

Abstract
Gene number can be considered a pragmatic measure of biological complexity, but reliable data is scarce. Estimates for vertebrates are 50-100,000 genes per haploid genome, whereas invertebrate estimates fall below 25,000. We wished to test the hypothesis that the origin of vertebrates coincided with extensive gene creation. A prediction is that gene number will differ sharply between invertebrate and vertebrate members of the chordate phylum. A gene number estimation method requiring limited sequence sampling of genomic DNA was developed and validated by using data for Caenorhabditis elegans. Using the method, we estimated that the invertebrate chordate Ciona intestinalis has 15,500 protein-coding genes (±3,700). This number is significantly lower than gene numbers of vertebrate chordates, but similar to those of invertebrates in distantly related phyla. The data indicate that evolution of vertebrates was accompanied by a dramatic increase in protein-coding capacity of the genome.
Data on gene numbers offer one route to testing the hypothesis that the origin of vertebrates coincided with extensive gene creation (1–3). However, in the near future, determination of eukaryotic gene numbers by whole-genome sequencing will be practicable only for Homo sapiens and a few model organisms. Estimation strategies also exist, but have drawbacks: counts of genetic loci fall far short of the real number of genes (4), estimates based on RNA reassociation studies are complicated by tissue-restricted expression (5), and extrapolation from the number of CpG islands (6) is only feasible for vertebrates, CpG islands being absent in invertebrates. Of the sequence-based approaches, clustering of expressed sequence tags (ESTs) (7, 8) requires massive commitment of resources. Extrapolation from the proportion of ESTs that match genes predicted from a set of sequenced cosmids containing genomic DNA, first used for Caenorhabditis elegans (4, 9), also requires large-scale sequencing of genomic DNA and cDNAs. The approach of Brenner et al. (10), though designed to estimate genome size, is also relevant. They sequenced more than 500 short random fragments of genomic Fugu rubripes DNA. The sequences were translated and searched against the protein database, and the fraction of coding sequence was calculated. Computing the equivalent fraction at that time for humans, and assuming Fugu and human share the same genes, they estimated the Fugu genome size to be 390 megabases, in agreement with independent estimates.
Here we report the development of a strategy for estimating the number of protein-coding genes requiring only limited sequencing, its validation on data from the nematode C. elegans, and application to the ascidian, Ciona intestinalis.
Following Brenner et al. (10), we generate random fragments of genomic DNA by sonication, then clone and sequence these fragments. Putative protein-coding sequences are identified by translating the sequence in all six frames and searching for homologs in the National Center for Biotechnology Information nonredundant protein database by using the blastx program (11), and processing of the blast output with custom-written unix scripts and programs (see Experimental and Computational Methods). The result is a value for the proportion (pg) of the bases in the genomic sample identified as protein coding by homology. This sample value can be taken as an estimate of the whole genome value, as the sample comprises multiple random fragments. As this technique fails to detect genes not represented in the databases, pg will be less than the fraction of the genome that codes for protein (fcode), the quantity we seek to estimate. The ideal solution would be to evaluate fcode as pg/pc, with pc denoting the proportion (counted codon by codon) of the organism’s complete set of proteins possessing database homologs. As the full set of proteins is not available, pc is estimated by sequencing members of a representative cDNA library, and searching for database homologs in the same manner as for genomic DNA. There are also several secondary factors, discussed below, which impinge on this approach to calculating fcode. From fcode the number of protein-coding genes can be inferred given the genome size and assuming a typical protein length. This latter assumption should constitute only a minor source of uncertainty, as estimates of average protein length show little variation in eukaryotes, being, for example, 484 and 442 amino acids for Saccharomyces cerevisiae and C. elegans, respectively (12), and 462 amino acids for H. sapiens proteins listed in the swiss-prot database (release 34) (13). The quantities pg and pc are subject to sampling error. Mathematical and numerical methods allow estimation of these errors and consequently the margin of error in fcode (see Experimental and Computational Methods). Being able to assess how the error margin scales with factors such as genome size and potential gene number allows selection of appropriate values for the number of genomic and cDNA sequences necessary to achieve an estimate with acceptable error bounds.
The strategy assumes that the likelihood of finding protein homologs by database searching is the same for exon sequences in genomic DNA and for cDNA. This assumption is not strictly valid, for three reasons. First, algorithms like blast are more effective at detecting similarities in long sequence stretches. The interruption of coding regions by introns in eukaryotes, coupled with the random location of the genomic fragments, means that even if an exon overlaps a genomic fragment, truncation caused by an intron or fragment boundary often will lead to it being short and so less likely to be detected. The result is underestimation of pg relative to pc. Our solution is to rescale pg via statistical modeling of the truncation effect. This method estimates the correction factor appropriate for any particular combination of exon size, fragment length, and hit score threshold θ. Given data on the distribution of exon lengths, a global correction factor γ then can be computed (see Experimental and Computational Methods), and the revised pg calculated as p′g = γpg. The other two biases influence fcode in opposing directions. They concern the use of cDNAs to provide a representative sample of proteins. First, the cDNAs may contain untranslated regions (UTRs); these regions ideally should be excluded before evaluating the fraction of translated cDNA with protein homologs. If this exclusion is not done, then pc will be underestimated. To minimize this bias we focus on 5′ end reads, as 5′ UTRs generally are shorter than 3′ UTRs (14) and many cDNAs are also incomplete. The second potential bias relates to the cDNA library. If cDNAs from highly expressed genes dominate it, then pc likely will be higher than if a gene’s representation in the library was independent of its expression level (the ideal case). To ameliorate the effect, the library should be normalized with respect to expression level as far as possible (4).
Experimental and Computational Methods
Generation of C. intestinalis DNA sequences.
Genomic DNA was sonicated, and fragments of around 500 bp were isolated. The fragment ends were repaired, cloned into the EcoRV site of pBluescript KS, and then transformed into SURE (Stratagene). Clones were sequenced once on an Applied Biosystems 373A Stretch automated sequencer, by using dye-terminator chemistry. After excluding reads <300 bp, 1,487 genomic sequences were obtained. The proportion of ambiguous bases was ≈0.5%.
Total RNA was prepared from adult C. intestinalis (without mantle) (15). Poly(A)+ RNA was isolated (Promega PolyATract Isolation System), and a cDNA library was made (Stratagene ZAP-cDNA kit). Clones were excised, then prepared and sequenced from the 5′ end by using the above method. A few also were sequenced from the 3′ end. Twenty two of the 83 final clones gave a strong signal when hybridized with total transcribed RNA probe—these highly expressed clones then were eliminated. A total of 76 ESTs were obtained.
Sequence Database Searches.
blastx searches were performed by using the blosum62 matrix. The mspcrunch program (16) was used to extract all of the hits from the blast files, then only those hits scoring above a threshold θ, set at 100, were retained. Excluding bases found to hit proteins associated with retroviral-like elements, i.e. reverse transcriptase, gag or pol proteins, and several database entries that contain erroneously translated rRNA, the number of bases lying in hits was computed.
Sampling Error Analysis.
Let n denote the number of sequences in each sample. The proportion px of bases in a sample that are covered by database hits, where x denotes g or c for genomic DNA or cDNA samples, respectively, can be written px = hl̄hit/l, where h is the fraction of sequences that shows database hits after the various screening steps, l̄hit is the mean number of bases per sequence covered by hits in the group of sequences possessing hits, and l is the mean sequence length. To estimate the sampling error in px, we treat each sequence as an independent trial, and take the sample proportion h to estimate the probability of any single sequence having database hits. Thus the number of sequences that show database hits (and consequently the proportion h) is modeled by binomial statistics. For all of the samples analyzed in the current work, h < 0.5, and nh > 30, so the binomial distribution is well approximated by the normal distribution. The quantity l̄hit also is subject to sampling variation; by the law of large numbers, the variance in l̄hit equals σlhit2/(nh), where σlhit2 denotes the variance of the distribution of lhit values from individual sequences, estimated directly from the sample data. For all of the samples in the current work, the coefficient of variation (σ/μ) associated with l̄hit is lower than that associated with h. Thus we made the approximation of ignoring the variation in l̄hit and so treated pg and pc as normally distributed variables. As fcode = pg/pc, we require the frequency distribution of the quotient of two normal variables. This distribution is given by ref. 17:
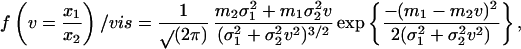 |
1 |
where m1, m2, σ1, and σ2 are the means and SDs, respectively of the normal distributions from which variates x1 and x2 are drawn, and it is assumed that x2 has a positive range (a valid assumption in the current context). It is then straightforward to compute confidence intervals for fcode by numerical integration of this distribution function.
Modeling the Truncation Effect.
Consider a genomic fragment of length lf, an exon of length le, and the criterion that exonic regions are only “recognizable” if they satisfy a length threshold T. Assume lf > le > T. There are lf + le − 1 alignments in which the fragment and exon overlap. As the fragment locations are random the alignments are equiprobable, so the average fraction of a fragment that overlaps exon, Q, is given by:
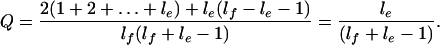 |
2 |
The average fragment fraction recognizable, Qr, is lower than this:
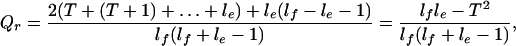 |
3 |
so the potential efficiency of coding-region recognition, Qr/Q, is (1 − T2/lfle). Similar reasoning shows that this result also holds when le > lf > T. The appropriate value of T (in bases) should be linked to the level of the hit score threshold θ by setting T = 3θ/Kθ, where Kθ is the average score per amino acid in the database hits detected against the genomic fragments using θ. The global correction factor γ then is computed as γedge/γvis, where γedge is the mean value of (1 − T2/lfle)−1 averaged over the range of exon sizes le and weighted with respect to the proportion of nucleotides in exons of each size. γvis is an estimate of the proportion of coding bases lying in exons of size >T: its inclusion compensates for the smallest exons being effectively invisible in high stringency homology searches of genomic sequence.
A second type of truncation effect arises because of the fragmentation of coding regions into conserved and nonconserved domains. However, unlike the effect caused by the exonic nature of genes, this type affects the hit rate against both genomic and cDNA sequences. As fcode is the ratio of these hit rates, the two influences should approximately cancel, so no explicit attempt is made to correct for this effect.
There is potentially another bias toward relative underestimation of pg. Consider, for example, two adjacent exons, each of which shows only weak homology to a database protein, such that the “matches” are rejected in the screening process. In the cDNA, however, the contiguity of the two coding regions would enhance the chance of a significant homology being detected. In practice, exploratory experiments with C. elegans data failed to find evidence of a significant contiguity bias (data not shown), therefore it was not considered further.
Construction and Analysis of C. intestinalis Cosmids.
Genomic DNA was obtained from adult C. intestinalis and partially digested with Sau3AI. After dephosphorylation of the digested DNA, fragments were size-selected on a sucrose gradient, cloned into SuperCos I (Stratagene), and packaged into XL1-Blue MR cells. The average insert size was ≈35,000 bp. Four cosmids were randomly selected for sequencing and analysis at the Sanger Centre; their GenBank accession numbers are Z80904, Z79640, Z83760, and Z83861.
Results and Discussion
Validation Using C. elegans Data.
To develop and test the approach, we conducted computational experiments on existing sequence data from C. elegans. This organism’s status as the target of a genome project makes it ideal for this purpose: large cDNA libraries exist and most of the genome has been sequenced and analyzed (9). As the genomic regions sequenced so far have tended to be gene-rich, it would be inappropriate to apply our method to estimating total gene number in C. elegans. Instead, we estimate the protein-coding fraction in a set of cosmids (repeating the experiment several times by using independent sequences) and compare this to the equivalent value for these cosmids as deduced by the sequencing team. These coding fraction values are 0.19 (SD .02) and 0.26, respectively (see Table 1)—similar but not equal. We attribute the slight discrepancy to underrepresentation of lowly expressed genes in the cDNA library leading to artificial inflation of pc. This belief is supported by the finding that re-evaluation of pc with a sample of proteins that should be unbiased with respect to expression level (1,000 of the putative proteins identified by the Sanger/St. Louis teams in genomic sequence) yielded pc = 0.261 and therefore a final fcode value of 23%. We conclude that it would be helpful to compensate for this almost inevitable bias. This adjustment was done in the subsequent study of Ciona by excluding highly abundant cDNA species (identified by hybridization) from the set of sequences used to derive pc. More generally, as each experiment required only 500 kb of genomic sequence, these tests with C. elegans data indicated that coding density estimation through limited sampling is a feasible strategy.
Table 1.
Estimation of the coding fraction for a set of C. elegans cosmids
| Sequence analysis statistics | ||
|---|---|---|
| Quantity | Sequence type
|
|
| Genomic | cDNA | |
| Length l (bp) | 500 ± 0 | 350 ± 3.6 |
| h | 0.095 ± .005 | 0.403 ± .03 |
| p | 0.048 ± .003 | 0.316 ± .02 |
| p′ | 0.060* ± .003 | — |
| Estimates of the coding fraction fcode | ||
| Method | Value | |
| Derived (p′g/pc) | 0.190 ± .02† | |
| Cosmid analysis‡ | 0.264 | |
A sample of genomic DNA sequences (1,000 fragments, each 500 bp) was randomly selected from the 1,316 cosmid sequences in the EMBL database by October 1996. A sample of cDNA sequences (100 5′ ESTs) was randomly selected from the EMBL entries from the normalized library of Yuji Kohara (National Institute of Genetics, Japan; library details available at http://www.nig.ac.jp/labs/nenpo-95e/G/G-d.html). To prevent bias, matches to C. elegans database sequences were ignored. This experiment was conducted 10 times using independent sequences to check the sampling error analysis: mean ± SD values over these runs are shown. h is the fraction of sequences in the sample with any database hits, p the proportion of bases in the sample covered by hits. p′ denotes p after correction, applicable for genomic DNA. Sensitivity to the value of θ is modest: repeat analyses with θ = 75 and 150 give mean fcode values of 0.217 and 0.171, respectively. We set θ = 100: lower values allow many false positive homologies, higher values make the method overly dependent on γ.
Evaluated by using the cosmid annotations to obtain the distribution of exon lengths used in computing γ; relevant values: K100 = 2.70, γedge = 1.11, and γvis = 0.885.
Consistent with the SD value predicted from the analysis of sampling error: 0.03.
Evaluated from the regions identified as coding in the cosmid sequence annotations.
Application to C. intestinalis.
Having validated the method, we then applied it to estimate the gene number of the ascidian, C. intestinalis, which is an invertebrate member of the phylum Chordata. Larval ascidians exhibit many vertebrate-like anatomical characteristics, and this fact plus molecular evidence (18) suggests that ascidians diverged from the chordate lineage, which subsequently led to the early vertebrates (2). This particular ascidian species was selected because its genome is known to be small. Renaturation kinetics studies (19) estimate the haploid genome size to be 35 times that of Escherichia coli. Combined with the known E. coli genome size, 4.64 megabases (20), this yields an estimate of 162 megabases. The results are shown in Table 2. In summary, using 1,487 genomic fragments, 76 ESTs, and exon size data obtained from four C. intestinalis cosmids sequenced and analysed at the Sanger Centre (see Experimental and Computational Methods), we estimate the fraction of the genome coding for protein to be 13.2% (90% confidence interval: 10.4–16.7%). Taking a figure of 460 for the mean number of amino acids per protein, this translates into an estimate of 15,500 ± 3,700 protein-coding genes. A second estimate can be derived from the 18 genes (excluding a likely reverse transcriptase) identified by homology searches and exon-prediction algorithms in the 150.7 kb of sequence in the four cosmids. Before extrapolating the sample gene density to the genome, it is usual to first assess whether the density in the cosmid sample is typical of the genome by seeing what proportion of a large cDNA library maps to the cosmid genes (4, 9). However, as our cosmids were randomly selected, we proceeded with direct extrapolation, which predicts 19,300 genes—somewhat higher than our main estimate.
Table 2.
Estimation of the coding fraction for C. intestinalis
| Sequence analysis statistics | ||
|---|---|---|
| Quantity | Sequence type
|
|
| Genomic | cDNA | |
| Length l (bp)* | 592 ± 83 | 409 ± 102 |
| h | 0.100 | 0.474 |
| p | 0.036 | 0.319 |
| p′ | 0.042† | — |
| Estimate of the coding fraction fcode | ||
| Derived (p′g/pc) | 0.132 | |
Analysis conducted by using 1,487 genomic sequences and 76 ESTs from cDNAs obtained as described in Experimental and Computational Methods. The analysis was performed by using θ = 100, ignoring all matches to C. intestinalis database sequences.
Data quoted as mean ± SD.
Evaluated by using 103 exons identified in the four C. intestinalis cosmids to approximate the distribution of exon lengths in computing the correction factor; relevant values: K100 = 2.41, γedge = 1.10, and γvis = 0.94.
We provide an estimate of gene number in an invertebrate member of the chordate phylum. It is notable that the value is in keeping with those of invertebrates in other phyla and below estimates for vertebrates, as illustrated in Fig. 1. The result therefore is consistent both with the hypothesis of a ceiling on invertebrate gene numbers (3, 25) and the hypothesis of genome duplication events occurring in the ancestral vertebrate lineage only after the divergence of the tunicates (1). Clearly, more stringent tests of these hypotheses await data from cephalochordates and primitive vertebrates such as Agnatha. In those future studies, the C. intestinalis gene number estimate reported here will serve as a reference value from a member of a basal chordate lineage.
Figure 1.

Gene number estimates. Shaded bars denote results based on completed genome projects. The error bars should be regarded as approximate indicators of uncertainty only, as statistically comparable values are generally not available. This is not a comprehensive summary of data from all available species. Current estimates for mouse (3, 21) and pufferfish (22) are not shown but fall within the range shown for human. The C. elegans estimate, 16,527 ± 260, is the most up-to-date figure as calculated by the method used in ref. 9. Other data are from refs. 3, 7, 20, 21, 23, and 24 and references therein.
Acknowledgments
We are grateful to Susan Tweedie and Jillian Charlton for help with preparing minipreps and DNA sequencing, to Matthew Jones, Barbara Harris, Richard Dobson, Nicola Lennard, and Jo Kershaw for cosmid sequencing, and to Joan Davidson and Aileen Greig for technical assistance. This work was supported by The Howard Hughes Medical Institute, The Wellcome Trust, The Medical Research Council (United Kingdom), and the Human Frontiers Science Programme.
Footnotes
References
- 1.Ohno S. Evolution by Gene Duplication. Heidelberg: Springer; 1970. [Google Scholar]
- 2.Holland, P. W. H., Garcia-Fernàndez, J., Williams, N. A. & Sidow, A. (1994) Development (Cambridge, U.K.), Suppl., 125–133. [PubMed]
- 3.Bird A P. Trends Genet. 1995;11:94–100. doi: 10.1016/S0168-9525(00)89009-5. [DOI] [PubMed] [Google Scholar]
- 4.Waterston R, Martin C, Craxton M, Huynh C, Coulson A, Hillier L, Durbin R, Green P, Shownkeen R, Halloran N, et al. Nat Genet. 1992;1:114–123. doi: 10.1038/ng0592-114. [DOI] [PubMed] [Google Scholar]
- 5.Cavalier-Smith T. The Evolution of Genome Size. Chichester: Wiley; 1985. [Google Scholar]
- 6.Antequera F, Bird A P. Proc Natl Acad Sci USA. 1993;90:11995–11999. doi: 10.1073/pnas.90.24.11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fields C, Adams M D, White O, Venter J C. Nat Genet. 1994;7:345–346. doi: 10.1038/ng0794-345. [DOI] [PubMed] [Google Scholar]
- 8.Schuler G D, Boguski M S, Stewart E A, Stein L D, Gyapay G, Rice K, White R E, Rodriquez-Tome P, Aggarwal A, Bajorek E, et al. Science. 1996;274:540–546. [PubMed] [Google Scholar]
- 9.Waterston R, Sulston J. Proc Natl Acad Sci USA. 1995;92:10836–10840. doi: 10.1073/pnas.92.24.10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brenner S, Elgar G, Sandford R, Macrae A, Venkatesh B, Aparicio S. Nature (London) 1993;366:265–268. doi: 10.1038/366265a0. [DOI] [PubMed] [Google Scholar]
- 11.Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 12.Netzer W J, Hartl F U. Nature (London) 1997;388:343–349. doi: 10.1038/41024. [DOI] [PubMed] [Google Scholar]
- 13.Bairoch A, Apweiler R. Nucleic Acids Res. 1996;24:21–25. doi: 10.1093/nar/24.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pesole G, Grillo G, Liuni S. Comput Chem. 1996;20:141–144. doi: 10.1016/s0097-8485(96)80016-7. [DOI] [PubMed] [Google Scholar]
- 15.Chomczyrski P, Sacchi N. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 16.Sonnhammer E L L, Durbin R. Comput Appl Biosci. 1994;10:301–307. doi: 10.1093/bioinformatics/10.3.301. [DOI] [PubMed] [Google Scholar]
- 17.Kendall M G, Stuart A. The Advanced Theory of Statistics. Vol. 1. London: Griffin; 1958. p. 270. [Google Scholar]
- 18.Satoh N, Jeffery W R. Trends Genet. 1995;11:354–359. doi: 10.1016/s0168-9525(00)89106-4. [DOI] [PubMed] [Google Scholar]
- 19.Laird C D. Chromosoma. 1971;32:378–406. doi: 10.1007/BF00285251. [DOI] [PubMed] [Google Scholar]
- 20.Blattner F R, Plunkett G, III, Bloch C A, Perna N T, Burland V, Riley M, Collado-Vides J, Glasner J D, Rode C K, Mayhew G F, et al. Science. 1997;277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 21.Miklos G L G, Rubin G M. Cell. 1996;86:521–529. doi: 10.1016/s0092-8674(00)80126-9. [DOI] [PubMed] [Google Scholar]
- 22.Elgar G. Hum Mol Genet. 1996;5:1437–1442. doi: 10.1093/hmg/5.supplement_1.1437. [DOI] [PubMed] [Google Scholar]
- 23.Fleischmann R D, Adams M D, White O, Clayton R A, Kirkness E F, Kerlavage A R, Bult C J, Tomb J F, Dougherty B A, Merrick J M, et al. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- 24.Goffeau A, Barrell B G, Bussey H, Davis R W, Dujon B, Feldmann H, Galibert F, Hoheisel J D, Jacq C, Johnston M, et al. Science. 1996;274:546–567. doi: 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]
- 25.Bird A, Tweedie S. Philos Trans R Soc London B. 1995;349:249–253. doi: 10.1098/rstb.1995.0109. [DOI] [PubMed] [Google Scholar]