

Abstract
We have analyzed DNA sequences from world-wide geographic strains of Plasmodium falciparum and found a complete absence of synonymous DNA polymorphism at 10 gene loci. We hypothesize that all extant world populations of the parasite have recently derived (within several thousand years) from a single ancestral strain. The upper limit of the 95% confidence interval for the time when this most recent common ancestor lived is between 24,500 and 57,500 years ago (depending on different estimates of the nucleotide substitution rate); the actual time is likely to be much more recent. The recent origin of the P. falciparum populations could have resulted from either a demographic sweep (P. falciparum has only recently spread throughout the world from a small geographically confined population) or a selective sweep (one strain favored by natural selection has recently replaced all others). The selective sweep hypothesis requires that populations of P. falciparum be effectively clonal, despite the obligate sexual stage of the parasite life cycle. A demographic sweep that started several thousand years ago is consistent with worldwide climatic changes ensuing the last glaciation, increased anthropophilia of the mosquito vectors, and the spread of agriculture. P. falciparum may have rapidly spread from its African tropical origins to the tropical and subtropical regions of the world only within the last 6,000 years. The recent origin of the world-wide P. falciparum populations may account for its virulence, as the most malignant of human malarial parasites.
Keywords: genetic polymorphism, demographic sweep, clonality, selective sweep, parasitic protozoa
There is an extensive literature indicating that the agent of malignant malaria, Plasmodium falciparum, is highly polymorphic. The studies have focused on antigenic determinants, drug-resistance phenotypes, allozymes (1–4), and chromosome sizes (5). Assessment of DNA sequence variation has been based almost exclusively on examination of several genes coding for antigenic determinants, where amino acid polymorphisms (nonsynonymous nucleotide polymorphisms) are common and likely to be affected by natural selection (6, 7). Our study seeks to ascertain certain population parameters by examining synonymous polymorphisms, which do not change the amino acid sequence of the encoded proteins and are less likely to be under selective pressure.
MATERIALS AND METHODS
DNA Sequences.
The 10 genes studied and the geographic origin of the strains are listed in Table 1. The genes are from isolates of P. falciparum collected from global malaria endemic regions. The Dhfr and Ts genes are found directly adjacent to one another on the parasite’s fourth chromosome and encode the bifunctional dihydrofolate reductase–thymidylate synthetase (DHFR–TS) domain. Certain mutations in the Dhfr gene have been widely associated with P. falciparum resistance to antifolate drugs, including pyrimethamine. Two other genes have been implicated with drug-resistant phenotypes of P. falciparum: the gene coding for dihydropteroate synthetase (Dhps) and the gene for multidrug resistance (Mdr1). The circumsporozoite protein (encoded by Csp1) is antigenic, and the rhoptry-associated protein (encoded by Rap1) may also be immunogenic. The other four genes in Table 1 are not known to be immunogenic or associated with resistance to any antimalarial drug currently in use. They code for calmodulin (Calm), glucose-6-phosphate dehydrogenase (G6pd), heat-shock protein 86 (Hsp86), and triose phosphate isomerase (Tpi).
Table 1.
Genes examined in the present survey of genetic variation among global populations of P. falciparum
| Location | Strain | Dhfr | Ts | Dhps | Mdr1 | Rap1 | Calm | G6pd | Hsp86 | Tpi | Csp1 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| South America | |||||||||||
| Honduras | HB3 | x | x | x | x | x | |||||
| Brazil | 7G8 | 2 | 2 | ||||||||
| Brazil | It.G2.F6 | x | |||||||||
| Brazil | It.D12 | x | |||||||||
| Brazil | IMTM22 | x | |||||||||
| El Salvador | Sal-1 | x | |||||||||
| Africa | |||||||||||
| Gambia | FCR3 | x | x | x | |||||||
| Gambia | FC27 | x | x | x | |||||||
| Sierra Leone | SL/D6 | x | x | ||||||||
| Ghana | NF7 | x | |||||||||
| Kenya | M24 | x | |||||||||
| Uganda | Palo Alto | x | x | x | |||||||
| Tanzania | I/CDC | x | |||||||||
| West Africa | Wellcome | x | |||||||||
| Asia/Oceania | |||||||||||
| Papua New Guinea | DR | 22† | |||||||||
| Papua New Guinea | MAD20 | x | |||||||||
| Thailand | K1 | 2 | 2 | x | x | x | x | x | |||
| Thailand | K39 | x | |||||||||
| Thailand | Cs1-2 | x | x | x | |||||||
| Thailand | GH2 | x | |||||||||
| Thailand | T4 | x | |||||||||
| Thailand | T9 | x | x | 3 | |||||||
| Thailand | Tak9 | x | |||||||||
| Thailand | TM335 | x | |||||||||
| Thailand | 806-946 | 13 | |||||||||
| Vietnam | V1 | x | x | x | |||||||
| Vietnam | V1/S | x | |||||||||
| India | Ind C | x | |||||||||
| India | W2 | x | |||||||||
| India | Ind-D | 2 | |||||||||
| India | Ind-J | 2 | |||||||||
| India | Dd2 | x | |||||||||
| Europe | |||||||||||
| Netherlands | CVD1 | x | |||||||||
| Netherlands | 3D7 | x | |||||||||
| Netherlands | NF54* | x | x | x | x | x | x |
The symbol x indicates a single DNA sequence per strain, whereas numbers indicate multiple isolates from the same strain; for Csp1, the Thailand sequences labeled 806-946 are from 13 strains. The GenBank accession numbers (or reference), successively from top to bottom, are as follows: Dhfr, J03772, J04643, J03772, J03028, J03772, M22159, J03772, J03772, J03772, and J03772; Tst, J03772, J04643, J03772, J03028, J03772, M22159, J03772, J03772, J03772, and J03772 (the authors of ref. 8 submitted Dhfr and Ts sequences for several isolates under a single accession number); Dhps, Z30665, Z30657, Z30659, Z30664, U07706, Z30655, Z31584, Z30656, Z30658, Z30660, Z30653, and Z30654; Mdr1, X56851, S53996, and L02513; Rap1, J02985, L10322, L10323, M32853, U41077, U41073, U41074, U41075, and U41076; Calm, X56950 (ref. 9), M99442 (ref. 9), and M59770 (ref. 9); G6pd, M80655 and X74988 (the authors of ref. 10 submitted the G6pd sequence for isolates K1 and 3D7 under a single accession number); Hsp86, L34028 and L34027; Tpi, L01654 and L01655; Csp1, K02194, U20969, M15505, M83172, M83174, M19752, M57499, M57498, M83173, M83149, M83150, M83156, M83158, M83161, M83163, M83164, M83165, M83166, M83167, M83168, M83169, M83170, M83886, X15363, and M22982.
NF54, CVD1, and 3D7 are laboratory strains isolated from a patient in the Netherlands who had not traveled in malaria endemic regions.
Ref. 11 and J. Reeder (personal communication).
Alignment and Phylogenetic Analysis.
We align each set of the gene sequences by means of a progressive multiple-sequence alignment algorithm, using the clustalw computer program (12), with corrections made by eye. Only the gene coding sequence is analyzed; introns and flanking regions are not considered, because of limited availability. The center region of the Csp1 gene contains several repeating units that cannot readily be aligned (see ref. 13) and, therefore, it has been removed from this analysis. We analyze separately the two regions, 5′ and 3′, that embrace the multiple-repeat middle region of Csp1.
We perform the phylogenetic analysis of the Dhfr gene by using the branch-and-bound search method with maximum parsimony optimization criterion of paup (14).
RESULTS
Table 2 summarizes the polymorphisms observed in the 10 genes studied. Amino acid replacement polymorphisms occur in the three drug-resistance genes Dhfr, Dhps and Mdr1, as well as in Csp1, the presumed immunogenic Rap1 gene, and G6pd. No silent polymorphisms are observed in these or any other genes. Silent polymorphisms are polymorphisms that do not affect the amino acid sequence coded. Silent polymorphisms occur in degenerate codons. We will distinguish 4-fold and 2-fold degenerate codons. The 6-fold degenerate codons for amino acids serine, leucine, and arginine are grouped according to their 4-fold or 2-fold codons. The 3-fold degenerate codon for isoleucine is included with the group of 4-fold degenerate codons for the present analysis.
Table 2.
Polymorphisms in 10 loci of P. falciparum
| Gene | Chr | Length, bp | ni | Dn | Ds | Synonymous sites per locus
|
Total synonymous sites analzyed
|
||
|---|---|---|---|---|---|---|---|---|---|
| 4-fold (li) | 2-fold (mi) | 4-fold (nili) | 2-fold (nimi) | ||||||
| Dhfr | 4 | 609 | 32 | 4 | 0 | 67 | 129 | 2,144 | 4,128 |
| Ts | 4 | 1,215 | 10 | 0 | 0 | 125 | 264 | 1,250 | 2,640 |
| Dhps | 8 | 1,269 | 12 | 5 | 0 | 128 | 227 | 1,536 | 2,724 |
| Mdr1 | 5 | 4,758 | 3 | 1 | 0 | 450 | 696 | 1,350 | 2,088 |
| Rap1* | — | 2,349 | 9 | 8 | 0 | 289 | 461 | 1,092 | 1,668 |
| Calm | 14 | 441 | 7 | 0 | 0 | 52 | 86 | 364 | 602 |
| G6pd | 14 | 2,205 | 3 | 9‡ | 0 | 242 | 468 | 726 | 1,404 |
| Hsp86 | 7 | 2,241 | 2 | 0 | 0 | 266 | 455 | 532 | 910 |
| Tpi | — | 597 | 2 | 0 | 0 | 90 | 131 | 180 | 262 |
| Csp1 5′end† | 3 | 387 | 25 | 7 | 0 | 30 | 90 | 688 | 2,010 |
| Csp1 3′end | 3 | 378 | 25 | 17 | 0 | 42 | 65 | 1,050 | 1,625 |
| Total | 16,449 | 130 | 51 | 0 | 1,781 | 3,072 | 10,912 | 20,061 | |
Chr is the chromosome on which the locus is located, when known; ni is the number of sequences; Dn and Ds are the observed number of nonsynonymous and synonymous polymorphisms, respectively; li and mi are the numbers of 4-fold and 2-fold synonymous sites examined.
Rap1 sequences are not complete in all nine isolates examined; values of mi and li are based on the complete gene sequence.
Two short indels occur among the 5′ sequences of Csp1; mi and li are given for the longest sequences (M15505 and M87173). We have excluded the central repeat region of the Csp1 gene, which codes for a variable number of repeats of motifs 4 amino acid long.
In addition to the nine nonsynonymous polymorphisms, there is a 3-bp indel at nucleotides 1,066–1,068.
We do not include β-tubulin (β-tub), of which three sequences are known (GenBank accession nos. M28398, M31205, and X16075), which exhibit three silent polymorphisms and 24 amino acid polymorphisms. The polypeptides α- and β-tubulin form the dimeric protein tubulin, which is a major structural component of microtubules. In most protozoans, including species of Trypanosoma and Leishmania, these genes occur in tandem, as repeated linear arrays along the chromosome. In P. falciparum, the α- and β-tub genes are located on different chromosomes, and each may be represented by at least two copies, as evidenced by multiple mRNA transcripts differing in their untranslated regions (15), although it has been suggested that these transcripts may result from posttranscriptional modification of a single mRNA (16). The large number of nonsilent polymorphisms in the three β-tub sequences suggests that they are not orthologous, i.e., alleles of the same locus. This is corroborated by noticing that the average genetic distance (incidence of amino acid replacements) between the three falciparum sequences is D = 0.040 ± 0.025, whereas between P. falciparum and Plasmodium berghei, D = 0.023 ± 0.010, although these two species diverged 55–129 million years (My) ago (17).
We first consider the hypothesis that the absence of silent polymorphism is a consequence of a recent population bottleneck (or demographic sweep), so that the extant world populations of P. falciparum can be traced to a single recent ancestor. If the population grew to a large size after the bottleneck, it is reasonable to assume that the genealogy of a sample of multiple strains collected from widely distributed localities would be a star-like phylogeny (Fig. 1) with the most recent common ancestor at the vertex of the star (18). Under this assumption, and ignoring the possibility of multiple hits at individual sites, the number of neutral polymorphisms that we observe in a sample of multiple strains will have a Poisson distribution with a mean that depends on the neutral mutation rate, the time elapsed, and the number of lineages examined. The expected number of polymorphisms is λ = μat ∑ nili + μbt ∑ nimi, where μa and μb are the neutral mutation rates at the third position of 4- and 2-fold degenerate codons, respectively; t is the time since the bottleneck; ni is the number of lineages sampled at the ith locus; and li and mi are, respectively, the number of 4-fold and 2-fold synonymous sites examined at the ith locus. This expression suggests an estimator of the time of the bottleneck, obtained by solving for t and replacing λ by S, the observed number of polymorphisms:
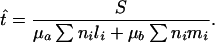 |
1 |
In our sample S = 0, so t̂ = 0. Because S is assumed to be Poisson-distributed, we can estimate an upper 95% confidence limit, t95, for the time of the bottleneck by finding the value of t for which the probability of no polymorphism (e−λ) equals 0.05. Because e−2.996 = 0.05, we calculate the t95 by writing 2.996 in the numerator of Eq. 1.
Figure 1.

Schematic representation of a star phylogeny; t represents the time elapsed between the population bottleneck (cenancestor) and the present.
To use Eq. 1, we need an estimate of the neutral mutation rate at third-position degenerate codons. We estimate this neutral mutation rate for Dhfr by comparing the P. falciparum gene sequences with those available for other species, the rodent parasites P. berghei, Plasmodium chabaudi, Plasmodium vinckei, and Plasmodium yoelii. Fig. 2 shows the phylogenetic relationships among these taxa, based on the Dhfr gene sequences. These relationships are in agreement with phylogenies based on other genes (6, 19). There are 146 amino acid differences between P. berghei and P. falciparum. Among the unchanged amino acid sites, there are 69 and 51 observed synonymous nucleotide differences among the 4-fold (n = 142) and 2-fold (n = 264) degenerate codons, respectively.
Figure 2.

Phylogeny of five Plasmodium species based on 592 coding nucleotides of the Dhfr gene. The tree is a majority-rule consensus, with midpoint rooting of 1,000 bootstrap replicates derived by the branch-and-bound method using the maximum parsimony optimality criterion of the paup 3.1.1 program (14). Bootstrap values are given above each branch. GenBank accession numbers of the sequences used are given in parentheses after the species names. Thick branches indicate the divergence between P. falciparum and P. berghei, which occurred no earlier than 129 My ago.
The observed number of nucleotide differences is probably less than the actual number of substitutions that have occurred since divergence, because multiple substitutions at individual sites would be observed as single differences or no difference. To correct for overlapping substitutions between sequences we use the Jukes–Cantor correction, which is conservative (i.e., it undercorrects) because it assumes that all changes occur with equal frequency, an unrealistic assumption, particularly for genomes such as that of Plasmodium spp. for which A+T richness is around 74% or greater (17). With the Jukes–Cantor method, we estimate that 111.2 4-fold and 64.5 2-fold synonymous substitutions have occurred between P. falciparum and P. berghei. The number of substitutions at 4-fold degenerate sites is estimated by 142(−3/4)ln[1 − (4/3) × (69/142)] = 111.2. At 2-fold degenerate sites a two-state model is assumed, and the number of substitutions is estimated by 264(−1/2)ln[1 − 2 × (51/264)] = 64.5. The conservative bias of the Jukes–Cantor model yields lower estimates of μ and correspondingly higher estimates of t95 than would be obtained with other methods. The radiation of the genus Plasmodium has been estimated to have occurred between 55 and 129 My ago (19). If we assume, conservatively, that the divergence between P. berghei and P. falciparum occurred 129 My ago, we obtain an estimate of the silent mutation rate in 4-fold degenerate codons, μa = 3.03 × 10−9 mutations per site per year; and in 2-fold degenerate codons, μb = 0.95 × 10−9. Alternatively, if 55 My for the divergence between P. falciparum and P. berghei is assumed, μa = 7.12 × 10−9 and μb = 2.22 × 10−9.
We can make a similar comparison of 708 silent sites between the Rap1 gene sequences of P. falciparum and Plasmodium reichenowi (chimpanzee malaria). If 7 My is assumed since the divergence of the two species, μa = 2.70 × 10−9 and μb = 1.33 × 10−9 mutations per site per year. Alternatively, based on a 5-My divergence of P. falciparum and P. reichenowi, μa = 3.78 × 10−9 and μb = 1.86 × 10−9. Average synonymous substitution rates estimated for other organisms are 3.5 × 10−9 and 15.6 × 10−9 per site per year, respectively for mammals and Drosophila (tables 7.1 and 7.6 in ref. 20).
Table 3 gives estimates of t95 and t50. The t50 values are obtained from Eq. 1 by making S = 0.693, because e−0.693 = 0.50. The conclusion is that the 95% confidence interval for the origin of the extant world populations of P. falciparum from a single ancestor is 0–24,511 years or 0–57,481 years, depending on the time of divergence assumed for the Plasmodium radiation. The falciparum–reichenowi divergence times yield 95% confidence intervals included within the previous ones. The t50 values refer to the estimated times for the origin of the falciparum populations, for which the probability of observing more variation is greater than 50% if the cenancestor would have been older than 5,670 or 13,296 years. We should note that all estimates in Table 3 are likely to be larger than actual values because we use the conservative Jukes–Cantor correction for overlapping substitutions. This means that the mutation rates are underestimated, thus yielding longer time intervals before one is 95% (or 50%) confident that at least one silent mutation should have been observed. For purposes of reference, we noted above that the average synonymous substitution rate in Drosophila has been estimated to be 15.6 × 10−9, based on more than 30 Drosophila genes (table 7.6 in ref. 20). The corresponding values for observing no synonymous substitutions among a sample size comparable to ours (n = 30,973 sites) are t95 = 6,281 and t50 = 1,453 years.
Table 3.
Estimated times (t95 and t50, in years) to the most recent common ancestor of the world populations of P. falciparum
| Assumption | Mutation rate (×10−9)
|
t95 | t50 | |
|---|---|---|---|---|
| 4-fold (μa) | 2-fold (μb) | |||
| Plasmodium radiation | ||||
| 55 My | 7.12 | 2.22 | 24,511 | 5,670 |
| 129 My | 3.03 | 0.95 | 57,481 | 13,296 |
| falciparum–reichenowi | ||||
| 5 My | 3.78 | 1.86 | 38,136 | 8,821 |
| 7 My | 2.70 | 1.33 | 53,363 | 12,343 |
t95 gives the upper boundary of the 95% confidence interval of the estimate; t50 represents the estimated time above which the probability is greater than 50% of observing more variation than actually observed.
DISCUSSION
Other hypotheses, besides a recent severe population bottleneck, might account for the absence of silent polymorphism in P. falciparum: (i) persistent low effective population size, (ii) low rates of spontaneous mutation, (iii) strong selective constraints on silent variation, and (iv) one or more recent selective sweeps affecting several chromosomes.
Hypothesis i can readily be excluded for the present given the worldwide distribution of P. falciparum that occurs in many millions of infected humans. If the effective worldwide population of P. falciparum would have been very small (tens or at most hundreds of individuals) for very many generations until not long ago, this would effectively amount to a population bottleneck.
There seems to be no reason to suspect that spontaneous mutation rates are exceptional in P. falciparum (hypothesis ii), and there are two arguments against it. One is the high incidence of polymorphisms at antigenic and drug-sensitivity sites, both in worldwide samples (3, 21–24) and in laboratory selection experiments with mice (25). The other argument is that there is divergence, in synonymous and nonsynonymous sites, between P. falciparum and other Plasmodium species (7, 26, 27).
Similarly, there seems to be no reason to suspect that there are strong selective constraints against silent variation in P. falciparum (hypothesis iii). P. falciparum has a 76.4% incidence of A+T, which is typical of other Plasmodium species; e.g., we have observed 75.8% in P. berghei. In addition, silent polymorphisms appear in the comparisons between P. falciparum and other Plasmodium species (Fig. 2 for Dhfr; see refs. 7 and 26 for other genes) at the typical rates observed for other organisms. Moreover, two clinical isolates of a related species, the Apicomplexan Cryptosporidium parvum, exhibit 29 silent polymorphisms among 1,530 bp of the Dhfr gene (sequences from ref. 28; GenBank accession nos. U41365 and U41366).
Natural selection (hypothesis iv) may account for the rapid spread of a favored genotype throughout populations, particularly when the population is large and/or the selection is strong. The repeated appearance throughout global malaria endemic regions of drug-resistant phenotypes, determined by nonsynonymous substitutions at the Dhfr, Dhps, and other loci, is most likely due to natural selection. Selection sweeps are known in other organisms, such as Drosophila melanogaster, where a single nucleotide sequence at the Sod locus is present in about 50% of all haplotypes throughout the world, without any silent substitutions along the 1,500-bp sequence, although there is one widespread nonsynonymous substitution that accounts for a fast–slow allozyme polymorphism (29, 30). Natural selection can account for the absence of synonymous variation at any of the 10 loci we have analyzed, if the particular gene sequence itself (or a gene with which it is linked) has been subject to a recent worldwide selective sweep, without sufficient time for the accumulation of new synonymous mutations. However, the 10 genes are located on, at least, six different chromosomes, and so six selective sweeps would need to have occurred more or less concurrently, which seems unlikely. A selective sweep simultaneously affecting all chromosomes could happen if the population structure of P. falciparum were predominantly clonal rather than sexual.
Some authors have argued that there is no evidence that P. falciparum propagates clonally in nature but rather predominantly propagates by crossing between dissimilar parasites (31). This inference is based on three observations: (i) high incidence of mixed infections in individual patients, (ii) presence of two unlike alleles in diploid oocysts isolated from mosquito midguts, and (iii) evidence of intragenic recombination. However, the question whether P. falciparum has an effectively clonal population structure is not incompatible with the occurrence of recombination in this obligatory sexual parasite. The issues are whether recombination is meiotic (rather than mitotic) and how often it occurs. Our analysis of polymorphisms in antigenic peptide repeats of the Csp1 gene indicates that these polymorphisms can be generated by somatic (mitotic), rather than sexual (meiotic), recombination. Moreover, the rate of intragenic recombination and the strength of linkage disequilibrium between nucleotide sites are independent of the distance between nucleotides, which is inconsistent with meiotic recombination but is consistent with a clonal population structure for P. falciparum (13). The evidence used for rejecting the hypothesis of clonality is based upon genetic polymorphisms at loci that code for antigenic determinants (32, 33). These loci may be under strong selection for avoiding the host’s immune responses, and it is, therefore, difficult to determine how frequently recombination takes place, because even rare recombinants could rapidly reach high frequency. For example, Escherichia coli is regarded as being clonal (34) and hence recombination is relatively rare; nonetheless the identification of recombinant segments within genes is not uncommon (35, 36). In light of this, the evidence currently available is not sufficient to reject the hypothesis that P. falciparum has, like Trypanosoma and Leishmania (37–43), a clonal population structure.
It may also be pointed out that a clonal population structure is not inconsistent with high levels of polymorphism, as observed in P. falciparum (for review, see ref. 31). Thus, the observation of 60 different phenotypes in 60 isolates provides no grounds for rejecting clonality out of hand, as done in ref. 2. One well known example (among many available ones) of extensive genetic variation in a clonal organism is E. coli, in which it is rare to find multiple independent isolates exhibiting the same multiple locus enzyme electrophoresis (MLEE) type; rather, a sample of 60 isolates is likely to contain 60 MLEE types (34, 44, 45). Genetic polymorphism is very extensive in Trypanosoma cruzi. On the basis of allozyme variation at 13 loci in a sample of 121 stocks, 43 different genotypes were found, 27 of which were present only once in the sample (37, 41, 43). Yet the clonal population structure of T. cruzi is well established (37–43). For P. falciparum, the data in Table 2 show that amino acid polymorphisms are common; thus, multiple multilocus genotypes will exist by accumulation of different amino acid polymorphisms independently arisen at different loci by natural selection. Arnot (46, 47) attributed the paucity of synonymous substitutions in P. falciparum to a codon-use bias. Codon-use bias is pervasive throughout the Plasmodium genus; however, it does not constrain silent polymorphisms between Plasmodium species or within species other than falciparum.
How could we account for a recent demographic sweep in P. falciparum, if it were not due to natural selection? One possible hypothesis is that P. falciparum has become a human parasite in recent times, by lateral transfer from some other host species. Recent human parasitism is frequently alleged as the explanation for the virulence of P. falciparum (48). This hypothesis demands that the Plasmodium parasites in the ancestral host species would be extremely similar to P. falciparum (i.e., that there be no synonymous or nonsynonymous substitutions between P. falciparum and the unknown parasite, except for amino acid replacements recently arisen in response to drugs or the host’s immune system). The closest known relative of P. falciparum is the chimpanzee parasite, P. reichenowi, which on the basis of nucleotide sequence differences is estimated to have diverged several My ago from P. falciparum (26, 49). An alternative hypothesis is that human parasitism by P. falciparum has long been highly restricted geographically and has dispersed throughout human populations in very recent times. Three (not mutually exclusive) possible scenarios may have led to this recent rapid dispersion: (i) changes in human behavior, (ii) genetic changes in the host–parasite–vector association that have altered the compatibility within the system, and (iii) widespread demographic changes (migration, abundance, etc.) of the human host, the mosquito vectors, and/or the parasite. Change in human behavior (scenario i), particularly the development of agricultural societies (50, 51), has been invoked as an explanation for the widespread occurrence of P. falciparum in human populations. The multiple independent origins of sickle-cell trait are cited as evidence in support of this hypothesis (8, 52). Genetic changes that have increased the affinity within the parasite–vector–host system (scenario ii) also seem a viable explanation for the recent expansion, given the high rate of phenotypic change in the parasite since the bottleneck (48, 53). In support of (scenario iii), Mario Coluzzi (54) has cogently argued that the worldwide distribution of P. falciparum has come about as a consequence of a recent dramatic rise in vectorial capacity due to repeated speciation events in Africa of the most anthropophilic members of the species complexes of the Anopheles gambiae and Anopheles funestus mosquito vectors.
The development of P. falciparum parasites in the mosquito vector is dependent on temperature, which affects zygote formation and the duration of the sporogonic cycle. The limiting temperature for P. falciparum lies around 18°C, but the optimal temperatures are above 26°C, at which temperature the sporozoites can reach the mosquito’s salivary glands in about 10 days, compatible with the life span of the vector (55). The modern spread of P. falciparum from tropical Africa into the Northern Hemisphere was, therefore, constrained by the Pleistocene glaciations. Climatic conditions approaching the present ones did not occur in the Mediterranean region and the Middle East until about 6,000 years ago, well after the end of the Würm glaciation (56, 57). The spread of P. falciparum and its vectors beyond tropical Africa may have occurred even later, as a consequence of extensive deforestation, the spread of agriculture, and the evolution of effective mosquito vectors (54–57). The molecular-based estimates presented herein, inferring that the cenancestor of the extant world populations of P. falciparum lived only several thousand years ago, are consistent with these considerations.
Acknowledgments
We thank Mario Coluzzi, Allan Dickerman, Victor DeFilippis, Walter Fitch, Anthony James, Benjamin Rosenthal, Steve Schrodi, Andrew Spielman, and Andrey Tatarenkov for helpful comments on the manuscript. We thank John Reeder for sharing unpublished Dhfr gene sequence data. This work was supported by National Institutes of Health Grant GM42397 to F.J.A.
ABBREVIATION
- My
Million year(s)
References
- 1.Babiker H A, Lines J, Hill W G, Walliker D. Am J Trop Med Hyg. 1997;56:141–147. doi: 10.4269/ajtmh.1997.56.141. [DOI] [PubMed] [Google Scholar]
- 2.Creasey A, Fenton B, Walker A, Thaithong S, Oliveira S, Mutambu S, Walliker D. Am J Trop Med Hyg. 1990;42:403–413. doi: 10.4269/ajtmh.1990.42.403. [DOI] [PubMed] [Google Scholar]
- 3.Kemp D J, Cowman A F, Walliker D. Adv Parasitol. 1990;29:75–149. doi: 10.1016/s0065-308x(08)60105-0. [DOI] [PubMed] [Google Scholar]
- 4.McConkey G A, Waters A P, McCutchan T F. Annu Rev Microbiol. 1990;44:479–498. doi: 10.1146/annurev.mi.44.100190.002403. [DOI] [PubMed] [Google Scholar]
- 5.Sinnis P, Wellems T E. Genomics. 1988;3:287–295. doi: 10.1016/0888-7543(88)90117-6. [DOI] [PubMed] [Google Scholar]
- 6.Hughes A L. Mol Biol Evol. 1992;9:381–393. doi: 10.1093/oxfordjournals.molbev.a040730. [DOI] [PubMed] [Google Scholar]
- 7.Hughes M K, Hughes A L. Mol Biochem Parasitol. 1995;71:99–113. doi: 10.1016/0166-6851(95)00037-2. [DOI] [PubMed] [Google Scholar]
- 8.Pagnier J, Mears J G, Dunda-Belkhodja O, Shaefer-Rego K E, Bedford C, Nagel R L, Labie D. Proc Natl Acad Sci USA. 1984;81:1771–1773. doi: 10.1073/pnas.81.6.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cowman A F, Galatis D. Exp Parasitol. 1991;73:269–275. doi: 10.1016/0014-4894(91)90098-h. [DOI] [PubMed] [Google Scholar]
- 10.O’Brien E, Kurdi-Haidar B, Wanachiwanawin W, Carvajal J L, Vulliamy T J, Cappadoro M, Mason P J, Luzzatto L. Mol Biochem Parasitol. 1994;64:313–326. doi: 10.1016/0166-6851(94)00028-x. [DOI] [PubMed] [Google Scholar]
- 11.Reeder J C, Rieckmann K H, Genton B, Lorry K, Wines B, Cowman A F. Am J Trop Med Hyg. 1996;55:209–213. doi: 10.4269/ajtmh.1996.55.209. [DOI] [PubMed] [Google Scholar]
- 12.Thompson J D, Higgins D G, Gibson T J. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rich S M, Hudson R R, Ayala F J. Proc Natl Acad Sci USA. 1997;94:13040–13045. doi: 10.1073/pnas.94.24.13040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Swofford D. Phylogenetic Analysis Using Parsimony. Washington, DC: Smithsonian Institution; 1991. [Google Scholar]
- 15.Sen K, Godson G N. Mol Biochem Parasitol. 1990;39:173–182. doi: 10.1016/0166-6851(90)90056-r. [DOI] [PubMed] [Google Scholar]
- 16.Delves C J, Alano P, Ridley R G, Goman M, Holloway S P, Hyde J E, Scaife J G. Mol Biochem Parasitol. 1990;43:271–278. doi: 10.1016/0166-6851(90)90151-b. [DOI] [PubMed] [Google Scholar]
- 17.Jukes T H, Cantor C R. In: Mammalian Protein Metabolism. Munro H T, editor. New York: Academic; 1969. pp. 21–132. [Google Scholar]
- 18.Slatkin M, Hudson R R. Genetics. 1991;129:555–562. doi: 10.1093/genetics/129.2.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Escalante A A, Ayala F J. Proc Natl Acad Sci USA. 1995;92:5793–5797. doi: 10.1073/pnas.92.13.5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li W-H. Molecular Evolution. Sunderland, MA: Sinauer; 1997. [Google Scholar]
- 21.Bickle Q, Anders R F, Day K, Coppel R L. Mol Biochem Parasitol. 1993;61:189–196. doi: 10.1016/0166-6851(93)90065-6. [DOI] [PubMed] [Google Scholar]
- 22.Hirawake H, Kita K, Sharma Y D. Biochim Biophys Acta. 1997;1360:105–108. doi: 10.1016/s0925-4439(97)00007-0. [DOI] [PubMed] [Google Scholar]
- 23.Kemp D J. Immunol Cell Biol. 1992;70:201–207. doi: 10.1038/icb.1992.25. [DOI] [PubMed] [Google Scholar]
- 24.Qari S H, Collins W E, Lobel H O, Taylor F, Lal A A. Am J Trop Med Hyg. 1994;50:45–51. doi: 10.4269/ajtmh.1994.50.45. [DOI] [PubMed] [Google Scholar]
- 25.Cowman A F, Lew A M. Mol Biochem Parasitol. 1990;42:21–30. doi: 10.1016/0166-6851(90)90109-y. [DOI] [PubMed] [Google Scholar]
- 26.Escalante A A, Barrio E, Ayala F J. Mol Biol Evol. 1995;12:616–626. doi: 10.1093/oxfordjournals.molbev.a040241. [DOI] [PubMed] [Google Scholar]
- 27.Hughes A L. Genetics. 1991;127:345–353. doi: 10.1093/genetics/127.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vasquez J R, Gooze L, Kim K, Gut J, Petersen C, Nelson R G. Mol Biochem Parasitol. 1996;79:153–165. doi: 10.1016/0166-6851(96)02647-3. [DOI] [PubMed] [Google Scholar]
- 29.Hudson R R. Proc Natl Acad Sci USA. 1994;91:6815–6818. doi: 10.1073/pnas.91.15.6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hudson R R, Saez A G, Ayala F J. Proc Natl Acad Sci USA. 1997;94:7725–7729. doi: 10.1073/pnas.94.15.7725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Babiker H, Walliker D. Parasitol Today. 1997;13:262–267. doi: 10.1016/s0169-4758(97)01075-2. [DOI] [PubMed] [Google Scholar]
- 32.Kerr P J, Ranford-Cartwright L C, Walliker D. Mol Biochem Parasitol. 1994;66:241–248. doi: 10.1016/0166-6851(94)90151-1. [DOI] [PubMed] [Google Scholar]
- 33.Marshall V M, Coppel R L, Martin R K, Oduola A M J, Anders R F, Kemp D J. Mol Biochem Parasitol. 1991;45:349–351. doi: 10.1016/0166-6851(91)90104-e. [DOI] [PubMed] [Google Scholar]
- 34.Ochman H, Selander R K. Proc Natl Acad Sci USA. 1984;81:198–201. doi: 10.1073/pnas.81.1.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hartl D L. Curr Opin Genet Dev. 1992;2:937–942. doi: 10.1016/s0959-437x(05)80119-4. [DOI] [PubMed] [Google Scholar]
- 36.Boyd E F, Hill C W, Rich S M, Hartl D L. Genetics. 1996;143:1091–1100. doi: 10.1093/genetics/143.3.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tibayrenc M, Ward P, Moya A, Ayala F J. Proc Natl Acad Sci USA. 1986;83:115–119. doi: 10.1073/pnas.83.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tibayrenc M, Kjellberg F, Ayala F J. Proc Natl Acad Sci USA. 1990;87:2414–2418. doi: 10.1073/pnas.87.7.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tibayrenc M, Kjellberg F, Arnaud J, Oury B, Brenière S F, Dardé M-L, Ayala F J. Proc Natl Acad Sci USA. 1991;88:5129–5133. doi: 10.1073/pnas.88.12.5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tibayrenc M, Neubauer K, Barnabé C, Guerrini F, Skarecky D, Ayala F J. Proc Natl Acad Sci USA. 1993;90:1335–1339. doi: 10.1073/pnas.90.4.1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tibayrenc M, Ayala F J. Evolution. 1988;42:277–292. doi: 10.1111/j.1558-5646.1988.tb04132.x. [DOI] [PubMed] [Google Scholar]
- 42.Tibayrenc M. Annu Rev Microbiol. 1996;50:401–429. doi: 10.1146/annurev.micro.50.1.401. [DOI] [PubMed] [Google Scholar]
- 43.Ayala F J. Biol Res. 1993;26:47–63. [PubMed] [Google Scholar]
- 44.Selander R K, Levin B R. Science. 1980;210:545–547. doi: 10.1126/science.6999623. [DOI] [PubMed] [Google Scholar]
- 45.Whittam T S, Ochman H, Selander R K. Proc Natl Acad Sci USA. 1983;80:1751–1755. doi: 10.1073/pnas.80.6.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arnot D. Parasit Today. 1990;6:64–65. doi: 10.1016/0169-4758(90)90210-u. [DOI] [PubMed] [Google Scholar]
- 47.Arnot D E. Acta Leiden. 1991;60:29–35. [PubMed] [Google Scholar]
- 48.Waters A P, Higgins D G, McCutchan T F. Proc Natl Acad Sci USA. 1991;88:3140–3144. doi: 10.1073/pnas.88.8.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Escalante A A, Ayala F J. Proc Natl Acad Sci USA. 1994;91:11373–11377. doi: 10.1073/pnas.91.24.11373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Livingston F B. Am Anthropol. 1958;60:533–560. [Google Scholar]
- 51.Weisenfeld S L. Science. 1967;157:1134–1140. doi: 10.1126/science.157.3793.1134. [DOI] [PubMed] [Google Scholar]
- 52.Stine OC, Dover G J, Zhu D, Smith K D. J Mol Evol. 1992;34:336–344. doi: 10.1007/BF00160241. [DOI] [PubMed] [Google Scholar]
- 53.Garnham P C C. Malaria Parasites and Other Haemosporidia. Oxford, U.K.: Blackwell; 1966. [Google Scholar]
- 54.Coluzzi M. Evoluzione Biologica & i Grandi Problemi della Biologia. Rome, Italy: Accademia dei Lincei; 1997. pp. 263–285. [Google Scholar]
- 55.Coluzzi M. Parassitologia. 1994;36:223–227. [PubMed] [Google Scholar]
- 56.Zulueta J de. Parassitologia. 1973;15:1–15. [Google Scholar]
- 57.Zulueta J de. Parassitologia. 1994;36:7–15. [PubMed] [Google Scholar]