP (·), TKHSV1 with AZT:ATP (−), TKHSV1 with dT:ATP (○), and TKHSV1 with IdU:ATP (+) are shown.
Fig. 3.
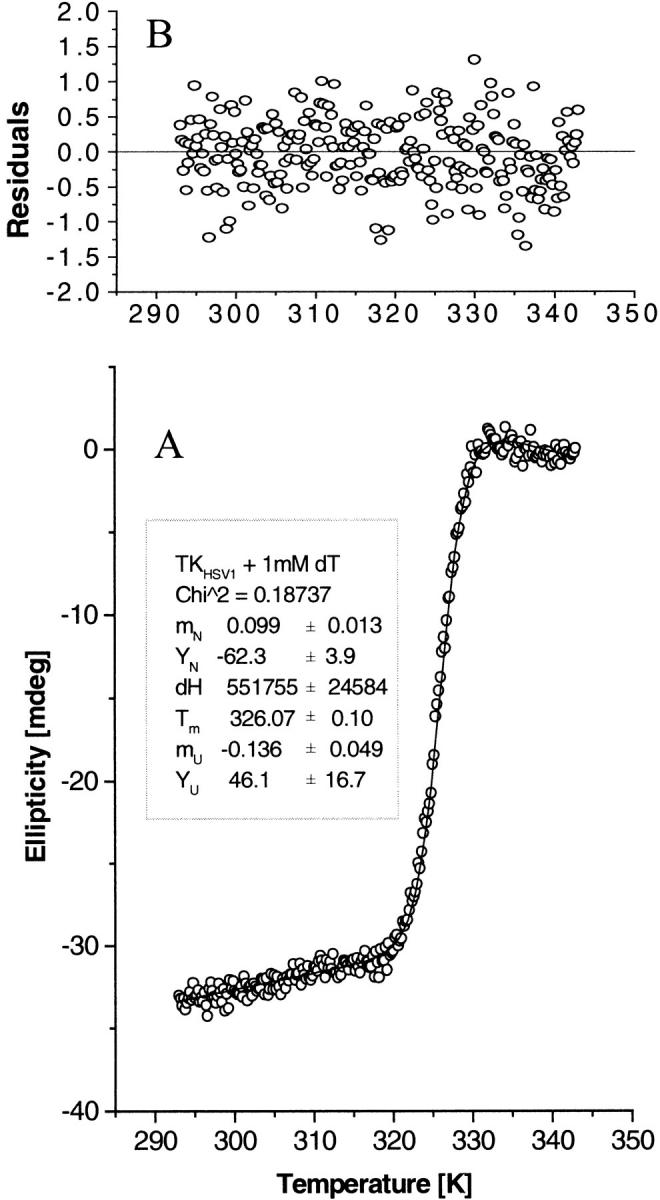
CD thermal denaturation profile analysis. Fit of the CD thermal denaturation profile of TKHSV1 (0.4 mg/mL) in TBSE containing 1 mM dT, as representative of the whole set of measured denaturation curves, to the two-state unfolding model described by Eq. 4b. (A) The open circles represent the CD signal of TKHSV1 at 223 nm as a function of temperature. The solid line is the result of a nonlinear fit routine, fitting the unfolding CD data using Eq. 4b. The inset shows the fit results for the slope mN and intercept YN of the native protein baseline, the melting temperature Tm, the enthalpy change ΔH between the unfolded and the native state (considered only as a mathematical fit parameter, not as thermodynamic value), and slope mU and intercept YU of the unfolded protein baseline. (B) Residuals of the data fit shown in A representing the difference between the theoretical function and the actual data points.
Substrate-free TKHSV1 has a melting temperature of 43.4 ± 0.1 °C. Addition of ATP has little effect on the melting temperature, whereas binding of the substrate dT leads to a significant increase of stability by shifting the midpoint of the denaturation curve up to 52.6 ± 0.5°C. Binding of both dT and ATP has an even more dramatic effect on the conformational stability of the protein, resulting in a Tm of 61.2 ± 0.3°C, which is almost 20°C higher than that observed for the substrate-free protein (Fig. 2A ▶; Table 1).
Table 1.
TKHSV1 substrate interactions: Km values and melting temperatures
| TK substrates | Km (μM) | Tmelt (°C) | Claculated Km (μM)a |
| IdU | 0.092 ± 0.004bc | 61.9 ± 0.5 | 0.10 |
| dT | 0.20 ± 0.05d | 61.2 ± 0.3 | 0.16 |
| AZT | 5.2 ± 1.70d | 55.8 ± 0.5 | 5.5 |
| GCV | 47.6e | 52.7 ± 0.1 | 44.4 |
| ACV | 200 ± 50bd | 50.3 ± 0.9 | 208 |
| HMTT | 30.9 ± 1.8bf | 50.3 ± 0.45 | 202.7 |
| HPT | 26.6 ± 0.4bf | 53.6 ± 0.1 | 23.9 |
a Theoretical Km values were calculated from the measured melting temperatures using the linear regression function derived from the TKHSV1 substrates calibration set (Fig. 4 ▶, Eq. 5).
b Measured as Ki value.
c Balzarini et al. 1989; d Pilger et al. 1999; e Kokoris et al. 1999; f U. Kessler, B.D. Pilger, O. Zerbe, L. Scapozza, and G. Folkers, unpubl. results.
In the presence of ATP and a set of various TKHSV1 substrates, including ACV, GCV, AZT, and 5-iodo-2′-deoxyuridine, the shape of all curves is conserved, whereas the midway points are all shifted toward higher temperatures compared to the TKHSV1 samples with ATP alone (Fig. 2B ▶; Table 1). Most strikingly, this thermal stabilization effect can be correlated by a logarithmic relationship with the substrate's affinity to the binding site expressed as apparent Km or the equivalent Ki value in the presence of the cosubstrate ATP (Fig. 4 ▶; Table 1), described by a linear regression analysis (Microcal Origin 5.0, Northampton, MA) with slope −3.52 ± 0.06 log μM, intercept 58.5 ± 0.1°C, and a correlation R = −0.9995 (P < .001).
Fig. 4.

Correlation between melting temperature and binding affinity by TKHSV1. Linear relationship between the melting temperature of TKHSV1 (0.4 mg/mL in TBSE) in complex with ATP and different TKHSV1 substrates (each at 1 mM), and the logarithm of the Km value of the substrates ranging over four orders of magnitude and determined by enzyme kinetics. Calibration set: IDU (Ki 0.09 μM; Balzarini et al. 1989), thymidine (Km 0.20 μM; Pilger et al. 1999), AZT (Km 5.2 μM; Pilger et al. 1999), GCV (Km 47.6 μM; Kokoris et al. 1999), and ACV (Km 200 μM; Pilger et al. 1999); test set: HPT (Ki ≅ Km 27 μM), HMTT (Ki ≅ Km 30.9 μM; U. Kessler, B.D. Pilger, O. Zerbe, L. Scapozza, and G. Folkers, unpubl. results). The solid line represents the linear regression calculated for the calibration set (intercept 58.47° ± 0.11°C, slope −3.52 ± 0.08 log μM, r2 = 0.998, P < .001). The resulting Eq. 5 is Tmelt (°C) = −3.52 * log Km (μM) + 58.47.
Fluorescence spectra
Comparison of the TKHSV1 intrinsic fluorescence spectra without addition of substrates with those of TKHSV1 in complex with thymidine and ATP reveals a decrease in fluorescence intensity and a blue shift of the maximum from 346 to 337 nm with ligand binding (Fig. 5 ▶). No interference of the ligand solutions under experimental conditions was observed. This shift suggests that at least one of the tryptophane residues becomes less solvent-exposed due to a subtle tertiary structure rearrangement.
Fig. 5.

Structural rearrangement monitored by fluorescence spectroscopy. Fluorescence spectra of ligand-free TKHSV1 (solid line) and TKHSV1 incubated with 250 μM ATP and 50 μM thymidine (gray line). Protein concentration was 0.2 mg/mL (5 μM) in TBSE containing 10 mM Tris HCl at pH 7.4, 150 mM NaCl, and 1 mM EDTA to suppress enzymatic activity. The excitation wavelength was set to 295 nm and emission fluorescence intensity was recorded at 25°C from 310 to 450 nm. Binding of ATP and dT to TKHSV1 leads to a decrease in intensity of the fluorescence signal and a shift of the maximum from 346 to 337 nm.
Structure of TK HSV1:HPT and TK HSV1:HMTT
TKHSV1:HPT
The structure of TKHSV1 in complex with HPT was determined at a resolution of 2.2 Å with an R factor of 21.7% (Rfree = 26.2%) (Table 2), 229 water molecules, and one sulfate ion in each of the two P-loops were added. The model is of good quality, because only the catalytically competent residue Arg163 of both subunits is in a disallowed region in the Ramachandran diagram, as observed in other TKHSV1 structures (Brown et al. 1995; Wild et al. 1995, 1997; Champness et al. 1998; Bennett et al. 1999; Prota et al. 2000; Vogt et al. 2000). The initial (2Fo-Fc) electron density map and the (Fo-Fc) difference electron density maps clearly indicated the presence of one substrate molecule in the active sites of each of the two subunits. The average B-factor is 28 Å2 (Table 2). The nucleobase lies between Met128 and Tyr172 in the typical sandwich-like complex and interacts with Gln125 forming a Watson-Crick-like hydrogen bond network as it is reported for the natural substrate dT (Wild et al. 1997; Champness et al. 1998). In addition, water-mediated hydrogen bonds between O2α and Arg176, and N1 and Tyr101-OH enhance the binding. The OH-group of the acyclic propyl chain is fixed similarly to the 5′-OH of dT by the interactions with Glu83, Arg222, and a water-mediated hydrogen bond to Glu225 (Fig. 6 ▶). An analogy to the natural substrate dT phosphorylation is possible in this position. The polypeptide model does not exhibit any significant deviation from the polypeptide of TKHSV1:dT (Wild et al. 1997; Champness et al. 1998).
Table 2.
Data collection and refinement statistics
| Data seta | TKHSV1 : HPT | TKHSV1 : HMTT |
| Diffraction data | ||
| X-ray source | Cu Kα | BW7B |
| Unit cell (Å) | a = 113.7 | a = 113.6 |
| b = 117.2 | b = 118.8 | |
| c = 108.5 | c = 108.5 | |
| Resolution range (Å) | 20–2.2 | 20–2.2 |
| Completeness (%) | 99 | 99 |
| Multiplicity | 3.8 | 3.9 |
| Unique reflections | 36554 | 37376 |
| Rsym2 (last shell) (%) | 10.6 (49) | 7.8 (62) |
| I/σ (last shell) | 10.7 (2.7) | 11.7 (2.0) |
| Refinement and final model | ||
| R-factor (Rfree) (%) | 21.7 (26.2) | 20.8 (26.4) |
| Average B-factor (Å)b | 28 | 44 |
| Number of non-H protein atoms | 4700 | 4719 |
| Substrate atoms | 26 | 42 |
| Water molecules | 229 | 161 |
| Sulfate ions | 2 | 2 |
a Crystals were of space group C2221. All data were collected at 100 K.
b Rsym = ΣhΣi|Ihi − <Ih>ΣhΣiIhi where h stands for the unique reflections and i counts through symmetry-related reflections.
Fig. 6.

Stereo view of HPT binding to TKHSV1 compared to dT in TKHSV1:dT complex. The conformation of HPT is well defined by the (2Fo–Fc) electron density contoured at a contour level of 1.3 σ. TKHSV1:HPT and dT are shown in dark and light gray ball and stick model, respectively. Hydrogen bonds are depicted as dashed lines, water molecules as gray balls. dT is displayed, but the residues of TKHSV1:dT (Champness et al. 1998) taking the same conformation as in the TKHSV1:HPT complex were omitted for clarity. The nucleobase lies between Met128 and Tyr172 in the typical sandwich-like complex and interacts with Gln125 forming a Watson-Crick-like hydrogen- bond network as it is reported for the natural substrate dT (Wild et al. 1997; Champness et al. 1998). In addition, water-mediated hydrogen bonds between O2α and Arg176 and N1 and Tyr101 enhance the binding. The acyclic side chain is fixed similarly to the 5′-OH of dT by the interactions of the OH-group with Glu83, Arg222, and an additional water-mediated hydrogen bond with Glu225.
TKHSV1:HMTT
To refine TKHSV1:HMTT, the same protocol as described for TKHSV1:HPT was used. The structure of TKHSV1 with HMTT was refined to 2.2 Å resolution with an R factor of 20.8% (Rfree = 26.4%) (Table 2), 161 water molecules, and 2 sulfate ions were added to the model. Again, one substrate molecule per subunit could be inserted unequivocally based on (Fo-Fc) difference electron density maps in each of the two active sites. The quality of the density map allowed us to exclude multiple binding modes for HMTT, although this possibility has been considered during the refinement of the structure. In subunit A residues 58–76 were completely rebuilt by inspecting the (2Fo-Fc) electron density and (Fo-Fc) difference electron density maps. The average B-factor is 44 Å2 (Table 2).
A superposition of TKHSV1:dT and TKHSV1:HMTT (Fig. 7 ▶) reveals significant conformational alterations in subunit A, whereas in subunit B these residues are fixed by crystal contacts (Table 3). This rearrangement occurs for the amino acid residues 60–75 of the P-loop and helix α1 in subunit A. Thus, we report the first structure of a TKHSV1:substrate complex displaying a substantially altered active site and completely ordered amino acid residues 70–74, which are missing in other models in the same subunit within the same space group (Brown et al. 1995; Champness et al. 1998).
Fig. 7.
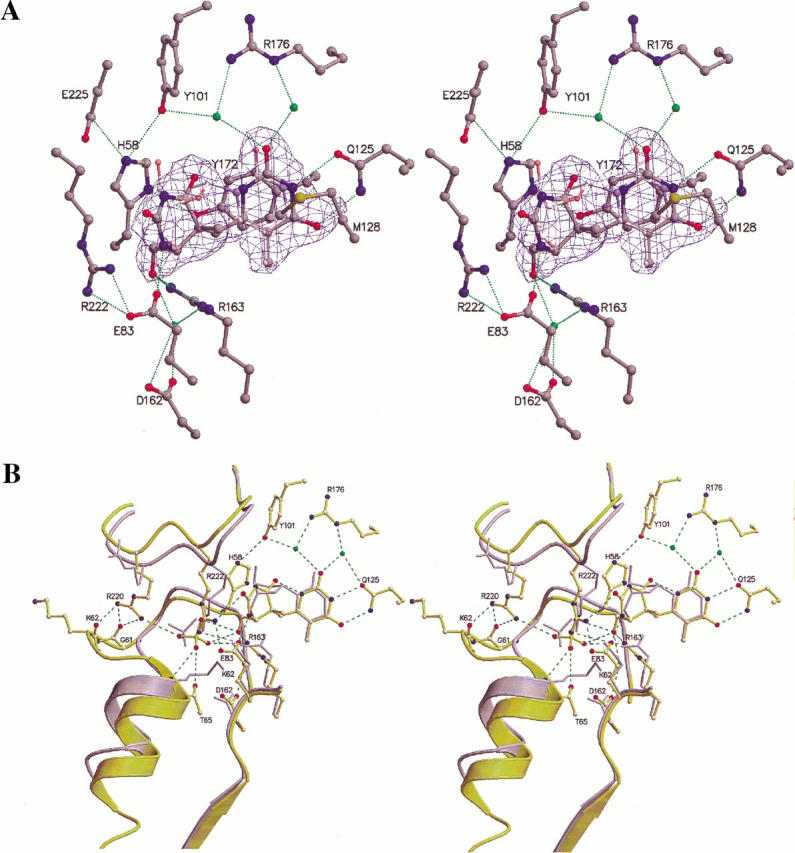
Superposition of TKHSV1:HMTT and TKHSV1:dT structures. (A) Substrate-binding site of the refined TKHSV1:HMTT complex (subunit A) superposed with TKHSV1:dT (Champness et al. 1998). For sake of clarity only the dT of the TKHSV1:dT complex is displayed. The conformation of HMTT is well defined by the (2Fo–Fc) electron, shown at a contour level of 1.3σ in blue. The carbon atoms of TKHSV1:HPT and dT are shown in dark and light gray, respectively, whereas the other atoms are color-coded (N: blue, O: red, S: yellow). Hydrogen bonds are shown as dashed lines, water molecules as green balls. (B) View into the active site showing a displacement induced by HMTT binding. TKHSV1:HMTT and TKHSV1:dT are shown in yellow and gray, respectively. All residues of TKHSV1:dT taking the same conformation as in TKHSV1:HMTT complex were omitted for clarity. Hydrogen bonds and water are displayed as in A. The thymine part of HMTT is positioned like the base of the natural substrate dT, whereas the sugar-mimicking moiety substituted at position 6 of the thymine ring is inserted much deeper into the active site. The 1.4-Å shift of the sulfate ion toward the substrate and the direct hydrogen bond between the hydroxyl group of HMTT mimicking the O5′ of dT is shown. The rearrangement occurring for the amino acid residues 60–75 of the P-loop and the displacement of the following helix α1 is depicted.
Table 3.
Crystal contacts of TKHSV1in space group C2221
| Contacta | Contact surfaceb (Å2) | Involved residuesc |
| I–II | 271 | aI–aII |
| I–III | 1133 | bI–bIII |
| I–IV | 498 | cI–dIV |
| I–V | 498 | dI–cV |
| I–VI | 645 | eI–fVI |
| I–VIII | 645 | fI–eVII |
a Reference dimer I (x, y, z) is in contact with six symmetry-related dimers, which are defined as follows: II (−x, y, −z+1/2), III (x, −y+1, −z), IV (−x+1/2, −y+1/2, z−1/2,), V (−x+1/2, −y+1/2, z+1/2), VI (x−1/2, −y+1/2, −z), VII (x+1/2, −y+1/2, −z).
b The solvent accessible surface of the reference dimer molecule, which is hidden in the crystal contact, calculated with XPLOR at a radius of 1.4 Å. The solvent accessible surface of a free TKHSV1 dimer is 45822 Å2.
c Residues that have a more than 1 Å2 decreased surface in the contact compared to the free molecule. The contact ensembles in subunits A and B are: a = B (338–339, 341–342, 372); b = A (146, 148–149, 240, 244, 247, 250–252, 280–282, 320–321, 323, 328, 331, 339, 342–343, 345–346, 348); c = A (177–180, 263–264, 293, 295, 298–302); d = B (260–264, 280–283, 286); e = B (63–64, 67–68, 71, 75, 219–220, 333); f = A (105, 211–212, 214–215, 218–219, 226–227, 229, 331–332).
The first thymine moiety of HMTT is positioned like the base of the natural substrate dT, whereas the second modified thymine that mimics the sugar and is attached at the first thymine ring in position 6 is lying deeper inside the binding site reserved for the sugar moiety. The observed orientation of the substrate induces a 1.4 Å shift of the sulfate molecule toward the substrate and a direct hydrogen bond between the hydroxyl group of HMTT mimicking the O5′ of dT and the sulfate molecule. The distance of the hydroxyl group of HMTT compared to 5′-OH of dT to Glu83 increases from 3.6 to 4.0 Å, whereas it comes 0.4 Å closer to Arg163-NH2. However, the complex structure is compatible with the phosphorylation of the substrate by TKHSV1 (U. Kessler, B.D. Pilger, O. Zerbe, L. Scapozza, and G. Folkers, unpubl. results). Thus, the anion binding P-loop is rearranged and the beginning of the following helix α1 is shifted, causing a displacement of the entire helix.
The novel loop structure is stabilized by hydrogen bonds between the side chain of Arg220 and the backbone oxygen of Gly61 and Lys62. Interestingly, the side chain of Lys62, which was reported to be essential for phosphoryl transfer during catalysis as deduced from adenylate kinase (Müller and Schulz 1992), now points away from the sulfate (Fig. 7B ▶). Furthermore, Arg220-NH2 interacts with the sulfate ion through an additional hydrogen bond. The structural peculiarities of TKHSV1: HMTT are pointed out in the analysis of the root mean square deviation (r.m.s.d.) of subunit A of TKHSV1:HMTT compared to TKHSV:dT (Fig. 7B ▶) (Champness et al. 1998). Fitting the backbone of all amino acids of the conserved region results in a r.m.s.d. of 0.5 Å, whereas the fit of the backbone of amino acid residues 60–75 shows a significant higher r.m.s.d. of 2.7 Å.
Discussion
TKHSV1 undergoes conformational changes with ligand binding
It has been reported earlier that segmental mobility is an intrinsic TK property and that residues forming the substrate-binding pocket as well as the LID domain have significantly higher mobility in the ligand-free structure than in TKHSV1:dT:ADP (Vogt et al. 2000). No structural information about the completely empty substrate-free apo-form of TKHSV1 is available yet, as the solved ``ligand-free'' TKHSV1 structure is complexed with sulfate. We examined the impacts of these movements caused by ligand binding on the structural stability of the enzyme.
Addition of ATP has little effect on the melting temperature, whereas binding of the substrate dT leads to a significant increase in stability by shifting the midpoint of the denaturation curve. These results are consistent with previous calorimetric and enzymatic studies of substrate binding to TKHSV1 (Kussmann-Gerber et al. 1999; Perozzo et al. 2000), showing that the free enzyme does not bind ATP and its binding site becomes only accessible in the TKHSV1:dT complex. Furthermore, values of ΔCp and TΔS determined by isothermal calorimetry measurements suggest large-scale conformational adaptation on the active site in sequential substrate binding (Perozzo et al. 2000). The significant increase in secondary structure with ligand addition showed by the far-UV CD spectra and the blue shift of the fluorescence maximum with ligand binding investigated through fluorescence spectroscopy provided further evidence for structural rearrangements, suggesting that the enzyme changes from a less ordered conformation to a more stable structured one. Confirmation and details of the indicated rearrangements are awaited from the crystal structure of a completely empty TKHSV1.
Quality of the thermal denaturation curve analysis
Notwithstanding that thermal TKHSV1 unfolding was not reversible (data not shown), indicating that no thermodynamic equilibrium has been achieved, it could be shown that thermal denaturation was independent from the heating rates, which in this case is the precondition for reproducibility of the melting temperature (a sample containing 0.4 mg/mL TK and 1 mM ATP and thymidine yielded Tm values of 60.81° ± 0.04°C, 61.89° ± 0.02°C, and 61.11° ± 0.16°C for heating rates of 30, 40, and 50°C/h, respectively). Thus, the data were fitted successfully to a sigmoid two-state unfolding model (Eq. 4b) and the fitting results for the parameter Tm were taken as indicator of relative protein stability, regarding ΔHm as a mathematical fit term and not as a thermodynamic parameter (Fig. 4 ▶).
Correlation of ligand-binding affinity and mode with thermal stability of TKHSV1 ligand complexes
A correlation between the thermal denaturation temperatures and the magnitude of the ligand affinity constants of the respective ligand, expressed as the Michaelis constant Km or Ki could be clearly depicted for TKHSV1. This concept has been previously described by Fukada and coworkers (1983) in a thermodynamic study of the binding of l-arabinose and d-galactose to the l-arabinose-binding protein of Escherichia coli. Furthermore, this is similar to a previously published correlation between thermal stability of class I MHC–peptide complexes and peptide-binding affinity, expressed by peptide dissociation constants (Morgan et al. 1997). One may argue on the use of Km as thermodynamic-binding constant, but underlying exceptional kinetic features of TKHSV1 have been described earlier (Kussmann-Gerber et al. 1999), showing that Km of substrates such as dT and ACV correspond with their dissociation constants Kd and thus, with their Ki value. Moreover, we see a general trend that the tighter the ligand is bound, the lower are the B-values of the amino acids involved in binding, which is in agreement with previously published work on Streptavidin (Weber et al. 1989).
Crystal structures of two novel TK substrates HMTT and HPT
The crystal structure of the TKHSV1:HMTT complex displays the rare effect of asymmetry in a homooligomeric protein due to crystal packing (Table 3). Although subunit B does not show significant deviations from previously reported complexes of HSV1 TK and various ligands, subunit A features alterations around the active site. The P-loop and the following helix α1 forming the anion-binding hole of the TKHSV1:HMTT complex are in a orientation not observed so far in any other TK:ligand complex. The loop contains a Lys at postion 62 that was reported to be essential for phosphoryl transfer during catalysis as deduced from adenylate kinases (Müller and Schulz 1992). Strikingly, this amino acid residue does not point toward the inner part of the ATP-binding site, but into the opposite direction. Nevertheless, HMTT is phosphorylated by TKHSV1, exhibiting good binding affinity (U. Kessler, B.D. Pilger, O. Zerbe, L. Scapozza, and G. Folkers, unpubl. results). These observations indicate the flexibility of the anion-binding site.
In consideration of the thermal stability data, the findings confirm the melting curves of TKHSV1 caused by the peculiar orientation of the novel substrate in the nucleoside-binding site. The melting point of TK incubated with 1.0 mM ATP and 1.0 mM HMTT is significantly lower than expected for a Ki value of 30.9 ± 1.8 μM as determined by enzyme kinetic measurements. In fact, the theoretical Ki value derived from the measured Tm value (Table 1) is calculated to be almost eight times higher, namely 202.7 μM, which means that the helix shift induced by the binding of HMTT destabilizes the overall protein conformation, resulting in a lower melting temperature for the TKHSV1:HMTT:ATP complex than expected for HMTT with a Ki value of 30.9 μM. The loss of stability depicted by the lower Tm value is in agreement with the decrease in Van der Waals interaction involving helix α1 showed by an enhanced solvent accessibility of amino acids 55–70 and with the loss of three n+4 hydrogen bonds characteristic of the helix, as a result of unwinding of the first helix turn that starts at Gly61 in TKHSV1:dT, caused by the shift within the P-loop. The increased instability measured for TKHSV1-binding HMTT is consistent with the increased flexibility of the polypeptide chain shown by a relatively high B-factor (Table 2) compared with TKHSV1:HPT and other structures of TKHSV1 in complex with several ligands solved in the same space group and under cryo conditions (Champness et al. 1998).
In contrast, the crystal structure of TKHSV1 in complex with HPT can be perfectly overlaid to the TKHSV1:dT:ATP structure with a r.m.s.d. of 0.5 Å over all backbone atoms. This coincides with all previously reported crystal structures of TK–substrate complexes, suggesting that HPT is a TK ligand that fits into the series of TK substrates showing linear relationship between log Ki and Tm. We could show in this study that the Ki value of 27 μM determined by enzyme kinetic is in perfect agreement with the Ki value of 23.9 μM derived from the melting temperature.
Conclusion
Despite the remarkably good correlation between the Km (Ki) values for a TK ligand and its corresponding TK:ligand: ATP melting temperature, some limitations must be considered envisaging the application of this system to predict binding constants for new TK ligands. It is conceivable that two ligands that bind to the same binding site with similar binding affinities could elicit different effects because they have a different structural distribution of binding interactions. In the case of TKHSV1, the measured Tm values result on the one hand from the additional binding free energy contributions of the ligand to the TK active site, but on the other hand from stability contributions of structural rearrangements to the overall protein fold. Therefore, Tm and Ki values can only be correlated for ligand classes showing the same binding mode and inducing the same ``closed'' conformation as thymidine. Nonetheless, this restriction may also yield profit in terms of computer-aided drug design. For new potential TK ligands, deviations between Ki values deduced from Tm and Ki values determined by enzyme kinetics would indicate a different binding mode. HMTT perfectly illustrates this mismatch. In terms of rational design, this will allow a better judgment of the working hypothesis based on a lead compound from which detailed structural information is missing.
Materials and methods
IPTG (isopropyl-2-d-thiogalactopyranoside), ATP (adenosine triphosphate), and reagents for enzyme assays were purchased from Boehringer Mannheim; AZT (3′-azido-deoxythymidine) and IdU (5-iodo-2′-deoxyuridine) from Sigma; ACV (aciclovir, 9–(2-hydroxyethoxymethyl) guanine) from Wellcome, London; and GCV (ganciclovir, 9–(1,3-dihydroxy-2-propoxymethyl) guanine) from Roche. DTT (1,4-dithio-dl-threitol), DNase I (deoxyribonuclease I from bovine pancreas) and thymidine (dT, 2′-deoxythymidine) were bought from Fluka. Glutathione agarose (12 atoms spacer, sulfhydryl attachment) and PMSF (phenylmethylsulfonylfluoride) were obtained from Sigma; PreScission protease from Pharmacia. HMTT and HPT were synthesized according to the method described elsewhere (U. Kessler, B.D. Pilger, O. Zerbe, L. Scapozza, and G. Folkers, unpubl. results).
E. coli strain BL21 served as host for expression. The plasmid pGEX-6P-2-HSV1TK encoding for the full-length TKHSV1 was constructed as described earlier (Prota et al. 2000; Vogt et al. 2000). All other reagents were purchased from Fluka.
Expression and purification of TKHSV1
TKHSV1 was expressed in E. Coli BL21 as PreScission cleavable amino-terminal GST fusion using the bacterial vector pGEX-6P-2-HSV1TK (Prota et al. 2000; Vogt et al. 2000). One liter of LB medium containing 0.1 mg/mL ampicillin was inoculated with an additional 50 mL overnight culture and expression was induced at 25°C by addition of 0.1 mM IPTG after cells reached an O.D.600 ≥0.7. After 20 h of induction, bacteria were harvested by centrifugation, frozen, thawed, and lyzed in lysis buffer (50 mM Tris at pH 7.5, 1 mM PMSF, 10 mM DTT, 10% glycerol, 1% Triton X-100) in the presence of 150 μg/mL lysozyme and 2000 units Dnase I (adding 10 mM MgCl2, 1 mM MnCl2, 10 mM EDTA for inactivation of Dnase I afterward) for 30 min at 4°C. The lysate was centrifuged at 12,000g for 15 min and the supernatant, containing the TK fusion protein and other periplasmic proteins, was applied three times to 4 mL of glutathione agarose, which was soaked over night at 4°C in TBS (Tris buffer saline containing 50 mM Tris/HCl at pH 7.4 and 150 mM NaCl), packed into a disposable polypropylene column and equilibrated with lysis buffer. The column was then successively washed with lysis buffer, 2 N NaCl in lysis buffer, 0.25 M phosphate buffer at pH 7.5 containing 10 mM ATP and 10 mM MgCl2, and 20 column volumes of TBSE buffer (10 mM Tris/HCl at pH 7.4, 150 mM NaCl, 1 mM EDTA) containing 1 mM DTT. The fusion protein was cleaved on column for 48 h at 4°C by adding 40 units of PreScission protease in 4 mL of TBSE containing 1 mM DTT to the column bed and gently resuspending the fusion protein-bound glutathione agarose. The cleaved TK was then eluted with TBSE as the full-length 376 amino acid protein as shown by amino-terminal sequencing, and dialyzed against TBSE for 24 h at 4°C.
Because Mg2+ is indispensable for the phosphorylation reaction catalyzed by TKHSV1, all measurements were done in the presence of 1 mM EDTA to suppress any enzymatic activity. All samples contained 150 mM NaCl to keep the ionic strength constant.
Expression and purification of the TK were followed by SDS-PAGE. Enzyme concentration was determined using a dye-binding assay (Bradford 1976). For CD analysis, the absorbance of the TKHSV1 sample at 280 nm was measured and the respective concentration was calculated by means of an extinction coefficient derived from the amino acid sequence by the program Sednterp (Sednterp version 1.03, program created by D.B. Hayes, T. Laue, and J. Philo, University of New Hampshire).
Protein crystallization and soaking
Ligand-free full-length HSV1 TK was crystallized as previously described (Prota et al. 2000; Vogt et al. 2000). Crystals were soaked in crystallization buffer containing 5 mM substrate, transferred to the mother liquor containing 30% (v/v) glycerol after 30 min and cryo-cooled to 100 K in a N2 gas stream.
Data collection
X-ray diffraction data of the cryo-cooled crystal were collected on a MAR300 image plate using 0.3-mm collimated Cu Kα radiation from a rotating anode generator (Rigaku, model RU2HC) at 45 kV and 120 mA for the complex TKHSV1:HPT and on EMBL beamline BW7B at DESY (Hamburg) for TKHSV1:HMTT using a MAR345 image plate. Data were processed using program MOSFLM (CCP4 1994). Further processing was carried out using programs SCALA and TRUNCATE (CCP4 1994).
Refinement
The initial model for both structures was the isomorphus ligand-free TKHSV1 without water molecules (Vogt et al. 2000). These coordinates were then submitted to rigid-body, positional and temperature-factor refinement, in alternation with modeling sessions. The refinement and difference-Fourier electron density map calculations were performed with program REFMAC (CCP4 1994) using the same protocol for both structures TKHSV1:HPT and TKHSV1:HMTT. Only in the first refinement cycle non-crystallographic restraints were applied to the dimeric protein model. Water molecules were added automatically using program ARPP (CCP4 1994) and individual B-factors were applied. Sulfate ions and substrate molecules were introduced manually. In the last refinement cycle the noncrystallographic restraints were completely removed. For model manipulations we used program O (Jones et al. 1991). The figures were produced with programs MOLSCRIPT (Kraulis 1991) and RASTER3D (Merrit and Bacon 1997). The coordinates and structure factors are deposited in the Protein Data Bank under accession codes 1E2M, and 1E2N for TKHSV1:HPT and TKHSV1:HMTT, respectively.
Far-UV CD spectra
All circular dichroism measurements were acquired using a JASCO J720 spectropolarimeter attached to a NESLAB 111 bath circulator. Far-UV spectra were recorded between 190 and 250 nm, using a 1-nm slit width and a 0.02-cm path length cell thermostated at 20°C. TKHSV1 was dialyzed into TBSE for 24 h at 4°C and the protein concentration was redetermined after filtration of the sample through a 0.2-μm filter device. Ligand stock solutions (ATP, dT) at 100 mM were prepared in dialysis buffer. Protein and ligand stocks were then combined and diluted to the following four final samples: (1) TKHSV1 0.8 mg/ml (19.5 μM); (2) TKHSV1 0.8 mg/mL with 0.10 mM dT; (3) TKHSV1 0.8 mg/mL with 0.10 ATP; and (4) TKHSV1 0.8 mg/mL with 0.10 mM dT and ATP, respectively. Samples were incubated at 4°C for 24 h before measurements. For control experiments, ligand stocks were diluted in dialysis buffer to yield 0.10 mM dT, 0.10 mM ATP, and 0.10 mM dT + ATP ligand reference solutions. For protein, dialysis buffer and ligand solutions, five scans were averaged (cuvette d = 0.01 cm, band width 1 nm, scan rate 20 nm/min, sensitivity 20 millidegree, response time 2 sec) and then smoothed by a fast Fourier transform routine (supplied by Jasco instrument software). Dialysis buffer and ligand spectra were subtracted from the respective protein spectra and the latter converted into mean residue molar ellipticity.
Thermal denaturation profiles monitored by CD
Thermal unfolding of TKHSV1 was monitored by measuring the CD signal at 223 nm of TK samples containing 0.4 mg/mL (9.7 μM) TKHSV1 in a temperature range from 20° to 70°C with a heating rate of 40°C/h (cell d = 0.5 cm). All measurements were done in TBSE containing 10 mM Tris/HCl at pH 7.4, 150 mM NaCl, and 1 mM EDTA, in the presence and absence of 1.0 mM TK substrate and 1.0 mM ATP. All samples were incubated for 30 min at 20°C before being measured. The average melting temperatures were based on at least three independent data sets. Reversibility was checked by recording the CD signal while cooling a sample from 70°C down to 20°C. To verify that the Tm values were not dependent on the heating rate, a sample containing 0.4 mg/mL TKHSV1 and 1 mM dT and ATP, respectively, was thermally unfolded at heating rates of 30°, 40°, and 50°C/h.
Fluorescence spectroscopy
Fluorescence emission spectra were obtained with a Spex FluoroMax spectrofluorimeter over a wavelength range of 310 to 450 nm at 20°C. Emission and excitation slits widths were 1 mm, and the integration time was 1 sec. We used 295 nm as excitation wavelength to excite only the tryptophane residues and to avoid interference by thymidine quenching at 278 nm. TKHSV1 samples at 0.2 mg/mL (5 μM) were incubated for 60 min before analysis at 25°C, either in TBSE or in TBSE containing 50 μM dT and 250 μM ATP. The data were corrected for buffer signals and represent unsmoothed scans.
Analysis of thermal unfolding data
The thermal unfolding profiles were fitted to a two-state model derived from a previously described model (Ramsay and Eftink 1994), requiring a heating rate independent thermal unfolding. We used a nonlinear least-square fit routine of the program Origin 5.0 (MicroCal Software, Inc., Northhampton).
In case of a transition between two physical states, any general spectroscopic signal observable Y at a particular temperature under a given set of conditions can be expressed as Y = fNYN + (1 − fN)Yu (Eq. 1), where fN is the fraction of molecules remaining in the native state and YN and YU are the signals contributed by the native and unfolded states, respectively. The value of fn is given by fN = 1/(1 + K) (Eq. 2) where K is the equilibrium constant for the unfolding transition K = exp[−ΔH(1 − T/Tm)/RT] (Eq. 3).
ΔH is the enthalpy change between the unfolded and the native states, and Tm is the temperature (in Kelvin) at which half of the material exists in the native state and the other half in the denatured state. Equations (1) to (3) can be combined to obtain the following relationship between the observable Y, in this case the CD signal at 223 nm and the temperature T (in K):
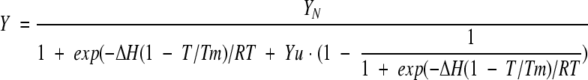 |
4a |
In the analysis of a thermal unfolding profile, the baseline of the observable is frequently observed to have a slope mN and mU, which must be included in the fitting process
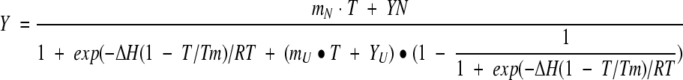 |
4b |
Acknowledgments
We are grateful to Dr. Remo Perozzo for providing us with substrate-free TKHSV1 crystals for soaking experiments. Ulrich Kessler was supported by Discovery Technologies Ltd.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ``advertisement'' in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
CD, circular dichroism
EDTA, ethylenediaminetetraacetic acid
GST, glutathione S-transferase
HMTT, (R,R)-6–(6-hydroxymethyl-5-methyl-2,4-dioxo-hexahydro-pyrimidin-5-ylmethyl)-5-methyl-1H- pyrimidin-2,4-dione
HPT, 6–(3-hydroxypropyl)thymine
TKHSV1, herpes simplex virus type 1 thymidine kinase
PAGE, polyacrylamide gel electrophoresis
r.m.s.d., root mean squared deviation
SDS, sodium dodecyl sulfate
Tris, tris(hydroxymethyl)amino-methane.
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/ps.27401.
References
- Balzarini, J., De Clercq, E., Baumgartner, H., Bodenteich, M., and Griengl, H. 1989. Carbocyclic 5-Iodo-2′-deoxyuridine (C-IDU) and carbocyclic (E)-5–(2-Bromovinyl)-2′-deoxyuridine (C-BVDU) as unique example of chiral molecules where the two enantiomeric forms are biologically active: interaction of the (+)- and (−)-enantiomers of C-IDU and C-BVDU with the thymidine kinase if herpes simplex virus type 1. Mol. Pharmacol. 37 395–401. [PubMed] [Google Scholar]
- Bennett, M.S., Wien, F., Champness, J.N., Batuwangala, T., Rutherford, T., Summers, W.C., Sun, H., Wright, G., and Sanderson, M.R. 1999. Structure to 1.9 A resolution of a complex with herpes simplex virus type-1 thymidine kinase of a novel, non-substrate inhibitor: X-ray crystallographic comparison with binding of aciclovir. FEBS Lett. 443 121–125. [DOI] [PubMed] [Google Scholar]
- Black, M.E., Newcomb, T.G., Wilson, H.M., and Loeb, L.A. 1996. Creation of drug-specific herpes simplex virus type 1 thymidine kinase mutants for gene therapy. Proc. Natl. Acad. Sci. 93 3525–3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonini, C., Ferrari, G., Verzeletti, S., Servida, P., Zappone, E., Ruggieri, L., Ponzoni, M., Rossini, S., Mavilio, F., Traversari, C., and Bordignon, C. 1997. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia [see comments]. Science 276 1719–1724. [DOI] [PubMed] [Google Scholar]
- Bradford, M.M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochem. 72 248–254. [DOI] [PubMed] [Google Scholar]
- Brown, D.G., Visse, R., Sandhu, G., Davies, A., Rizkallah, P.J., Melitz, C., Summers, W.C., and Sanderson, M.R. 1995. Crystal structures of the thymidine kinase from herpes simplex virus type-1 in complex with deoxythymidine and ganciclovir. Nature Structural Biol. 2 876–881. [DOI] [PubMed] [Google Scholar]
- Caruso, M. and Bank, A. 1997. Efficient retroviral gene transfer of a Tat-regulated herpes simplex virus thymidine kinase gene for HIV gene therapy. Virus Res. 52 133–143. [DOI] [PubMed] [Google Scholar]
- CCP4. 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D50 760–763. [DOI] [PubMed] [Google Scholar]
- Champness, J.N., Bennett, M.S., Wien, F., Visse, R., Summers, W.C., Herdewijn, P., de Clerq, E., Ostrowski, T., Jarvest, P.L., and Sanderson, M.R. 1998. Exploring the active site of herpes simplex virus type 1 thymidine kinase by x-ray crystallography of complexes with aciclovir and other ligands. Proteins 32 350–361. [DOI] [PubMed] [Google Scholar]
- Christians, F.C., Scapozza, L., Crameri, A., Folkers, G., and Stemmer, W.P. 1999. Directed evolution of thymidine kinase for AZT phosphorylation using DNA family shuffling. Nature Biotechnol. 17 259–264. [DOI] [PubMed] [Google Scholar]
- Culver, K.W., Ram, Z., Wallbridge, S., Ishii, H., Oldfield, E.H., and Blaese, R.M. 1992. In vivo gene transfer with retroviral vector-producer cells for treatment of experimental brain tumors [see comments]. Science 256 1550–1552. [DOI] [PubMed] [Google Scholar]
- Culver, K.W., Van Gilder, J., Link, C.J., Carlstrom ,T., Buroker, T., Yuh, W., Koch, K., Schabold, K., Doornbas, S., and Wetjen, B. 1994. Gene therapy for the treatment of malignant brain tumors with in vivo tumor transduction with the herpes simplex thymidine kinase gene/ganciclovir system. Hum. Gene Therapy 5 343–379. [DOI] [PubMed] [Google Scholar]
- Drake, R.R., Wilbert, T.N., Hinds, T.A., and Gilbert, K.M. 1999. Differential ganciclovir-mediated cell killing by glutamine 125 mutants of herpes simplex virus type 1 thymidine kinase. J. Biol. Chem. 274 37186–37192. [DOI] [PubMed] [Google Scholar]
- Elion, G.B., Furman, P.A., Fyfe, J.A., de Miranda, P., Beauchamp, L., and Schaeffer, H.J. 1977. Selectivity of action of an antiherpetic agent, 9–(2-hydroxyethoxymethyl) guanine. Proc. Natl. Acad. Sci. 74 5716–5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukada, H., Sturtevant, J.M., and Quiocho, F.A. 1983. Thermodynamics of the binding of L-arabinose and D-galactose to the L-arabinose-binding protein of Escherichia coli. J. Biol. Chem. 258 13193–13198. [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A47 110–119. [DOI] [PubMed] [Google Scholar]
- Keller, P.M., Fyfe, J.A., Beauchamp, L., Lubbers, C.M., Furman, P.A., Schaeffer, H.J., and Elion, G.B. 1981. Enzymatic phosphorylation of acyclic nucleoside analogs and correlations with antiherpetic activities. Biochem. Pharmacol. 30 3071–3077. [DOI] [PubMed] [Google Scholar]
- Kokoris, M., Sabo, P., Adman, E., and Black, M. 1999. Enhancement of tumor ablation by a selected HSV-1 thymidine kinase mutant. Gene Ther. 6 1415–1426. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT—A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Kussmann-Gerber, S., Kuonen, O., Folkers, G., Pilger, B.D., and Scapozza, L. 1998. Drug resistance of herpes simplex virus type 1—Structural considerations at the molecular level of the thymidine kinase. Eur. J. Biochem. 255 472–481. [DOI] [PubMed] [Google Scholar]
- Kussmann-Gerber, S., Wurth, C., Scapozza, L., Pilger, B.D., Pliska, V., and Folkers, G. 1999. Interaction of the recombinant herpes simplex virus type 1 thymidine kinase with thymidine and aciclovir: A kinetic study. Nucleosides & Nucleotides 18 311–330. [DOI] [PubMed] [Google Scholar]
- Merrit, E.A. and Bacon, D.J. 1997. Raster 3D: Photorealistic molecular graphics. Methods Enzymol. 277 505–524. [DOI] [PubMed] [Google Scholar]
- Moolten, F.L. 1994. Drug sensitivity (``suicide'') genes for selective cancer chemotherapy. Cancer Gene Ther. 1 279–287. [PubMed] [Google Scholar]
- Morgan, C.S., Holton, J.M., Olafson, B.D., Bjorkman, P.J., and Mayo, S.L. 1997. Circular dichroism determination of class I MHC-peptide equilibrium dissociation constants. Protein Sci. 6 1771–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller, C.W. and Schulz, G.E. 1992. Structure of the complex between adenylate kinase from Escherichia coli and the inhibitor Ap5A refined at 1.9 A resolution. A model for a catalytic transition state. J. Mol. Biol. 224 159–177. [DOI] [PubMed] [Google Scholar]
- Perozzo, R., Jelesarov, I., Bosshard, H.R., Folkers, G., and Scapozza, L. 2000. Compulsory order of substrate binding of HSV 1 thymidine kinase—A calorimetric study. J. Biol. Chem. 275 16139–16145. [DOI] [PubMed] [Google Scholar]
- Pilger, B.D., Perozzo, R., Alber, F., Wurth, C., Folkers, G., and Scapozza, L. 1999. Substrate diversity of herpes simplex virus thymidine kinase—Impact of the kinematics of the enzyme. J. Biol. Chem. 274 31967–31973. [DOI] [PubMed] [Google Scholar]
- Prota, A., Vogt, J., Perozzo, R., Pilger, B.D., Wurth, C., Marquez, V., Russ, P., Schulz, G., Folkers, G., and Scapozza, L. 2000. Kinetics and crystal structure of the wild-type and the engineered Y101F mutant of Herpes simplex virus type 1 thymidine kinase interacting with (North)-methanocarba-thymidine, Biochemistry 39 9597–9603. [DOI] [PubMed] [Google Scholar]
- Ramsay, G.D. and Eftink, M.R. 1994. Analysis of multidimentional spectroscopic data to monitor unfolding of proteins. Methods Enzymol. 240 615–645. [DOI] [PubMed] [Google Scholar]
- Sacco, M.G., Mangiarini, L., Villa, A., Macchi, P., Barbieri, O., Sacchi, M.C., Monteggia, E., Fasolo, V., Vezzoni, P., and Clerici, L. 1995. Local regression of breast tumors following intramammary ganciclovir administration in double transgenic mice expressing neu oncogene and herpes simplex virus thymidine kinase. Gene Ther. 2 493–497. [PubMed] [Google Scholar]
- Tong, X.W., Agoulnik, I., Blankenburg, K., Contant, C.F., Hasenburg, A., Runnebaum, L.B., Stickeler, E., Kaplan, A.L., Woo, S.L., and Kieback, D.G. 1997. Human epithelial ovarian cancer xenotransplants into nude mice can be cured by adenovirus-mediated thymidine kinase gene therapy. Anticancer Res. 17 811–813. [PubMed] [Google Scholar]
- Verzeletti, S., Bonini, C., Marktel, S., Nobili, N., Ciceri, F., Traversari, C., and Bordignon, C. 1998. Herpes simplex virus thymidine kinase gene transfer for controlled graft-versus-host disease and graft-versus-leukemia: Clinical follow-up and improved new vectors. Hum. Gene Ther. 9 2243–2251. [DOI] [PubMed] [Google Scholar]
- Vogt, J., Perozzo, R., Pautsch, A., Prota, A., Schelling, P., Pilger, B., Folkers, G., Scapozza, L., and Schulz, G.E. 2000. Substrate binding and diversity of HSV 1 1TK studied by X-ray crystallography. Proteins 41 545–553. [DOI] [PubMed] [Google Scholar]
- Vonrhein, C., Schlauderer, G.J., and Schulz, G.E. 1995. Movie of the structural changes during a catalytic cycle of nucleoside monophosphate kinases. Structure 3 483–490. [DOI] [PubMed] [Google Scholar]
- Weber, P.C., Ohlendorf, D.H., Wendoloski, J.J., and Salemme, F.R. 1989. Structural origins of high-affinity biotin binding to streptavidin. Science 243 85–88. [DOI] [PubMed] [Google Scholar]
- Wild, K., Bohner, T., Aubry, A., Folkers, G., and Schulz, G.E. 1995. The three-dimensional structure of thymidine kinase from herpes simplex virus type 1. FEBS Lett. 368 289–292. [DOI] [PubMed] [Google Scholar]
- Wild, K., Bohner, T., Folkers, G., and Schulz, G.E. 1997. The structures of thymidine kinase from herpes simplex virus type 1 in complex with substrates and a substrate analogue. Protein Sci. 6 2097–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]