

Abstract
Whole-genome duplication approximately 108 years ago was proposed as an explanation for the many duplicated chromosomal regions in Saccharomyces cerevisiae. Here we have used computer simulations and analytic methods to estimate some parameters describing the evolution of the yeast genome after this duplication event. Computer simulation of a model in which 8% of the original genes were retained in duplicate after genome duplication, and 70–100 reciprocal translocations occurred between chromosomes, produced arrangements of duplicated chromosomal regions very similar to the map of real duplications in yeast. An analytical method produced an independent estimate of 84 map disruptions. These results imply that many smaller duplicated chromosomal regions exist in the yeast genome in addition to the 55 originally reported. We also examined the possibility of determining the original order of chromosomal blocks in the ancestral unduplicated genome, but this cannot be done without information from one or more additional species. If the genome sequence of one other species (such as Kluyveromyces lactis) were known it should be possible to identify 150–200 paired regions covering the whole yeast genome and to reconstruct approximately two-thirds of the original order of blocks of genes in yeast. Rates of interchromosome translocation in yeast and mammals appear similar despite their very different rates of homologous recombination per kilobase.
Comparison of gene order among genomes can be used for two purposes: inferring the phylogenetic relationships of species, and estimating the number and type of genomic rearrangements that have occurred since two genomes last shared a common ancestor. Three mechanisms of rearrangement are usually considered: inversion, transposition, and reciprocal translocation (1–3). Gene order comparisons have been made on sequenced organelle and viral genomes (4–8), and on more sparsely mapped mammalian and plant nuclear chromosomes (1, 8–10).
The genome of baker’s yeast (Saccharomyces cerevisiae) contains approximately 55 large duplicated chromosomal regions, as described by our laboratory (11), Mewes et al. (12), and Coissac et al. (13). We proposed that these duplicated regions (“blocks”) are traces of ancient tetraploidy in S. cerevisiae that remain detectable after widespread deletion of superfluous duplicate genes, sequence divergence of the remaining duplicates, and successive genomic rearrangements. Patterns and characteristics of the duplicated blocks should contain information about the original order of the blocks and the number of rearrangements that have taken place since genome duplication, as well as information about the extent of gene retention versus deletion after the original genome duplication. Analysis of the layout of duplicated blocks in the genome points to reciprocal translocation as the main form of large-scale genome rearrangement in yeast because, of the 55 blocks reported by Wolfe and Shields (11), only 5 have different orientations relative to the centromere in the two copies. This conservation of orientation with respect to the centromere is characteristic of reciprocal translocations and would not be expected if a significant role were played by either inversion or transposition of large chromosomal regions.
In this study we tried to estimate properties of the yeast genome prior to the whole-genome duplication, and to reconstruct gene order evolution in its aftermath. We assumed that the model proposed in our original study—duplication of the whole genome in a single event—is correct, even though other models (such as the duplication of many but not all chromosomes) cannot be ruled out absolutely (11–13). Our aim in this study was to estimate the number of reciprocal translocations that occurred, the original number of genes in the genome, and the original order of the blocks that are now duplicated. The methods used are based on comparative genomics, but they differ from most previous gene order studies because the two genomes we are comparing are not distinct but are indistinguishable, fragmented, and fused within the same nucleus.
We began by making computer simulations to model yeast genome evolution. A genome was duplicated, genes were deleted at random, and reciprocal translocations were made between chromosomes. An algorithm equivalent to that used to find duplicated blocks in the real yeast data (11) was applied to the simulated genomes. These sets of blocks were then analyzed in two ways. The first method involved reversing reciprocal translocations to bring the genome back to a symmetrical configuration (as would be expected immediately after genome duplication), and using parsimony to choose between alternative series of translocations. The simulations showed that this method cannot regenerate the original block order or provide an accurate estimate of the number of translocations when this number is large. The second method involved adjusting the parameters of the simulation (number of duplicate genes retained and number of translocations) to find the parameter ranges that yielded simulated genomes that were similar to the yeast data in terms of number of blocks, extent of the genome placed inside blocks, and number of duplicate genes identified in blocks. It was possible to find parameters for the simulations that produced duplicated block patterns very similar to those in the real genome.
An analytic approach was developed based on the method of Nadeau and Taylor (1) for estimating the number of rearrangements between the human and mouse genetic maps. The analytic approach was used to estimate the number of reciprocal translocations in the real yeast data, given the proportion of the genome that is spanned by known duplicated chromosomal blocks. The estimate produced by this approach falls within the range of estimates produced independently by simulation. This estimate in turn permits estimation of a rate of chromosomal translocation in yeast and its comparison with other species. Last, we investigated whether genome data from additional species would allow us to determine the original order of genes in yeast.
General Methods
Unit of Length.
We took the distance between two genes to be the number of genes located between them, rather than the actual distance in kilobases, despite the fact that complete sequence data are available for the genome. The number of genes is a more natural unit when discussing the distribution of reciprocal translocation sites because the probability of a translocation event having been fixed between two points is likely to be influenced most by the amount of noncoding DNA in the interval, which is expected to be correlated more strongly with the number of genes than with the physical separation of the points along the chromosome. This unit is also more natural when discussing the distribution of duplicates that have been retained after diploidization because the probability of deletion of a gene should not be strongly influenced by its physical size.
Simulations and Identification of Duplicated Segments.
In simulations we assumed that there were no inversions, transpositions, or any other type of rearrangement except reciprocal translocations; that translocations occur at random intergenic locations; that gene deletion occurs by random deletion of single genes; that sequence similarity is detected only between genes duplicated during the tetraploidization; and that natural selection does not impose any functional constraints on gene order. In our model an original genome with eight chromosomes was duplicated and genes were deleted randomly until the current configuration (5,790 genes on 16 chromosomes) remained. This is a rough approximation of the process associated with genome duplication and subsequent diploidization (14). Reciprocal translocations were then made between randomly chosen points in the genome, and blocks of duplicated genes were identified by using criteria similar to those in our original study (11). It was possible to fully automate the block-finding process because all the duplicate genes in the simulated data resulted from genome duplication (there were no multigene families) and as a result blocks were easily identifiable by a simple program. The blocks produced were not very sensitive to the value chosen for the maximal distance between intervening genes once this was greater than about 20 genes. A maximal distance of 45 was used in practice. A minimum of three retained duplicates was required for the identification of a block. The program used to locate the blocks in the simulated data was adapted for use on the real data to permit direct comparison of the results with those in ref. 11. Subtelomeric regions were ignored altogether because the level of noise was too great for the identification of blocks within these regions by this simple method. The threshold for identifying duplicate genes in the real data was a BLASTP score of 200. The resulting blocks were almost identical to the blocks reported by Wolfe and Shields (11), which were identified by using a criterion of three duplicate genes per 50 kb.
Transformation of the Genome to a Symmetrical Configuration.
By “symmetrical configuration” we mean a configuration of blocks in which the chromosomes can be grouped into two identical sets. The computer program written to transform the arrays of blocks to a symmetrical configuration is based on a simple search method in which a symmetry improving operation is chosen at each step. It does not find the shortest or most parsimonious path by which a symmetrical configuration can be achieved. Each point in Fig. 1 was constructed by choosing the shortest of just 10 such paths to symmetry. It is unnecessary to search further because from Fig. 1 we can see that we are already achieving symmetry in fewer steps than were involved in the simulation. The most parsimonious path tells us little about the actual evolutionary path taken.
Figure 1.

Simulations of rearranging a duplicated yeast genome and then reconstructing its original structure. The number of steps taken by our program to bring about symmetry in a configuration of blocks is plotted against the number of reciprocal translocations in the simulation that brought about the original block configuration. Each point represents the shortest of 10 simulations of a 5,790-gene genome with 446 pairs of retained paralogs. Five runs were carried out for each value on the x-axis. Circles indicate the average fraction of the genome that could be assigned to duplicated blocks in simulations (using a minimum of three duplicated genes per block); this fraction declines as more reciprocal translocations are made.
Making the Genome Symmetrical by Reversing Reciprocal Translocations
Inspection of the map of duplicated regions (11) shows three points where the symmetry of the map could be increased by reversing apparent reciprocal translocations. These points involve duplicated chromosomal blocks 14/23/37/50, 38/39/50/52, and 5/6/32/33 (see ref. 11). In each of these cases four blocks can be reduced to two larger blocks by undoing a translocation. This observation suggests that it might be possible to “unscramble” the yeast genome by making a series of reversals of reciprocal translocations until a completely symmetrical genome remains. We speculated that the shortest series of reverse translocations leading to symmetry might correspond to the evolutionary path taken by the yeast genome after its duplication, and we investigated this possibility by computer simulation. The problem of finding the minimal number of translocations to transform the gene order of one genome into another has been studied extensively (5, 7, 8, 15). Here rather than calculating the translocation distance between two genomes we wish to examine sets of translocations that relocate the paralogous blocks within a single genome so that the chromosomes form two identical sets.
Genomes were simulated undergoing duplication, gene deletion, and multiple reciprocal translocations. Duplicated blocks (containing three or more duplicate genes) in the simulated genomes were then identified, and a search was made for series of reciprocal translocations that would rearrange these blocks into a symmetrical configuration. A search routine in which translocations were chosen by a hill-climbing approach (continually increasing the symmetry of the genome) was developed. In simulations with 20 or fewer translocations this search method usually returned the blocks to a perfectly symmetrical configuration in the same number of steps as were performed to bring about the configuration (Fig. 1). As the number of translocations in the simulation is increased the number of steps required to bring about symmetry levels off and begins to fluctuate widely. The fraction of the genome that can be placed in duplicated blocks decreases as the number of translocations increases, and many smaller blocks are not detected (Fig. 1). The effect of failing to detect some blocks (or deleting some blocks from a data set) is to reduce the number of steps required to return the remaining blocks to a symmetric configuration (see also ref. 16). It then becomes possible to return to a symmetrical genome in fewer steps than the original number of translocations.
The shortest solution we found for the real yeast data (in a nonexhaustive search) returned the blocks to a perfectly symmetrical configuration in 41 steps [after three initial inversions to correct the five blocks whose orientation with respect to the centromere is opposite to that of their copies (11)]. Forty-one reciprocal translocations would give rise to 2R + C = 90 pairs of duplicated chromosomal regions, where R is the number of reciprocal translocations and C is the original preduplication number of chromosomes (eight). Because we have discovered only 55 duplicated blocks and because only half of the genome is placed in blocks (11) we can be confident that there are many smaller duplicated regions that have not been discovered. Because the effect of deleting blocks only decreases the number of steps required to return to a symmetrical configuration we can deduce that it is likely that there have been more than 41 reciprocal translocations since genome duplication.
Numerical Estimate of the Number of Reciprocal Translocations Since Duplication
Even when the number of reciprocal translocations in simulations is so large that saturation has been reached for the number of reverse steps required to achieve symmetry (Fig. 1), the fraction of the genome that is assigned to duplicated blocks continues to decrease almost linearly. This observation suggests that approaches based on the latter measure might be more effective ways to estimate the number of translocations than the reverse-translocation approach taken above, when the number of translocations is large.
We repeated the simulations, varying two parameters to reproduce the observed state of the yeast genome. These simulations used reciprocal translocation as the sole mechanism of chromosomal rearrangement, and the block layouts they produced were similar to the structure of the real genome (Fig. 2). The parameters varied were the number of reciprocal translocations fixed since whole genome duplication, and the number of genes retained in duplicate (paralogs†) after genome duplication (Fig. 3). We do not have an exact value for the number of paralogs in the whole (real) yeast genome because similar genes can be identified as paralogs only by their occurrence in the correct position within a regional chromosomal duplication, and we do not have a duplication map for all parts of the genome. Similarly, in the simulated genomes, the number of pairs of paralogs recovered in blocks is less than the actual number of paralogs present (Fig. 3).
Figure 2.

The duplicated chromosomal regions in a simulated genome with 446 pairs of paralogs retained and 75 reciprocal translocations since duplication. These simulations gave rise to patterns and densities of duplicated blocks that are similar to those mapped in the real data (11). Circles indicate centromeres; bars show duplicated blocks. The scale indicates numbers of genes.
Figure 3.

Genome structure simulations in which the number of reciprocal translocations and the number of retained paralogs were varied. Each cell shows values for the number of blocks discovered in the genome (top), the proportion of the genome that is in blocks (middle), and the number of pairs of duplicated genes discovered within blocks (bottom). Mean values among 200 replicates are shown. The standard error on the number of blocks was ≤4; that on the fraction of genome in blocks was ≤0.03; that on the number of paralogs identified was ≤9. Shaded cells are within two standard errors of the value for the yeast data. Fifty-five blocks covering 51% of the genome and containing 365 pairs of paralogs have been mapped in the yeast data (11).
Each cell of Fig. 3 shows characteristics of the genomes produced from 200 simulations for a given combination of input parameters. The values, in the yeast data, of the three genome characteristics shown in Fig. 3 are 55 blocks, 0.51 of the genome in blocks, and 365 pairs of paralogs in blocks. Only input parameters in the region of 8% of duplicate genes retained (400–450 pairs) and 70–100 translocations give results similar to the real data.
Analytic Estimate of the Number of Reciprocal Translocations Since Duplication
It is also possible to convert the fraction of the genome in blocks (Fig. 1) into an estimate of the number of translocations, without computer simulation, by using an analytic method analogous to that of Nadeau and Taylor (1). In their approach to the similar problem of determining the combined rate of rearrangements in mice and humans, Nadeau and Taylor examined the average lengths of conserved linkage groups. In yeast, because we have complete sequence information, we can use the fraction of the genome that is spanned by paralogous chromosomal blocks instead.
We wish to estimate the underlying number of chromosomal regions (“Segments”) that were demarcated by reciprocal translocations, rather than the number of duplicated regions that can now be identified (“Blocks”). Each Block that has been identified (11) is part of a larger Segment. Because we have assumed that paralogs are scattered randomly throughout the genome, the number contained in a Segment of length x is described by a Poisson distribution as in ref. (1). The probability that a Segment of length x contains three, or more, paralogs and so would be identified is
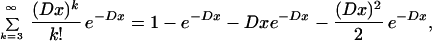 |
where D is the density of paralogs in the whole genome. We do not know the value of D exactly because only the paralogs that occur in the correct position within a Block can be identified as paralogs. We have a lower limit on D because we know the number of paralogs that are contained in Blocks. We can estimate the correct value of D in two ways. We can use the simulations (Fig. 3) and note that there is a relatively small window of densities for which our parameter values come close to modeling the real data. The simulations suggest a density of about 0.15 paralog per gene (i.e., 2 × 7.5%), but this method has the undesirable effect of linking the analytic and simulative methods of calculating the result. To avoid this linking we can examine the number of cases where two genes that are homologs (i.e., a significant “simple” BLASTP pair as defined in ref. 11) are both located anywhere in the half of the genome that has been mapped into Blocks, but their locations are such that they are not considered to be paralogs. The number of such internal duplicates should be approximately the same as the number of strictly external nonparalog hits (i.e., with both genes occurring outside Blocks) because the areas inside and outside Blocks are approximately the same in extent. Any excess in hits outside Blocks represents likely paralogs that have not been identified because they are not contained in Blocks of three or more. This method yields a density of 0.155 paralog per gene.
Because the probability of having a region of length x with no translocation point is e−x/L, where L is the average length of all Segments, the probability density of identified Segment lengths is
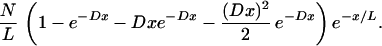 |
The constant N/L is introduced for normalization, where N is the total number of Segments.
The total fraction of the genome covered by identified Segments, F, is then expected to be
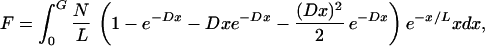 |
where G is the total length of the genome.
If L is small compared with G, the integral evaluated at G approaches 0, so we evaluate the integral at 0 only:
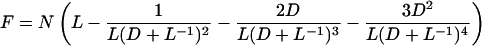 |
or
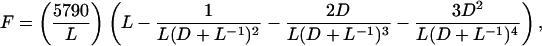 |
1 |
where 5,790 is the number of genes in the genome. F is the proportion of the genome spanned by identified Segments—i.e., the Segments containing the known Blocks.
If m is the expected length of a Segment that contains n paralogs separated by a total distance r, then m = r(n + 1)/(n − 1) as in Nadeau and Taylor (1). We can modify our figure for the fraction of the yeast genome covered by Blocks to approximate the fraction of the genome spanned by identified Segments. We calculate the expected length of each Segment from the range of the paralogs it contains, and sum the Segments. The fraction of the genome covered by Blocks is 0.496, and the estimated fraction of the genome in identified Segments is 0.686 (not including some telomeric genes that could not be confidently placed in Blocks or outside the blocked region because of a high level of intertelomeric similarity). The value of L required to give this result in Eq. 1 is 16.45 genes. This gives N = 5790/16.45 = 352 Segments (organized as 176 pairs). From 2R + C = 176 pairs of Segments, and C = 8 chromosomes, the number of reciprocal translocations (R) is approximately 84. In simulations the standard deviation of the fraction of the genome under Blocks was ≤0.03. This gives us an estimate of 84 ± 15 (for approximately two standard deviations) for the number of reciprocal translocations that have been fixed in yeast since genome duplication.
We can make a prediction of the number of additional Blocks that we would expect to find if we relaxed the block-finding criteria to include Segments containing only two paralogs. The probability of a Segment of length x containing y paralogs is [(Dx)y/y!]e−Dx. The expected number of Segments of length x is (1/L)e−x/LN. Therefore the expected number of Segments containing y paralogs is
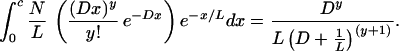 |
On the basis of a model with 446 pairs of retained duplicates and 84 reciprocal translocations the expected value of the number of Segments containing two paralogs is 26 ± 5 (the error was calculated from simulations; Table 1). The number of additional two-member blocks found in the real data is 34. This high value leads us to suspect that it could be difficult to distinguish between genuine small duplicated regions and statistical noise. The predicted number of one-member blocks is 36 (Table 1).
Table 1.
Theoretical predictions, results of simulations, and values from the real data, of the number of blocks containing a given number of paralogs
| Number of paralogs (P) | Number of blocks having P paralogs
|
||
|---|---|---|---|
| Theoretical prediction | Simulation | Real data | |
| 0 | 49.6 | 49.4 | NA |
| 1 | 35.6 | 35.9 ± 5.9 | NA |
| 2 | 25.6 | 26.2 ± 5.2 | NA |
| 3 | 18.4 | 18.5 ± 4.3 | 10 |
| 4 | 13.2 | 13.2 ± 3.5 | 10 |
| 5 | 9.5 | 9.6 ± 3.1 | 6 |
| 6 | 6.8 | 6.8 ± 2.5 | 4 |
| 7 | 4.9 | 4.8 ± 2.1 | 6 |
| 8 | 3.5 | 3.3 ± 1.7 | 6 |
| 9 | 2.5 | 2.4 ± 1.4 | 1 |
| 10 | 1.8 | 1.8 ± 1.2 | 4 |
| 11 | 1.3 | 1.3 ± 1.1 | 2 |
| 12 | 0.9 | 0.9 ± 1.0 | 1 |
| 13 | 0.7 | 0.6 ± 0.7 | 4 |
| 14 | 0.5 | 0.4 ± 0.6 | 0 |
| 15 | 0.3 | 0.3 ± 0.6 | 0 |
| 16–20 | 0.8 | 0.6 ± 2.6 | 1 |
| 21–25 | 0.1 | 0.0 | 0 |
The simulation results (mean ± 2 SD from 2,000 replicates) are from a model with 446 retained paralogs and 84 reciprocal translocations. NA, not applicable.
Possible Clustering of Duplicates
The approach of trying to reverse reciprocal translocations produced symmetrical arrangements of the 55 known blocks in 41 steps (after three initial inversions). In Fig. 1, 41 reverse steps is on the lower extreme of the scatter when the graph has become saturated, which is the region of interest because the other methods show that there have been approximately 84 reciprocal translocations since genome duplication. However, in other simulations in which the number of blocks was fixed from the outset at 55 (results not shown), 41 was close to the mean number of operations required for the return to a symmetrical configuration. This discrepancy arises because the simulations that produced the closest match to the real values of genome parameters (Fig. 3) tended to have slightly larger numbers of duplicated blocks than were discovered in the yeast data. In many cases this difference was only about 1–2 standard deviations and may not be systematic, but a nonrandom distribution of paralogs could also explain the shortage of discovered blocks (20). If paralogs tend to be located in clusters more Segments than might be expected could fail to contain three paralogs, the requirement for identification. It was not possible to test for clustering of paralogs over the whole genome because they cannot be identified outside blocks. No clustering was discovered when all duplicates were taken into account. Some clustering of duplicates may occur for functional reasons—for example, the frequent duplication of pairs of adjacent ribosomal protein genes transcribed divergently from a shared promoter. If there is an excess of undiscovered blocks because of clustering of retained duplicates we would expect this to affect the numbers of smaller blocks. Using the analytic method to predict the number of blocks of a given size that we would expect to find in the data, we find that the expectation is consistently higher than the actual value for blocks containing six paralogs or fewer (Table 1). This is what we would expect if clustering of duplicates is preventing the discovery of some smaller blocks.
Establishing the Original Gene Order
We considered the possibility that the approach of finding the most parsimonious path to genome symmetry might reveal some aspects of the original (pre-duplication) block order, even though the number of steps in this series is too few, as explained above. However, there are a great many equally parsimonious paths returning the data to a symmetrical configuration in the shortest number of steps. This degeneracy is intrinsic. The two operations in Fig. 4 have the same effect on symmetry. Because reverse translocations are commutative (except possibly those involving more than one operation on a single chromosome arm) whole sets of stepwise equivalent operations can give rise to vastly different configurations of blocks in the same number of steps. For example, almost any series of 20 reverse translocations, each of which maximally improves the symmetry at each step, could be used to return a simulation involving 20 reciprocal translocations to symmetry. Because each of these translocations has an alternative that improves the symmetry to exactly the same degree (Fig. 4) we have 220 sets of possible final block orders brought about by 20 operations on the data. We cannot distinguish between these block orders without further information.
Figure 4.

An example of the two indistinguishable solutions to the problem of reversing a single reciprocal translocation in S. cerevisiae. The translocation involves duplicated chromosomal blocks 14, 23, 50, and 37 (see ref. 11).
This degeneracy, in the case of yeast, could in principle be resolved by the inclusion of information from one other species that diverged from S. cerevisiae at around the time of genome duplication. We find in simulations that we can completely reconstruct the order of the blocks in a duplicated yeast-like genome, using as an outgroup a second species that diverged from it immediately prior to duplication, if fewer than 40 reciprocal translocations have been fixed in the duplicated genome (results not shown). Above this number of translocations the solution begins to decay, because if both copies of a block have been shifted by reciprocal translocation we no longer have any information from the duplicated genome about their original locations. In simulations with a realistic number (75) of reciprocal translocations, approximately two-thirds of the duplicated chromosomal blocks that contained sufficient numbers of paralogs for identification could be placed in their original order by using a species that diverged shortly before genome duplication (results not shown).
Similar gene order reconstructions are possible even without genome duplication, but sequence information from a third species is required. Using sequence information from three species that diverged at around the same time to determine ancestral gene order is more reliable than the method described above because in this case all the genes in the genome, not just the paralogs making up blocks, can be used to infer the original gene order.
Discussion
Our simulations involved several assumptions. Chromosomal inversions and gene transposition were ignored as possible mechanisms of gene order change. This assumption is reasonable because inversions and transpositions on a scale large enough to produce blocks containing at least three paralogs are evidently uncommon (11). We assumed also that reciprocal translocations are evenly distributed even though this is open to debate (21, 22). If reciprocal translocation sites are not random our result for the number of translocations since genome duplication is likely to be an underestimate. Our estimates of the proportion of genes retained in duplicate, and the number of genes in the original genome, are sensitive to the sequence similarity threshold used in the analysis (BLASTP > 200, which is quite stringent). The choice of similarity threshold should not, however, affect the estimate of the number of translocations; detecting additional paralogs in yeast is analogous to mapping additional genes in humans and mice, and the inclusion of these extra pieces of data should not substantially alter the estimates of the extent of rearrangement (1, 23).
The 70–100 reciprocal translocations estimated to have occurred would have produced 148–208 paired duplicated chromosomal blocks if each breakpoint was unique. One-third of these (55 blocks) were large enough to be detected in our original study (11), and the remainder must correspond to blocks containing two, one, or even no duplicated genes. We estimate that 36 one-member blocks and 26 two-member blocks exist (Table 1), but it will be difficult to identify them because of statistical noise. It may be possible to map more of the yeast genome into blocks by using different sequence similarity cutoffs or search methods, or by including other data such as tRNA gene locations. In our previous analysis (11) we made the naive assumption that, because we discovered about 400 paralogs in half the genome, there would be 800 in the whole genome. In fact, the block-finding approach preferentially finds the most duplicate-rich regions of the genome. As shown in Fig. 3, we now envisage that the ancestral yeast genome had about 5,350–5,400 protein-coding genes, not 5,000 (11). We have identified only 1/3 of the blocks, but these contain about 80% of the paralogs (Fig. 3).
The most effective way to study how the yeast genome has evolved after its duplication would be to sequence the genome of a second, closely related, ascomycete species. This would reveal most of the original order of the duplicated yeast blocks, and should enable us to identify the 49 anticipated “zero-membered” blocks (Table 1). These are segments of the yeast genome that are “sisters” derived from genome duplication, but where no paralogous genes have been retained. A second genome sequence would also provide a definitive test of whether the entire yeast genome was duplicated (11), or just large portions of it. Genome sequencing projects, or “single-pass” sequencing surveys, are in progress for Schizosaccharomyces pombe, Candida albicans, Kluyveromyces lactis, Ashbya gossypii (24), and several species in the Saccharomyces sensu stricto group. The extent of gene order conservation between yeast and either Sch. pombe or C. albicans is probably too low to permit reconstruction of much of the original yeast genome (25), but the others should be useful. Because the Saccharomyces sensu stricto species share the genome duplication (25) it may be possible to determine their phylogenetic relationships by using gene order information alone. For example (see Fig. 4), if we can determine the ancestral order of blocks 14, 23, 50, and 37 by using, say, information from K. lactis, we can identify which pair of adjacent blocks represents the derived state and then search the other Saccharomyces sensu stricto yeasts for synapomorphy.
Our estimate that 70–100 reciprocal translocations have occurred in roughly 100 million years (Myr) (11) since yeast genome duplication results in an estimate of the rate of genomic rearrangement in yeast that is quite similar to the rate in human/mouse comparisons [about 100–180 rearrangements, also in approximately 100 Myr (1, 23, 26, 27)]. This is surprising, given their very different genome sizes (12 Mb in yeast; 3,000 Mb in humans) and rates of homologous recombination (1 centimorgan corresponds to ≈3 kb in yeast but ≈1 Mb in humans). The two organisms have similar genome sizes in centimorgans, suggesting that the ratio (expressed in terms of rearrangements per centimorgan per year) between rates of translocation and homologous recombination may be similar in the two taxa. Estimates of rates of genomic rearrangement in plants indicate that they too may be similar to mammals (10, 28), but whether there is really a molecular clock for chromosomal rearrangement as proposed by Paterson et al. (10) will not be clear without better maps for many taxa.
Acknowledgments
We thank Denis Shields for discussion and comments on the manuscript. This work was supported by the European Union Biotechnology Programme (BIO4-CT95–0130) and Forbairt.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
We use the word “paralogs” here specifically to refer to duplicate genes produced by whole-genome duplication, and not to any other sort of paralogs (17). Spring (18) proposed “tetralogs” as a name for the four-member gene sets resulting from putative ancient octoploidy of vertebrate genomes, and “homeologs” has also been used (19).
References
- 1.Nadeau J H, Taylor B A. Proc Natl Acad Sci USA. 1984;81:814–818. doi: 10.1073/pnas.81.3.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sankoff D. Biochimie. 1993;75:409–413. doi: 10.1016/0300-9084(93)90174-q. [DOI] [PubMed] [Google Scholar]
- 3.Blanchette M, Kunisawa T, Sankoff D. Gene. 1996;172:GC11–GC17. doi: 10.1016/0378-1119(95)00878-0. [DOI] [PubMed] [Google Scholar]
- 4.Palmer J D, Osorio B, Thompson W F. Curr Genet. 1988;14:65–74. [Google Scholar]
- 5.Sankoff D, Leduc G, Antoine N, Paquin B, Lang B F, Cedergren R. Proc Natl Acad Sci USA. 1992;89:6575–6579. doi: 10.1073/pnas.89.14.6575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boore J L, Collins T M, Stanton D, Daehler L L, Brown W M. Nature (London) 1995;376:163–165. doi: 10.1038/376163a0. [DOI] [PubMed] [Google Scholar]
- 7.Hannenhalli S, Chappey C, Koonin E V, Pevzner P A. Genomics. 1995;30:299–311. doi: 10.1006/geno.1995.9873. [DOI] [PubMed] [Google Scholar]
- 8.Bafna V, Pevzner P A. Mol Biol Evol. 1995;12:239–246. [Google Scholar]
- 9.Nadeau J H. In: Advanced Techniques in Chromosome Research. Adolph K W, editor. New York: Dekker; 1991. pp. 269–296. [Google Scholar]
- 10.Paterson A H, Lan T H, Reischmann K P, Chang C, Lin Y R, Liu S C, Burow M D, Kowalski S P, Katsar C S, DelMonte T A, et al. Nat Genet. 1996;14:380–382. doi: 10.1038/ng1296-380. [DOI] [PubMed] [Google Scholar]
- 11.Wolfe K H, Shields D C. Nature (London) 1997;387:708–713. doi: 10.1038/42711. [DOI] [PubMed] [Google Scholar]
- 12.Mewes, H. W., Albermann, K., Bähr, M., Frishman, D., Gleissner, A., Hani, J., Heumann, K., Kleine, K., Maierl, A., Oliver, S. G., et al. (1997) Nature (London) 387, Suppl., 7–65. [DOI] [PubMed]
- 13.Coissac E, Maillier E, Netter P. Mol Biol Evol. 1997;14:1062–1074. doi: 10.1093/oxfordjournals.molbev.a025712. [DOI] [PubMed] [Google Scholar]
- 14.Ohno S. Evolution by Gene Duplication. London: Allen & Unwin; 1970. [Google Scholar]
- 15.Hannenhalli S. In: Combinatorial Pattern Matching, 6th Annual Symposium. Galil Z, Ukkonen E, editors. Berlin: Springer; 1995. pp. 162–176. [Google Scholar]
- 16.Ferretti V, Nadeau J H, Sankoff D. In: Combinatorial Pattern Matching, 7th Annual Symposium. Hirschberg D, Myers G, editors. Berlin: Springer; 1996. pp. 159–167. [Google Scholar]
- 17.Fitch W M. Syst Zool. 1970;19:99–113. [PubMed] [Google Scholar]
- 18.Spring J. FEBS Lett. 1997;400:2–8. doi: 10.1016/s0014-5793(96)01351-8. [DOI] [PubMed] [Google Scholar]
- 19.Morizot D C. In: Isozymes: Structure, Function, and Use in Biology and Medicine. Ogita Z-I, Markert C L, editors. New York: Wiley–Liss; 1990. pp. 207–234. [Google Scholar]
- 20.Sankoff D, Parent M-N, Marchand I, Ferretti V. In: Combinatorial Pattern Matching, 8th Annual Symposium. Apostolico A, Hein J, editors. Berlin: Springer; 1997. pp. 262–274. [Google Scholar]
- 21.Lundin L G. Genomics. 1993;16:1–19. doi: 10.1006/geno.1993.1133. [DOI] [PubMed] [Google Scholar]
- 22.Sankoff D, Ferretti V. Genome Res. 1996;6:1–9. doi: 10.1101/gr.6.1.1. [DOI] [PubMed] [Google Scholar]
- 23.Copeland N G, Jenkins N A, Gilbert D J, Eppig J T, Maltais L J, Miller J C, Dietrich W F, Weaver A, Lincoln S E, Steen R G, et al. Science. 1993;262:57–66. doi: 10.1126/science.8211130. [DOI] [PubMed] [Google Scholar]
- 24.Altmann-Jöhl R, Philippsen P. Mol Gen Genet. 1996;250:69–80. doi: 10.1007/BF02191826. [DOI] [PubMed] [Google Scholar]
- 25.Keogh, R. S., Seoighe, C. & Wolfe, K. H. (1998) Yeast, in press. [DOI] [PubMed]
- 26.DeBry R W, Seldin M F. Genomics. 1996;33:337–351. doi: 10.1006/geno.1996.0209. [DOI] [PubMed] [Google Scholar]
- 27.Sankoff D, Ferretti V, Nadeau J H. J Comput Biol. 1997;4:559–565. doi: 10.1089/cmb.1997.4.559. [DOI] [PubMed] [Google Scholar]
- 28.Wolfe K H. In: Molecular Genetics of Photosynthesis. Barber J, Andersson B, editors. Oxford: IRL; 1996. pp. 45–57. [Google Scholar]