

Abstract
The structures of a number of processive enzymes have been determined recently. These proteins remain attached to their polymeric substrates and may perform thousands of rounds of catalysis before dissociating. Based on the degree of enclosure of the substrate, the structures fall into two broad categories. In one group, the substrate is partially enclosed, while in the other class, enclosure is complete. In the latter case, enclosure is achieved by way of an asymmetric structure for some enzymes while others use a symmetrical toroid.In those cases where the protein completely encloses its polymeric substrate, the two are topologically linked and an immediate explanation for processivity is provided. In cases where there is only partial enclosure, the structural basis for processivity is less obvious. There are, for example, pairs of proteins that have quite similar structures but differ substantially in their processivity. It does appear, however, that the enzymes that are processive tend to be those that more completely enclose their substrates. In general terms, proteins that do not use topological restraint appear to achieve processivity by using a large interaction surface. This allows the enzyme to bind with moderate affinity at a multitude of adjacent sites distributed along its polymeric substrate. At the same time, the use of a large interaction surface minimizes the possibility that the enzyme might bind at a small number of sites with much higher affinity, which would interfere with sliding. Proteins that can both slide along a polymeric substrate, and, as well, recognize highly specific sites (e.g., some site-specific DNA-binding proteins) appear to undergo a conformational change between the cognate and noncognate-binding modes.
Keywords: Processivity, sliding, toroid, groove, cellulases
Polymers are essential to all living organisms. Within a cell, genetic information is stored and propagated by DNA, a polymer of nucleic acids. The related polymer, RNA, transcribes genetic information and catalyzes chemical reactions. RNA serves as a template for the synthesis of proteins, which, in turn, are polymers of amino acids. Cellulose, a polymer of glucosyl units, is responsible for the structural integrity of plants. As such, it is the earth's most abundant biopolymer (Varrot et al. 1999a). Another polymer of glucose, starch, stores surplus biochemical energy. Hyaluron, composed of disaccharide subunits, is a high-molecular-weight polymer found in the extracellular matrix, especially in joints, where it creates a viscous solution that absorbs shocks (Li et al. 2000).
Because polymers are so prevalent, it is not surprising that many enzymes have evolved to synthesize, modify, or degrade them. A number of these enzymes are processive, which can be defined as remaining attached to their substrates and performing multiple rounds of catalysis before dissociating (von Hippel et al. 1994). Nucleic-acid polymerases are well-known examples. Other processive enzymes include some proteins involved in DNA repair. These enzymes bind to and slide along DNA, scanning for aberrant chemical moieties. They are processive in that they react with adjacent sites without diffusing from DNA, but must traverse stretches of unreactive polymer to find each site. Thus, they utilize facilitated diffusion (Dowd and Lloyd 1990), also known as sliding or facilitated target location (von Hippel and Berg 1989) to achieve processivity. Sliding can be defined as remaining attached to the substrate and binding at multiple sites before dissociating. As such, it can be closely related to processivity and examples are included in this review. Sliding along a linear polymer such as DNA reduces a three-dimensional search for a target site to one in one dimension. The general value of reduction of dimensionality in biological search and diffusion processes has long been recognized (Adam and Delbruck 1968), but also questioned (Stanford et al. 2000).
Processive enzymes differ greatly in the "extent" of their processivity, i.e., in the average number of subunits covered before dissociation (Table 1). Some enzymes, like restriction endonucleases and DNA-repair enzymes, are considered quasiprocessive, as they are able to slide along 200 (Carey and Strauss 1999) to 2000 (Higley and Lloyd 1993) base pairs. At the other extreme, certain polymerase holoenzymes, responsible for replicating genetic material, seem virtually unlimited in this respect. For example, bacteriophage T4 polymerase, with its accessory proteins, is capable of polymerizing over 20,000 nucleotides before dissociating (Kornberg and Baker 1992).
Table 1.
Processive enzymes discussed in this review
| Enzyme | Substratea | Substrate- binding site | Extent of processivity | Reference |
| Uracil DNA glycosylase (UDG) | dsDNA | Groove | 1500 to 2000 | Higley and Lloyd 1993 |
| AP-endonuclease (APE-1) | dsDNA | Groove | 200 | Carey and Strauss 1999 |
| BamHI | dsDNA | Groove | 1330 (scanning length) | Nardone et al. 1986 |
| TATA-binding protein, TBP | dsDNA | Groove | Not available | — |
| Reverse transcriptase | ssRNA, ssDNA | Groove | <50 | Avidan and Hizi 1998 |
| T7 RNA polymerase (RNAP) | dsDNA | Groove | Thousands | Muller et al. 1988 |
| Taq RNAP | dsDNA | Groove | 10,000b | Mooney and Landick 1999 |
| Yeast RNAP II | dsDNA | Groove | 1,000,000b | Mooney and Landick 1999 |
| Helicase PcrA | dsDNA | Groove | 20 | Soultanas et al. 1998 |
| Hyaluronate lyase (SagHL) | Hyaluronan | Groove | Not available | — |
| Cellulase Cel6A | Cellulose | Groove | Not available | — |
| Cellulase Cel7A | Cellulose | Groove | Not available | — |
| Cellulase Cel48F | Cellulose | Groove | Not available | — |
| Cellulase E4 | Cellulose | Groove | Not available | — |
| Exonuclease I (exol) | ssDNA | Closed, asymmetric | >900 | Brody et al. 1986 |
| Lytic transglycosylase Slt35 | Peptidoglycan | Closed, asymmetric | Not available | — |
| Lytic transglycosylase Slt70 | Peptidoglycan | Closed, asymmetric | Not available | — |
| RNA-dependent RNAP | ssRNA | Closed, asymmetric | ∼2500c | Lohmann et al. 1997 |
| T7 DNA polymerase (DNAP) (with thioredoxin) | dsDNA | Closed, asymmetric | Thousands | Tabor et al. 1987 |
| β-protein (in Pol III holoenzyme) | dsDNA | Closed, toroid | >5000 | Kornberg and Baker 1992 |
| T4 gp45 (in polymerase holoenzyme) | dsDNA | Closed, toroid | >2,0000 | Kornberg and Baker 1992 |
| PCNA (in polymerase δ) | dsDNA | Closed, toroid | >13,000 | McConnell et al. 1996 |
| λ-exonuclease | dsDNA | Closed, toroid | ∼3000 | Carter and Radding 1971 |
| Polynucleotide phosphorylase | ssRNA | Closed, toroid | Not available | — |
| Helicase T7gp4 | dsDNA | Closed, toroid | 40,000c | Patel & Picha 2000 |
| 20S proteasome | Polypeptide | Closed, toroid | ∼140d | Akopian et al. 1997 |
a dsDNA, double-stranded DNA; ssDNA, single-stranded DNA.
b May have other factors present besides core polymerase.
c This is a tentative estimate. Processivity has not been strictly demonstrated.
d Degradation results in peptides, not individual amino acids.
Processive enzymes have been the focus of a number of structural studies. These molecules fall into two classes. First, there are those for which the polymer-binding site forms a groove, sometimes saddle- or hand-shaped, and appear to partially enclose their substrates. Second, there are those that are shaped like a toroid or ring and appear to completely enclose their substrates. This suggests that such partial or complete enclosure is a requirement for processivity. At the same time, structural studies of closely related processive and nonprocessive enzymes have demonstrated that shape alone is not sufficient for processivity. There also are many enzymes that possess a large groove but are not processive. It is not immediately obvious whether it is the chemical identity of the residues lining the polymer-binding site, or their geometry, that is critical in determining whether or not the enzyme can slide.
This review will discuss the two structural classes of processive enzymes and provide examples of each class. Additionally, we will discuss the insights that structural studies have given into the mechanisms of sliding. Not included in this review are the processive motor proteins like kinesin and myosin, which have a different basis of processivity. Likewise, the ribosome is not discussed.
Structural classes of processive enzymes
Class I. Partially enclosing structures
The structures of the largest class of processive enzymes have a well-developed groove or cleft that partially encloses the substrate. In the most fully developed cases, the groove is very pronounced and the protein can be described as having the shape of a saddle. Several examples of partially enclosing processive enzymes that bind DNA are shown in Figure 1 ▶.
Fig. 1.

Representative, processive DNA-binding proteins with nonenclosed active sites. All enzymes are drawn with DNA-binding sites at the top of the figure. (a) Uracil DNA glycosylase (PDB code 1SSP). (b) Apurinic endonuclease (PDB code 1DEW). (c) BamHI (PDB code 1ESG). (d) Human immunodeficiency virus reverse transcriptase (PDB code 1RTD). The enzymes in (a) and (b) have very narrow grooves. This is consistent with cocrystal structures showing that these enzymes bind the phosphate backbone of a single strand of duplex DNA.
An interesting example of this class is uracil DNA glycosylase (UDG) (Fig. 1a ▶), responsible for processively scanning and removing misincorporated uracil bases from DNA (Higley and Lloyd 1993). Cocrystal structures of UDG with DNA that contains uracil bases show that a strand from the double helix of DNA is bound in a cleft on the surface of UDG (Parikh et al. 1998). It is thought that UDG binds DNA nonspecifically in the cleft and then processively searches for uracil (Parikh et al. 1998). UDG then removes the uracil and probably remains bound to the potentially toxic `abasic' site until it is displaced by the next enzyme in the DNA-repair pathway, AP-endonuclease (APE-1) (Parikh et al. 2000). APE-1 (Fig. 1b ▶), which introduces a nick in the damaged DNA strand (Wilson and Thompson 1997), also is processive (Carey and Strauss 1999; Mol et al. 2000). Although the grooves on the surface of both UDG and APE-1 are required for binding of cognate DNA, as demonstrated by the cocrystal structures, they also permit processive scanning of noncognate DNA.
Some restriction endonucleases scan DNA in search of their cognate DNA sequences. Although these enzymes have not been demonstrated to be processive (i.e., performing multiple rounds of catalysis before diffusing from their polymeric substrate), we include them in this review, as they provide structural insight into sliding. For example, facilitated linear diffusion has been demonstrated for BamHI (Fig. 1c ▶) (Nardone et al. 1986). This molecule has a large groove used for sliding along noncognate DNA (Viadiu and Aggarwal 2000). Another example of a sliding enzyme is TBP, the TATA-box binding protein (not shown), which is required for accurate initiation of transcription in eukaryotes. This transcription factor slides along nonspecific DNA (Coleman and Pugh 1995). Its structure resembles a saddle (Chasman et al. 1993) and it binds in the minor groove of DNA (Kim and Burley 1994).
Enzymes that polymerize double-stranded nucleic acids from a single-strand template, like reverse transcriptase (Fig. 1d ▶) (Huang et al. 1998; Najmudin et al. 2000) and T7 RNA polymerase (not shown) (Sousa et al. 1993), often assume a saddle shape. This saddle or hand shape is thought to help grip the substrate and aid in processivity. Likewise, the structure of Taq RNA polymerase (not shown) (Zhang et al. 1999) suggests that a saddle (or claw) shape allows this molecule to remain bound to its substrate. Similar observations may be made for yeast RNA polymerase II (not shown) (Cramer et al. 2000). This molecule has a "clamp" subunit, critical for processivity of transcription, which is thought to close down over the DNA template, partially encircling the substrate.
Processive DNA helicases, like PcrA (not shown), fall into this structural class as they have a saddle-shaped groove to bind single-stranded DNA (ssDNA) (Velankar et al. 1999). Helicases constitute a special case of processive enzymes as, unlike many of the molecules covered in this review, they couple the hydrolysis of ATP to the physical movement along their polymer substrate. Helicases use the energy of ATP hydrolysis to open the hydrogen-bonded double helix of DNA, a prerequisite for their movement along the DNA strand.
Further examples of processive enzymes containing a large groove, and which degrade oligosaccharide or cellulose substrates, are shown in Figure 2 ▶. The first example is hyaluronate lyase from Streptococcus pneumoniae (spnHL) (Fig. 2a ▶). The active site of this enzyme resides in a long positively charged groove, which provides a suitable complement to negatively charged hyaluronan. Although this enzyme has not been shown to be processive, the closely related hyaluronate lyase from Streptococcus agalactiae (sagHL) (Jedrzejas 2000) processively degrades hyaluronan (Pritchard et al. 1994). SpnHL, like sagHL, produces only disaccharides as products, as is expected from processive degradation. Therefore, it is presumed that spnHL is processive, consistent with its saddle-shape.
Fig. 2.

Representative processive and nonprocessive oligosaccharide-degrading enzymes. As in Figure 1 ▶, all the structures are aligned with the active-site cleft at the top of the figure. (a) Hyaluronate lyase (coordinates courtesy of S. Li and M. Jedrzejas). (b) Processive endocellulase Cel48F (code 1FAE). This molecule has an elaborately formed tunnel using two sets of loops. (c) Processive cellulase Cel6a (code 1QK2). (d) Related nonprocessive endoglucanase, E2 (code 1TML). (e) Processive cellulase Cel7a (code 8CEL). (f) Related nonprocessive endogluconase Cel7b (code 2A39). The cellulases shown in (c) (Cel6A) and (d) (E2) have similar structures. Cel6A, however, has additional loops that tend to form a tunnel and may account for the observation that this enzyme is processive whereas E2 is not. A similar situation occurs for Cel7A (shown in [e]) and Cel7B (shown in [f]). The two enzymes have similar folds but Cel7A has additional tunnel-forming loops that may account for its processivity. The structures in (d) and (f) show that a substrate-binding groove, per se, is not sufficient to confer processivity.
Many cellulases also contain large grooves. Five examples are included in Figure 2 ▶. Three of these enzymes (Cel6A, Cel7A, and Cel48F; Fig. 2 ▶, c, e, and b) do not simply remove cellulose subunits from the end of the polymer (see Armand et al. 1997, for example). Rather, they can catalyze both endo- and exocleavage. The structures offer an explanation as to how they achieve this dual specificity. The enzymes have loops that fold over their active sites, resulting in an enclosed substrate-binding tunnel. Structural studies of inhibitors bound to Cel6A have shown that these loops can move away from the active site, exposing it to solvent (Varrot et al. 1999b; Zou et al. 1999). This alternate conformation allows these enzymes to bind and cleave at sites within the cellulose strand. Once the initial endotype cleavage has occurred, the loops close, converting the enzyme to a processive exocellulase. The functional significance of these loops has been underscored by other crystallographic studies. Cel6A has homology to the endoglucanase E2 from Thermomononsporum fusca, an enzyme that catalyzes internal cuts to cellulose (Zou et al. 1999). In this related enzyme, the tunnel-forming loops are missing (Fig. 2d ▶) (Spezio et al. 1993). Likewise, Cel7A is homologous to the endoglucanase Cel7B (Fig. 2f ▶) (Zou et al. 1999). Cel7B also lacks the tunnel-forming loops (MacKenzie et al. 1998), which presumably make Cel7A processive. Interestingly, E2 and Cel7B, while missing the tunnel-forming loops, still have grooves in which cellulose binds. Therefore, the presence of a surface groove is not sufficient to ensure processivity.
The processive endocellulase E4 (not shown) does not use mobile loops to achieve both endo- and exoactivities. Instead, the active site of this enzyme resides in a long, open cleft (Sakon et al. 1997). This cleft extends from the main catalytic domain onto an adjacent cellulose-binding domain. It is assumed that a cellulose chain binds in the long cleft where the initial cleavage occurs. One segment of the cleaved cellulose strand then dissociates, while the other remains loosely associated with the saddle-shaped cellulose-binding domain, and is processively degraded.
Class II. Closed structures
Class II(a). Asymmetric structures
There are some processive enzymes that use a single polypeptide chain to create an asymmetric structure that is capable of completely enclosing a polymeric substrate. In two cases (Fig. 3a,b ▶), the bulk of the enzyme forms a well-defined groove within which the substrate appears to bind. In addition, there is a single polypeptide strand that extends across the mouth of the groove. The first example (Fig. 3a ▶) is exonuclease I from E. coli, which degrades ssDNA in a 3′ to 5′ direction (Thomas and Olivera 1978; Breyer and Matthews 2000). The second enzyme (Fig. 3b ▶) is Slt35, also from E. coli, which degrades peptidoglycan within the bacterial-cell wall (Holtje 1996; van Asselt et al. 1999). The unusual nature of the structure suggests that it is the bridging polypeptide strand that is critical for processivity. In this model, the polypeptide strand stretching across the mouth of the groove is required to keep the substrate from dissociating. This has, however, not been demonstrated rigorously. Perhaps the long-binding groove alone is sufficient to confer processivity. To date, the structures shown in Figure 3 ▶, a and b, are the only known instances in which this strategy is employed to achieve processivity. This may be, in part, because of the inherent difficulty in creating a hole or tunnel within a folded monomeric protein. Also, the bridging polypeptide may be susceptible to proteolysis.
Fig. 3.
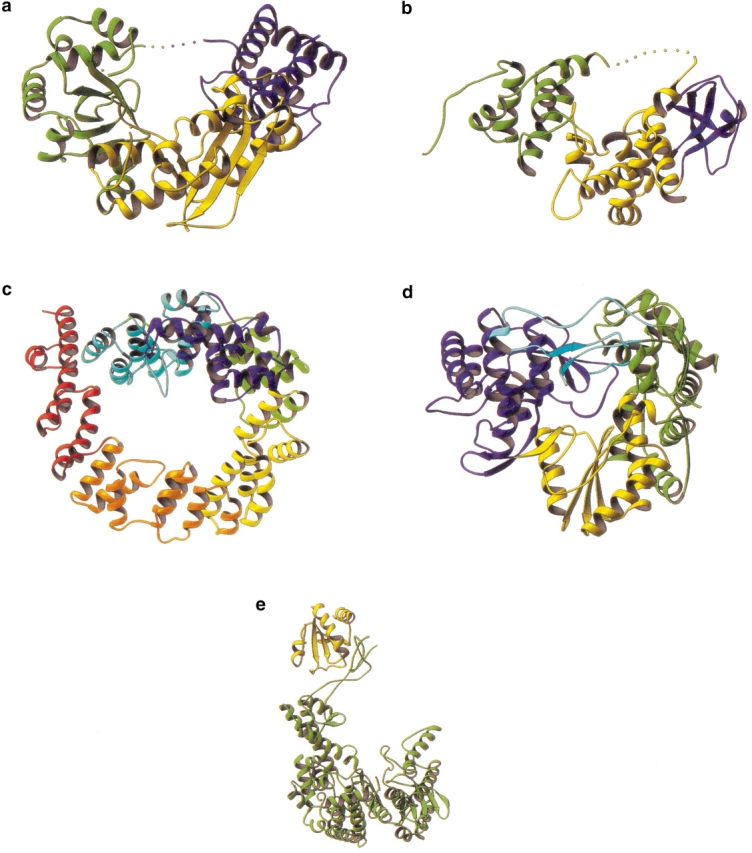
Closed asymmetric structures. (a) Exonuclease I (code 1FXX). (b) Lytic transglycosylase Slt35 (code 1QUT). (c) Lytic transglycosylase Slt70 (code 1Q5A). (d) RNA-dependent RNA polymerase from hepatitis C virus (code 1C2P). This molecule is a saddle-shaped enzyme with large inserted loops that allow full encircling of the substrate. (e) Phage T7 DNA polymerase with processivity factor, E. coli thioredoxin (code 1T7P). Although in the "open" conformation in the crystal structure, it is thought that thioredoxin links the two fingers and thumb of the polymerase to form a closed, processive polymerase complex.
Several other enzymes that form closed asymmetric structures are shown in Figure 3 ▶, c–e. The lytic transglycosylase Slt70 from E. coli (Fig. 3c ▶) processively degrades bacterial-cell wall (Holtje 1996). It has an unusual structure in which an asymmetric ring is created from 22 α-helices (Thunnissen et al. 1994). Attached to the ring, by a linker domain, is the catalytic domain, which has some structural homology to goose egg-white lysozyme. It is thought that the ring allows the protein to encircle the polysaccharide strands that constitute the cell wall and thus confer processivity (Holtje 1996).
The RNA-dependent RNA polymerase from hepatitis C virus also classifies as a closed asymmetric structure (Fig. 3d ▶) (Lesburg et al. 1999). Like other polymerases, it is formed by palm, fingers, and thumb domains. Extensive loops from the fingers and thumb domains create an encircled-active site. Topologically speaking, this molecule is very similar to the cellulase structures that use loops to form a tunnel enclosing the active site. Depending on the degree of enclosure provided by the loops, these molecules can be considered to be within either class I or class II(a).
The final, somewhat unusual example of this class is the T7 DNA polymerase (Fig. 3e ▶). In this case, the native enzyme has a saddlelike structure and is not very processive (Doublie et al. 1998). It requires a host protein, thioredoxin, to enable processive polymerization of DNA (Modrich and Richardson 1975). A crystal structure of the T7 DNA replication complex shows template DNA bound in the groove of the polymerase (Doublie et al. 1998). Thioredoxin is associated with one edge of the groove. Although the structure has an open conformation (Fig. 3e ▶), it suggests that during processive replication the thioredoxin molecule bridges between the two stirrups of the saddle, completely enclosing the substrate.
Class II(b). Toroids
Some processive proteins encircle their substrates by forming symmetric, oligomeric toroids (Fig. 4 ▶). This class includes the sliding clamp proteins that associate with many DNA polymerases to enable processive replication. The E. coli β protein (Fig. 4a ▶) (Kong et al. 1992), the eukaryotic protein PCNA (Fig. 4b ▶) (Krishna et al. 1994), and bacteriophage T4 gp45 (not shown) (Moarefi et al. 2000) all have pseudosixfold symmetry. Intriguingly, PCNA (Fig. 4b ▶) and gp45 each are a trimer of subunits that contains two homologous domains that are related by a 60° rotation. When three monomers associate, the resulting molecule has pseudosixfold symmetry. In contrast, the β protein (Fig. 4a ▶) is a dimer of subunits with three homologous domains, each related by a 60° rotation. Assembly of two monomers to form β results in a molecule with pseudosixfold symmetry. Sliding clamps require accessory proteins, the clamp loaders, to encircle DNA. These enzymes are ATPases, and are thought to transiently separate the monomers that constitute the clamp to slip them around double-stranded DNA (dsDNA).
Fig. 4.

Closed, symmetric structures. (a) E. coli protein (code 2POL). (b) PCNA (code 1PLQ). (c) λ-exonuclease (code 1AVQ). (d) Streptomyces antibioticus PNPase (code 1E3P). (e) Hexameric helicase portion of the gene 4 protein from bacteriophage T7 (code 1E0J). (f) 20S proteasome (code 1PMA).
Other toroidal molecules include the bacteriophage λ-exonuclease (Fig. 4c ▶) that processively degrades one strand of dsDNA in a 5′ to 3′ direction (Fig. 4d ▶) (Kovall and Matthews 1997). Phage λ uses this exonuclease to create 3′ overhangs on its genomic dsDNA to facilitate genetic recombination with the E. coli chromosome through the double-stranded break repair and single-stranded annealing pathways. λ-exonuclease is a trimeric protein with the three subunits arranged to form a toroid (Kovall and Matthews 1997). The channel formed by λ-exonuclease is tapered such that dsDNA may enter one side but only ssDNA may exit from the other side (Kovall and Matthews 1997).
Polynucleotide phosphorylase (PNPase) processively removes nucleotides from the 3′ end of single-stranded RNA (ssRNA). PNPase plays a critical role in the degradation of mRNA, which, in turn, affects gene expression. The structure of PNPase from Streptomyces antibioticus is toroidal (Fig. 4d ▶) (Symmons et al. 2000). Experimental evidence suggests that ssRNA threads through the center of PNPase, demonstrating that the toroidal shape of the molecule is responsible for its processivity.
Hexameric helicases, like bacteriophage T7 gene 4 protein (Singleton et al. 2000), also form toroids (Fig. 4e ▶). A single strand of the unwound DNA is threaded through the hexamer (Yu et al. 1996). The toroidal shape allows the helicase to processively unwind DNA and ensures that it remains associated with the substrate.
Also found in this category are self-compartmentalized intracellular proteases. The 20S proteasome from Thermoplasma acidophilum (Fig. 4f ▶) processively cuts a protein into small peptides before beginning to degrade another protein (Akopian et al. 1997). This oligomeric enzyme is barrel shaped, with 28 subunits arranged in four adjacent rings of seven subunits (Lowe et al. 1995). The active sites are on the inside of the barrel. It is thought that the barrel shape allows the proteasome to sequester a protein for processive degradation.
Structural insights into sliding
Tight binding of a macromolecule to its substrate is achieved through complementary interfaces. For sliding, however, an enzyme must strike the right energetic balance so as to remain associated with its polymeric substrate, while retaining the ability to move from site to site. This problem of energetic balance is solved in many different ways and has been elucidated by structural studies, among other techniques. In the following section, three different solutions to the problems of sliding are presented.
Example 1. BamHI — conformational rearrangement
The restriction endonuclease BamHI can locate its target DNA faster than the limit of three-dimensional diffusion. This enzyme binds nonspecific, duplex DNA and slides along the DNA until it locates a restriction site (Nardone et al. 1986). A recent crystal structure of BamHI bound to noncognate DNA suggests how sliding may occur (Viadiu and Aggarwal 2000). The enzyme is a saddle-shaped dimer and binds DNA in a groove formed by the two monomers (Fig. 5 ▶) (Newman et al. 1995). BamHI, in the absence of DNA, is a twofold symmetric dimer (Fig. 5a ▶) (Newman et al. 1994). Each monomer is composed of a central β-sheet flanked by α-helices. BamHI's dimerization domain consists of a parallel four-helix bundle, with each monomer contributing two helices.
Fig. 5.

Comparison of BamHI structures. Structures of free (A), nonspecific (B), and specific (C) DNA-bound forms of BamHI. Regions that undergo local conformational changes are shown in yellow color. The enzyme becomes progressively more closed around the DNA as it goes from the nonspecific to the specific DNA-binding mode. Residues 79–92 are unstructured in the free enzyme but become ordered in both the nonspecific and specific DNA complexes, albeit in different conformations. The C-terminal residues unwind in the specific complex to form partially disordered arms, whereas in the nonspecific complex they remain α-helical. (Reproduced from Viadiu and Aggarwal 2000, with permission.)
When bound to target DNA, most of the recognition contacts occur within the major groove (Newman et al. 1995). The N-terminal ends of the parallel-bundle helices penetrate the major groove and contribute the majority of the amino acids required for target recognition. The cocrystal structure also shows that BamHI undergoes a large rearrangement. The monomers rotate toward each other and two C-terminal α-helices (one from each monomer) unfold from the molecule and bind in the minor groove. Thus the molecule almost completely encircles DNA (Fig. 5c ▶).
The structure of BamHI bound to noncognate DNA shows marked differences from that of BamHI bound to its cognate site (Fig. 5b ▶) (Viadiu and Aggarwal 2000). In general, the structure resembles that of BamHI in the absence of DNA. The C-terminal helices that bind in the minor groove of the target DNA remain folded within the BamHI structure. Thus, the DNA is not encircled by BamHI, but instead protrudes from the cleft. Because the DNA is somewhat withdrawn from the protein, none of the amino acids responsible for sequence-specific recognition are within hydrogen-bonding distance of the DNA. Without the structural rearrangement, the DNA-binding surface in the groove of the molecule also lacks the positive charge that predominates in the specific complex. In fact, the noncognate complex shows a loss of all base-specific interactions in addition to a loss of all contacts to the DNA backbone. It appears that BamHI is able to bind nonspecific DNA through weak electrostatic (helix-dipole) interactions. In common with many other DNA-binding proteins (e.g., Berg et al. 1982), facilitated diffusion of BamHI is dependent upon salt concentration; in the presence of high salt concentration, facilitated diffusion is lost. This also suggests that the attractive forces that are used in sliding primarily are electrostatic.
Thus, the structural studies suggest that BamHI maintains its native structure during sliding, relying on electrostatic interactions with the DNA. Once the specific target site is reached, rather large structural rearrangements occur and other, more intimate, interactions are formed between the two partners. Similar conclusions were reached from structural studies of a Cro repressor and its complexes with DNA (Albright et al. 1998).
Example 2. Cellulase 48F. Conformational change of side chains
Processive cellulases slide along cellulose polymers, catalyzing multiple rounds of hydrolysis before dissociating from their substrates. A recent structural study of Cel48F, a processive endocellulase from Clostridium cellulolyticum, provides clues as to how this is achieved. In vivo, Cel48F is found within the cellulosome, a large complex of cellulases secreted by the bacteria that act synergistically to degrade cellulose. Cel48F is composed of an N-terminal catalytic domain and a C-terminal docking domain that attaches to components within the cellulosome.
The crystal structure of the catalytic domain, initially solved in the presence of a thiooligosaccharide inhibitor (Parsiegla et al. 1998), showed that Cel48F is composed of an (αα)6 barrel. At one end of the barrel, long connections between the α-helices form a tunnel that leads to a cleft (Fig. 6 ▶). The active site is at the end of the tunnel before the cleft. It is the tunnel that is responsible for retaining the cellulose strand during processive degradation while the product, cellobiose, exits by way of the cleft.
Fig. 6.

Scheme of the active site of Cel48F with the positions of inhibitor (bold broken lines) and substrate (normal lines) presented as ellipsoids or, in subsite-1, as a box. The possible aromatic stacking partners are shown and their interactions at the subsites within the tunnel are indicated by open arrows. The position of the cleavage site is marked by a filled arrow with the relative positions of the proton donor Glu55 and the possible catalytic bases Asp230 and Glu44 also shown. (Reproduced from Parsiegla et al. 2000, with permission.)
The structure of the Cel48F catalytic domain also has been determined in the presence of a different inhibitor (Parsiegla et al. 2000). Additionally, an active site mutation (E55Q) allowed for cocrystallization in the presence of two different substrate molecules, cellotetraose and cellohexaose. In all four complexes, density was seen in the tunnel, although the binding sites were not coincident. The sites occupied by the substrate molecules in the inactive mutant agree well (rmsd 0.17Å) and constitute one set of cellulose-binding sites. Within these sites, glucosyl moieties form stacking interactions with three tryptophans and one tyrosine. Hydrogen bonds also were observed to water molecules and polar/charged residues lining the tunnel. The inhibitors, however, occupy a second set of sites that are somewhat less well defined. This second set of sites results in a different set of interactions with the hydrophobic residues lining the cavity, including different stacking interactions with the aromatic residues. Comparison of the two sets of glucose-binding sites shows that the set occupied by the substrate molecules is shifted half the length of a glucosyl residue towards the active site relative to the inhibitor set (Fig. 6 ▶).
Because the binding of the substrates is well defined, it suggests that these sites might be designed to hold the substrate in a specific alignment appropriate for catalysis. At the same time, by having an interleaving set of binding sites (at which the inhibitors bind), it reduces the energy barrier to translocation of the substrate and so promotes sliding.
Example 3. Sliding clamps or topological tethers
Sliding clamps form `topological' links with dsDNA. These molecules presumably are designed to ensure tight but nonspecific interaction with dsDNA. The inner diameters of these molecules are 30–35Å (Fig. 4a,b ▶). Hence, they easily accommodate B-form dsDNA, which has a diameter of ∼24Å. The large diameter of the cavity might reduce direct interaction with the DNA, but also might be required for loading the protein onto the DNA. Tight interactions with DNA also are avoided because α-helices lining the inner surface of the toroid are orthogonal to the major and minor grooves. This prevents residues from entering the grooves and making specific interactions with the base pairs.
The molecules also have an unusual charge distribution. Most of the surface is negatively charged, with the exception of the inner cavity, which bears a positive electrostatic potential. The positive surface may reduce the energetic cost of loading the protein onto the DNA. Once loaded, however, the overall acidic character of the protein may help to keep the DNA as far away as possible, i.e., in the center of the toroid.
Possible exceptions
Not all processive proteins fit neatly into the classification outlined above. One intriguing "outlier" is the UL42 processivity factor from herpes simplex virus, which is required for processive DNA replication. The toroidal sliding clamps shown in Figure 4 ▶, a and b, show no ability to form stable complexes with linear dsDNA. Unlike these molecules, UL42 forms a stable but nonspecific complex with such a DNA substrate. Unexpectedly, however, the structure determination of UL42 (Zuccola et al. 2000) showed it to be a monomer of two domains, each of which has a fold similar to a domain of PCNA. The two domains are, however, related by a rotation of only 40°, resulting in a flat surface that is replete with positive charges and is presumed to be responsible for binding dsDNA. The authors postulate that, in the absence of the polymerase, UL42 is held in close proximity by electrostatic attraction but moves along the dsDNA in what is analogous to a one-dimensional random walk. Polymerase, when present, provides directionality to the movement.
Conclusions
The essence of a processive enzyme is to retain affinity for its polymeric substrate during multiple rounds of catalysis. At the same time, the enzyme should bind with comparable affinity at all sites along the polymer; in other words, it should bind nonspecifically. One way in which this can be achieved is by a toroidal structure as exemplified by the sliding clamps (Fig. 4a,b ▶). In general, however, high affinity and low specificity can be achieved by having a large interaction surface. As the area of interaction is increased, additional favorable interactions can be included and the affinity increased essentially without limit. At the same time, an extended binding surface minimizes the chance that there will be perfect complementarity between the two partners. When one of the interacting partners is a polymer, the binding surface can be designed to take advantage of the repetitive nature of the target. With DNA, for example, electrostatic interactions can be repeated along the phosphate backbone. With cellulose, hydrophobic interactions can be repeated with successive glucosyl groups (Fig. 6 ▶). Such repeated interactions are likely to result in more-or-less equal affinity at different locations along the polymeric substrate. It could be that the extended, concave, binding sites seen in many of the processive enzymes (Figs. 1, 2 ▶ ▶) results from a tendency to increase the area of interaction between the enzyme and its substrate. In some contexts, a large area of interaction between two interacting partners can be used to generate both high affinity and high specificity. In this situation, the interacting surfaces on the two partners are highly complementary, each being designed to perfectly match the other. In the context of processivity, however, a large interaction surface can be used to generate moderate affinity at a multitude of different binding sites, and to avoid very tight binding at any specific site.
While an appropriately shaped active site of an enzyme appears desirable for processivity, it is not sufficient. As demonstrated by the endoglucanases Cel7B and E2, a large substrate-binding cleft similar to that found in class I enzymes does not confer processivity. Processivity (or sliding) requires affinity as well as shape complementarity. The early studies of lac repressor led to the suggestion that electrostatic interactions could be used to generate a complex between the protein and the DNA that was of relatively high affinity, but not sequence-specific (Berg et al. 1982). Such a complex could explain the sliding of the protein along the DNA. Subsequent structural studies of many processive enzymes provide much support for these early ideas. The complex of BamHI with noncognate DNA, for example (Fig. 5b ▶), confirms the role of electrostatic interactions. At the same time, structural studies show other ways in which sliding and/or processivity can be achieved. One alternative is to completely encircle the substrate (Figs. 3, 4 ▶ ▶). Another is to make use of repeated hydrophobic or stacking interactions (Fig. 6 ▶). The structural studies also show how an enzyme such as BamHI can switch between two distinct conformations, one appropriate for sliding and the other for tight binding at a sequence-specific site. Thus, two distinct modes of substrate interaction can be combined in the same protein.
Table 1 summarizes the available data on the extent of processivity for the proteins discussed in this review. In many cases, the measurements were made to demonstrate that a given protein was processive, not necessarily to quantitate the degree of processivity. Very tentatively, the available data are consistent with the hypotheses that the proteins that completely enclose their substrates are more processive than those that rely on partial enclosure. Further studies, however, are needed to determine if this is actually the case.
Acknowledgments
We thank Drs. Songlin Li and Mark Jedrzejas for sending the coordinates of hyaluron lyase, and Dr. Richard Kingston for helpful comments on the manuscript. This work was supported in part by NIH grant GM20066 to B.W.M.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/ps.10301.
References
- Adam, G. and Delbruck, M. 1968. Reduction of dimensionality in biological diffusion processes. In Structural chemistry and molecular biology (eds. A. Rich and N. Davidson), pp. 198–215. W.H. Freeman and Company, San Francisco.
- Akopian, T.N., Kisselev, A.F., and Goldberg, A.L. 1997. Processive degradation of proteins and other catalytic properties of the proteasome from Thermoplasma acidophilum. J. Biol. Chem. 272 1791–1798. [DOI] [PubMed] [Google Scholar]
- Albright, R.A., Mossing, M.C., and Matthews, B.W. 1998. Crystal structure of an engineered Cro monomer bound nonspecifically to DNA: Possible implications for nonspecific binding by the wild-type protein. Protein Sci. 7 1485–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armand, S., Drouillard, S., Schülein, M., Henrissat, B., and Driguez, H. 1997. A bifunctionalized fluorogenic tetrasaccharide as a substrate to study cellulases. J. Biol. Chem. 272 2709–2713. [DOI] [PubMed] [Google Scholar]
- Avidan, O. and Hizi, A. 1998. The processivity of DNA synthesis exhibited by drug-resistant variants of human immunodeficiency virus type-1 reverse transcriptase. Nucleic Acids Res. 26 1713–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg, O.G., Winter, R.B., and von Hippel, P.H. 1982. How do genome-regulatory proteins locate their DNA target sites? Trends Biochem. Sci. 7 52–55. [Google Scholar]
- Breyer, W.A. and Matthews, B.W. 2000. Structure of Escherichia coli exonuclease I suggests how processivity is achieved. Nature Struct. Biol. 7 1125–1128. [DOI] [PubMed] [Google Scholar]
- Brody, R.S., Doherty, K.G., and Zimmerman, P.D. 1986. Processivity and kinetics of the reaction of exonuclease I from Escherichia coli with polydeoxyribonucleotides. J. Biol. Chem. 261 7136–7143. [PubMed] [Google Scholar]
- Carey, D.C. and Strauss, P.R. 1999. Human apurinic/apyrimidinic endonuclease is processive. Biochemistry 38 16553–16560. [DOI] [PubMed] [Google Scholar]
- Carter, D.M. and Radding, C.M. 1971. The role of exonuclease and β protein of phage λ in genetic recombination. II. Substrate specificity and the mode of action of λ exonuclease. J. Biol. Chem. 246 2502–2512. [PubMed] [Google Scholar]
- Chasman, D.I., Flaherty, K.M., Sharp, P.A., and Kornberg, R.D. 1993. Crystal structure of yeast TATA-binding protein and model for interaction with DNA. Proc. Natl. Acad. Sci. 90 8174–8178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman, R.A. and Pugh, B.F. 1995. Evidence for functional binding and stable sliding of the TATA binding protein on nonspecific DNA. J. Biol. Chem. 270 13850–13859. [DOI] [PubMed] [Google Scholar]
- Cramer, P., Bushnell, D.A., Fu, J., Gnatt, A.L., Maier-Davis, B., Thompson, N.E., Burgess, R.R., Edwards, A.M., David, P.R., and Kornberg, R.D. 2000. Architecture of RNA polymerase II and implications for the transcription mechanism. Science 288 640–649. [DOI] [PubMed] [Google Scholar]
- Doublie, S., Tabor, S., Long, A.M., Richardson, C.C., and Ellenberger, T. 1998. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2Å resolution. Nature 391 251–258. [DOI] [PubMed] [Google Scholar]
- Dowd, D.R. and Lloyd, R.S. 1990. Biological significance of facilitated diffusion in protein-DNA interactions. Applications to T4 endonuclease V-initiated DNA repair. J. Biol. Chem. 265 3424–3431. [PubMed] [Google Scholar]
- Higley, M. and Lloyd, R.S. 1993. Processivity of uracil DNA glycosylase. Mutat. Res. 294 109–116. [DOI] [PubMed] [Google Scholar]
- Holtje, J.V. 1996. Lytic transglycosylases. EXS 75 425–429. [DOI] [PubMed] [Google Scholar]
- Huang, H., Chopra, R., Verdine, G.L., and Harrison, S.C. 1998. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: Implications for drug resistance. Science 282 1669–1675. [DOI] [PubMed] [Google Scholar]
- Jedrzejas, M.J. 2000. Structural and functional comparison of polysaccharide-degrading enzyme. Crit. Rev. Biochem. Mol. Biol. 35 221–251. [DOI] [PubMed] [Google Scholar]
- Kim, J.L. and Burley, S.K. 1994. 1.9Å resolution refined structure of TBP recognizing the minor groove of TATAAAAG. Nat. Struct. Biol. 1 638–653. [DOI] [PubMed] [Google Scholar]
- Kong, X.P., Onrust, R., O'Donnell, M., and Kuriyan, J. 1992. Three-dimensional structure of the β-subunit of E. coli DNA polymerase III holoenzyme: A sliding DNA clamp. Cell 69 425–437. [DOI] [PubMed] [Google Scholar]
- Kornberg, A. and Baker, T. 1992. DNA Replication, 2nd ed. W.H. Freeman and Company, New York.
- Kovall, R. and Matthews, B.W. 1997. Toroidal structure of λ-exonuclease. Science 277 1824–1827. [DOI] [PubMed] [Google Scholar]
- Krishna, T.S., Kong, X.P., Gary, S., Burgers, P., and Kuriyan, J. 1994. Crystal structure of the eukaryotic DNA polymerase processivity factor PCNA. Cell 79 1233–1243. [DOI] [PubMed] [Google Scholar]
- Lesburg, C.A., Cable, M.B., Ferrari, E., Hong, Z., Mannarino, A.F., and Weber, P.C. 1999. Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat. Struct. Biol. 6 937–943. [DOI] [PubMed] [Google Scholar]
- Li, S., Kelly, S.J., Lamani, E., Ferraroni, M., and Jedrzejas, M.J. 2000. Structural basis of hyaluronan degradation by Streptococcus pneumoniae hyaluronate lyase. EMBO J. 19 1228–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmann, V., Korner, F., Herian, U., and Bartenschlager, R. 1997. Biochemical properties of hepatitis C virus NS5B RNA-dependent RNA polymerase and identification of amino acid sequence motifs essential for enzymatic activity. J. Virol. 71 8416–8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe, J., Stock, D., Jap, B., Zwickl, P., Baumeister, W., and Huber, R. 1995. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4Å resolution. Science 268 533–539. [DOI] [PubMed] [Google Scholar]
- MacKenzie, L.F., Sulzenbacher, G., Divne, C., Jones, T.A., Wöldike, H.F., Schülein, M., Withers, S.G., and Davies, G.J. 1998. Crystal structure of the family 7 endoglucanase I (Cel7B) from Humicola insolens at 2.2Å resolution and identification of the catalytic nucleophile by trapping of the covalent glycosyl-enzyme intermediate. Biochem. J. 335 409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell, M., Miller, H., Mozzherin, D.J., Quamina, A., Tan, C.K., Downey, K.M., and Fisher, P.A. 1996. The mammalian DNA polymerase delta-proliferating cell nuclear antigen-template-primer complex: Molecular characterization by direct binding. Biochemistry 35 8268–8274. [DOI] [PubMed] [Google Scholar]
- Moarefi, I., Jeruzalmi, D., Turner, J., O'Donnell, M., and Kuriyan, J. 2000. Crystal structure of the DNA polymerase processivity factor of T4 bacteriophage. J. Mol. Biol. 296 1215–1223. [DOI] [PubMed] [Google Scholar]
- Modrich, P. and Richardson, C.C. 1975. Bacteriophage T7 deoxyribonucleic acid replication in vitro. A protein of Escherichia coli required for bacteriophage T7 DNA polymerase activity. J. Biol. Chem. 250 5508–5514. [PubMed] [Google Scholar]
- Mol, C.D., Izumi, T., Mitra, S., and Tainer, J.A. 2000. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination. Nature 403 451–456. [DOI] [PubMed] [Google Scholar]
- Mooney, R.A. and Landick, R. 1999. RNA polymerase unveiled. Cell 98 687–690. [DOI] [PubMed] [Google Scholar]
- Muller, D.K., Martin, C.T., and Coleman, J.E. 1988. Processivity of proteolytically modified forms of T7 RNA polymerase. Biochemistry 27 5763–5771. [DOI] [PubMed] [Google Scholar]
- Najmudin, S., Cote, M.L., Sun, D., Yohannan, S., Montano, S.P., Gu, J., and Georgiadis, M.M. 2000. Crystal structures of an N-terminal fragment from Moloney murine leukemia virus reverse transcriptase complexed with nucleic acid: Functional implications for template-primer binding to the fingers domain. J. Mol. Biol. 296 613–632. [DOI] [PubMed] [Google Scholar]
- Nardone, G., George, J., and Chirikjian, J.G. 1986. Differences in the kinetic properties of BamHI endonuclease and methylase with linear DNA substrates. J. Biol. Chem. 261 12128–12133. [PubMed] [Google Scholar]
- Newman, M., Strzelecka, T., Dorner, L.F., Schildkraut, I., and Aggarwal, A.K. 1994. Structure of restriction endonuclease BamHI and its relationship to EcoRI. Nature 368 660–664. [DOI] [PubMed] [Google Scholar]
- Newman, M., Strzelecka, T., Dorner, L.F., Schildkraut, I., and Aggarwal, A.K. 1995. Structure of BamHI endonuclease bound to DNA: Partial folding and unfolding on DNA binding. Science 269 656–663. [DOI] [PubMed] [Google Scholar]
- Parikh, S.S., Mol, C.D., Slupphaug, G., Bharati, S., Krokan, H.E., and Tainer, J.A. 1998. Base excision repair initiation revealed by crystal structures and binding kinetics of human uracil-DNA glycosylase with DNA. EMBO J. 17 5214–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh, S.S., Putnam, C.D., and Tainer, J.A. 2000. Lessons learned from structural results on uracil-DNA glycosylase. Mutat. Res. 460 183–199. [DOI] [PubMed] [Google Scholar]
- Parsiegla, G., Juy, M., Reverbel-Leroy, C., Tardif, C., Belaich, J-P., Driguez, H., and Haser, R. 1998. The crystal structure of the processive endocellulase CelF of Clostridium cellulolyticum in complex with a thiooligosaccharde inhibitor at 2.0Å resolution. EMBO J. 17 5551–5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsiegla, G., Reverbel-Leroy, C., Tardif, C., Belaich, J.P., Driguez, H., and Haser, R. 2000. Crystal structures of the cellulase Cel48F in complex with inhibitors and substrates give insights into its processive action. Biochemistry 39 11238–11246. [DOI] [PubMed] [Google Scholar]
- Patel, S.S. and Picha, K.M. 2000. Structure and function of hexameric helicases. Ann. Rev. Biochem. 69 651–697. [DOI] [PubMed] [Google Scholar]
- Pritchard, D.G., Lin, B., Willingham, T.R., and Baker, J.R. 1994. Characterization of the group B streptococcal hyaluronate lyase. Arch. Biochem. Biophys. 315 431–437. [DOI] [PubMed] [Google Scholar]
- Sakon, J., Irwin, D., Wilson, D.B., and Karplus, P.A. 1997. Structure and mechanism of endo/exocellulase E4 from Thermomonospora fusca. Nat. Struct. Biol. 4 810–818. [DOI] [PubMed] [Google Scholar]
- Singleton, M.R., Sawaya, M.R., Ellenberger, T., and Wigley, D.B. 2000. Crystal structure of T7 gene 4 ring helicase indicates a mechanism for sequential hydrolysis of nucleotides. Cell 101 589–600. [DOI] [PubMed] [Google Scholar]
- Soultanas, P., Dillingham, M.S., and Wigley, D.B. 1998. Escherichia coli ribosomal protein L3 stimulates the helicase activity of the Bacillus stearothermophilus PcrA helicase. Nucleic Acids Res. 26 2374–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa, R., Chung, Y.J., Rose, J.P., and Wang, B.C. 1993. Crystal structure of bacteriophage T7 RNA polymerase at 3.3Å resolution. Nature 364 593–599. [DOI] [PubMed] [Google Scholar]
- Spezio, M., Wilson, D.B., and Karplus, P.A. 1993. Crystal structure of the catalytic domain of a thermophilic endocellulase. Biochemistry 32 9906–9916. [DOI] [PubMed] [Google Scholar]
- Stanford, N.P., Szczelkun, M.D., Marko, J.F., and Halford, S.E. 2000. One- and three-dimensional pathways for proteins to reach specific DNA sites. EMBO J. 19 6546–6557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symmons, M.F., Jones, G.H., and Luisi, B.F. 2000. A duplicated fold is the structural basis for polynucleotide phosphorylase catalytic activity, processivity and regulation. Structure 8 1215–1226. [DOI] [PubMed] [Google Scholar]
- Tabor, S., Huber, H.E., and Richardson, C.C. 1987. Escherichia coli thioredoxin confers processivity on the DNA polymerase activity of the gene 5 protein of bacteriophage T7. J. Biol. Chem. 262 16212–16123. [PubMed] [Google Scholar]
- Thomas, K. and Olivera, B. 1978. Processivity of DNA exonucleases. J. Biol. Chem. 253 424–429. [PubMed] [Google Scholar]
- Thunnissen, A.M., Dijkstra, A.J., Kalk, K.H., Rozeboom, H.J., Engel, H., Keck, W., and Dijkstra, B.W. 1994. Doughnut-shaped structure of a bacterial muramidase revealed by x-ray crystallography. Nature 367 750–753. [DOI] [PubMed] [Google Scholar]
- van Asselt, E.J., Dijkstra, A.J., Kalk, K.H., Takacs, B., Keck, W., and Dijkstra, B.W. 1999. Crystal structure of Escherichia coli lytic transglycosylase Slt35 reveals a lysozyme-like catalytic domain with an EF-hand. Structure Fold. Des. 7 1167–1180. [DOI] [PubMed] [Google Scholar]
- Varrot, A., Hastrup, S., Schülein, M., and Davies, G.J. 1999a. Crystal structure of the catalytic core domain of the family 6 cellobiohydrolase II, Cel6A, from Humicola insolens, at 1.92Å resolution. Biochem. J. 337 297–304. [PMC free article] [PubMed] [Google Scholar]
- Varrot, A., Schulein, M., and Davies, G.J. 1999b. Structural changes of the active site tunnel of Humicola insolens cellobiohydrolase, Cel6A, upon oligosaccharide binding. Biochemistry 38 8884–8891. [DOI] [PubMed] [Google Scholar]
- Velankar, S.S., Soultanas, P., Dillingham, M.S., Subramanya, H.S., and Wigley, D.B. 1999. Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell 97 75–84. [DOI] [PubMed] [Google Scholar]
- Viadiu, H. and Aggarwal, A.K. 2000. Structure of BamHI bound to nonspecific DNA: A model for DNA sliding. Mol. Cell 5 889–895. [DOI] [PubMed] [Google Scholar]
- von Hippel, P.H. and Berg, O.G. 1989. Facilitated target location in biological systems. J. Biol. Chem. 264 675–678. [PubMed] [Google Scholar]
- von Hippel, P.H., Fairfield, F.R., and Dolejsi, M.K. 1994. On the processivity of polymerases. Ann. NY Acad. Sci. 726 118–131. [DOI] [PubMed] [Google Scholar]
- Wilson, D.M. III and Thompson, L.H. 1997. Life without DNA repair. Proc. Natl. Acad. Sci. 94 12754–12757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, X., Hingorani, M.M., Patel, S.S., and Egelman, E.H. 1996. DNA is bound within the central hole to one or two of the six subunits of the T7 DNA helicase. Nat. Struct. Biol. 3 740–743. [DOI] [PubMed] [Google Scholar]
- Zhang, G., Campbell, E.A., Minakhin, L., Richter, C., Severinov, K., and Darst, S.A. 1999. Crystal structure of Thermus aquaticus core RNA polymerase at 3.3Å resolution. Cell 98 811–824. [DOI] [PubMed] [Google Scholar]
- Zou, J., Kleywegt, G.J., Stahlberg, J., Driguez, H., Nerinckx, W., Claeyssens, M., Koivula, A., Teeri, T.T., and Jones, T.A. 1999. Crystallographic evidence for substrate ring distortion and protein conformational changes during catalysis in cellobiohydrolase Ce16A from Trichoderma reesei. Structure Fold. Des. 7 1035–1045. [DOI] [PubMed] [Google Scholar]
- Zuccola, H.J., Filman, D.J., Coen, D.M., and Hogle, J.M. 2000. The crystal structure of an unusual processivity factor, herpes simplex virus UL42, bound to the C terminus of its cognate polymerase. Mol. Cell 5 267–278. [DOI] [PubMed] [Google Scholar]