

Abstract
The tenth fibronectin type III (FN3) domain of human fibronectin (FNfn10), a prototype of the ubiquitous FN3 domain, is a small, monomeric β-sandwich protein. In this study, we have bisected FNfn10 in each loop to generate a total of six fragment pairs. We found that fragment pairs bisected at multiple loops of FNfn10 show complementation in vivo as tested with a yeast two-hybrid system. The dissociation constant of these fragment pairs determined in vitro were as low as 3 nM, resulting in one of the tightest fragment complementation systems reported so far. Furthermore, we show that the affinity of fragment complementation is correlated with the stability of the uncut parent protein. Exploring this correlation, we screened a yeast two-hybrid library of one fragment and identified mutations that suppress the effect of a destabilizing mutation in the other fragment. One of the identified mutations significantly increased the stability of the uncut wild-type protein, proving that fragment complementation can be used as a novel strategy for the selection of proteins with enhanced stability.
Keywords: protein engineering, reconstitution, library screening, β-sheet, surface loops
The association of protein fragments into the native fold and concomitant restoration of its function have been reported for a number of proteins (Sancho and Fersht 1992; Shiba and Schimmel 1992; de Prat Gay and Fersht 1994; Kobayashi et al. 1995; Ostermeier et al. 1999; Berggard et al. 2000; Berggard et al. 2001; Ojennus et al. 2001). This phenomenon of fragment complementation illustrates that an amino acid sequence can be separated without a loss of the information necessary to code for the native structure. Classic examples of restoration of enzyme function by fragment complementation include ribonuclease S-protein (Kato and Anfinsen 1969) and β-galactosidase (Ullmann et al. 1967). Fragment complementation has been used as a tool to understand the mechanism of protein folding (de Prat Gay et al. 1995a,b; Kobayashi et al. 1995; Honda et al. 1999; Jourdan and Searle 2000) and in vivo protein–protein interactions (Johnsson and Varshavsky 1994; Rossi et al. 1997; Karimova et al. 1998; Pelletier et al. 1998; Magliery et al. 2005). Residual structures within the individual fragments may correspond to folding nucleation sites (Dyson et al. 1992a,b; Sancho et al. 1992; de Prat-Gay 1996). Biophysical studies of such noncovalent complexes from fragments have also been reported (Sancho and Fersht 1992; Neira et al. 1996; Ladurner et al. 1997; Honda et al. 1999; Kobayashi et al. 1999), although few high-resolution structures are available (Yu et al. 2000; Ojennus et al. 2001).
To facilitate productive association between complementary fragments, one needs to identify a successful bisection site in the protein of interest. Creating a point of discontinuity within an α-helix or a β-strand is expected to significantly destabilize the native fold. In contrast, cleavage of a protein within a surface loop or turn often leads to the generation of associating fragments. In this paper, we refer to such a cleavage site as a “permissible” site. For example, a combinatorial investigation of Escherichia coli glycinamide ribonucleotide formyltransferase illustrated that permissible sites mainly cluster in loops (Ostermeier et al. 1999). However, small, single-domain proteins often have very few permissible loops, maybe because interactions involving surface loops and turns are more critical for the stability of such proteins (Hamill et al. 2000). The knowledge on the locations of permissible sites is also useful for protein engineering, because such sites highlight potential sites for modifications.
In this work we studied the fragment complementation of the tenth type III domain of human fibronectin (FNfn10). FNfn10 is a small β-sandwich protein with seven β-strands (Fig. 1 ▶). Here, we define “fragment complementation” as the restoration of the native fold of the parent protein, because FNfn10 does not have an easily detectable function. Our group has established FNfn10 as a scaffold for producing novel binding proteins (Koide et al. 1998, 2002). The loops of FNfn10 have been extensively mutated to engineer such binding proteins. We have systematically examined all the loops for their tolerance toward insertions of residues (Batori et al. 2002). These studies show that most of the loops of FNfn10 allowed insertion of residues without significant destabilization of the protein. In this work, we use the term “loop” to inclusively refer to a loop and a turn. Here, we examined each of FNfn10 loops as a possible cleavage site for generating complementary fragments. We found that fragment pairs generated by bisection in four out of the six loops reconstituted. Among these, fragments generated by cleaving FNfn10 in the loop connecting the C and D strands reconstituted with a very high affinity. Furthermore, we describe an application of this fragment complementation system to engineering proteins with enhanced stability.
Figure 1.

(A) Schematic drawing of the structure of FNfn10 (Dickinson et al. 1994). (B) The amino acid sequence of FNfn10 in its secondary structure context. Residues in a β-strand are shown as white circles. Those residues whose side chain forms the hydrophobic core are enclosed with a thicker line. Loop residues are shown shaded. The arrows mark the position in the loops where FNfn10 was separated to generate complementary fragments.
Results
Nomenclature
Loops in FNfn10 are named with respect to the β-strands that they connect. For example, the loop connecting the C and D strands is referred to as the CD loop (Fig. 1 ▶). Fragments generated by cleaving FNfn10 in a loop are named according to the β-strands they contain, starting with the letter A for the N-terminal fragments and ending with the letter G for the C-terminal fragments. For example, the N- and C-terminal fragments generated by cleaving FNfn10 in the CD loop are referred to as ABC and DEFG, respectively. The C-terminal fragment generated by cleaving FNfn10 variant I59A in the CD loop is referred to as DEFG(I59A).
Fragment complementation of FNfn10 examined with yeast two-hybrid
Earlier studies with FNfn10 indicated that most of the loops in the protein (Fig. 1 ▶) can tolerate an insertion of Gly residues without significant destabilization (Batori et al. 2002). We developed a yeast-two hybrid system to test whether FNfn10 fragments associate in vivo. We chose this yeast two-hybrid approach, because yeast mating allows for convenient testing of all possible combinations of fragments (Finley and Brent 1994). We found that complementary fragments bisected in the BC, CD, and DE loops, respectively, interacted strongly in our yeast two-hybrid system (Fig. 2 ▶). Furthermore, fragments with overlapping segments, such as ABC and CDEFG, and ABCD and DEFG also showed strong interaction. In contrast, the two largest fragments, ABCDEF and BCDEFG, did not show any detectable association with their respective complementary fragments or even overlapping fragments such as FG and AB, respectively.
Figure 2.
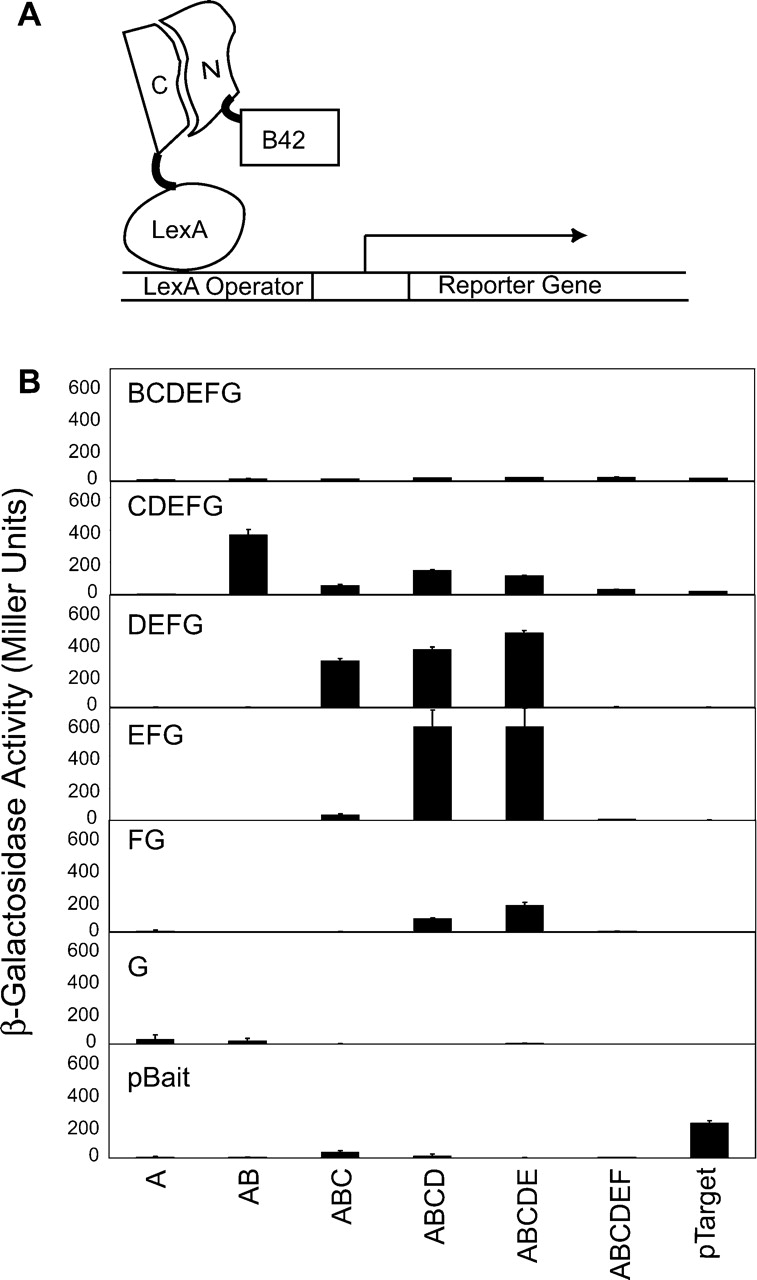
(A) Schematic drawing of the yeast two-hybrid system used to monitor the reconstitution of the N- and C-terminal fragments. The C-terminal fragment is fused to the LexA DNA binding domain and the N-terminal fragment is fused to the B42 activation domain. (B) Yeast two-hybrid β-galactosidase assay for all combinations of the N-and C-terminal fragments of FNfn10 produced in this work. The identity of a C-terminal fragment is indicated in each panel, and those of N-terminal fragments at the bottom of the figure. pTarget and pBait are two control proteins (Origene), which interact with each other.
Fragment complementation of FNfn10 in vitro
Next, we performed in vitro measurements to determine dissociation constant (Kd) of complementary FNfn10 fragment pairs. Fragment pairs from bisection in the CD and EF loop were chosen for in vitro Kd measurements, because they showed different extent of complementation in yeast two-hybrid (Fig. 2 ▶). We made mutant proteins in which the sequence, GGMGG, was introduced into a loop (Fig. 1 ▶), and purified the proteins and cleaved them with cyanogen bromide to produce fragments.
FNfn10 has a single tryptophan (Trp22 in the B strand) (Fig. 1 ▶) whose fluorescence is quenched in the folded state. Thus, we used tryptophan fluorescence of the ABC and ABCDE fragments to monitor the reconstitution reaction. Figure 3B ▶ shows the quenching of fluorescence of the ABCDE fragment as it was mixed with increasing concentrations of the FG fragment. The Kd value obtained was 1.2 ± 0.28 μM, indicating modest binding affinity between these two fragments.
Figure 3.

Fluorescence emission intensity of the ABC fragment plotted versus the concentration of the DEFG fragment (A) and that of the ABCDE fragment versus the concentration of the FG fragment (B). In A, the solution contained a 0.5-μM ABC fragment, 1 M urea, and 750 mM glycerol. In B, the ABCDE fragment concentration was 1 μM, and the solution did not contain urea or glycerol. (C) Urea dependence of the dissociation constant of the reconstitution reaction of the ABC and DEFG fragments. Data with 750 mM glycerol are shown. (D) Glycerol dependence of the dissociation constant of the reconstitution reaction of the ABC and DEFG fragments. Data in the presence of 2 M urea are shown.
Initial efforts in determining Kd for the reconstitution of the ABC and DEFG fragments were unsuccessful, because we found a gradual decrease of the concentration of the ABC fragment during titration experiments, probably due to adsorption to the quartz cuvette wall and/or aggregation. The addition of glycerol at a final concentration of 750 mM to the reaction mixture prevented this time-dependent decrease. Thus, further experiments were carried out in the presence of glycerol. Initial titration experiments of the ABC fragment with the DEFG fragment indicated that the affinity was so high that an accurate determination of Kd was not possible using submicromolar concentrations of fragments required in our assay. We carried out titrations in different concentrations of urea, where Kd fell in a medium to high nanomolar range (Fig. 3A ▶). Extrapolation of the Kd values to zero concentration of urea, but still in the presence of 750 mM glycerol, gave a value of ~1.5 nM (Fig. 3C ▶). Titrations were then carried out at different concentrations of glycerol at a fixed concentration of urea to see the dependence of Kd on glycerol concentration (Fig. 3D ▶). The second extrapolation resulted in a dissociation constant of 3.6 nM in the absence of both urea and glycerol. This value was independent of the order of the extrapolation.
Since this value of dissociation constant was obtained from extrapolation at different concentrations of urea and glycerol, we also measured binding of DEFG fragment to a ubiquitin fusion protein of the ABC fragment. We were able to obtain the dissociation constant without addition of glycerol or urea at low pH. The Kd value obtained was 14.4 ± 0.23 nM at pH 2.6, confirming the strong binding between these fragments. Due to the remarkably high affinity between the ABC and DEFG fragments, subsequent experiments were carried out with this pair.
The reconstituted complex forms the native fold
The conformation of the reconstituted complex was investigated using multidimensional NMR spectroscopy. Since the wild-type ABC fragment showed poor solubility, we used a more soluble mutant of this fragment having surface mutations D7K and E9Q (Koide et al. 2001). The 1H, 15N-heteronuclear single-quantum correlation (HSQC) spectrum (Kay et al. 1992) of this mutant in the uncleaved form was similar to that of wild-type FNfn10, indicating that the native fold was maintained. The ABC and DEFG fragments in isolation showed poorly dispersed HSQC peaks indicative of disordered structure (Fig. 4A,C ▶). An addition of the unlabeled counterpart to the labeled fragment resulted in a drastic increase in the spectral dispersion (Fig. 4B,D ▶). The composite of these two spectra (Fig. 4F ▶) is nearly identical to that of the uncut protein (Fig. 4E ▶), thus confirming that the reconstituted complex has the same global fold as the original uncut protein.
Figure 4.
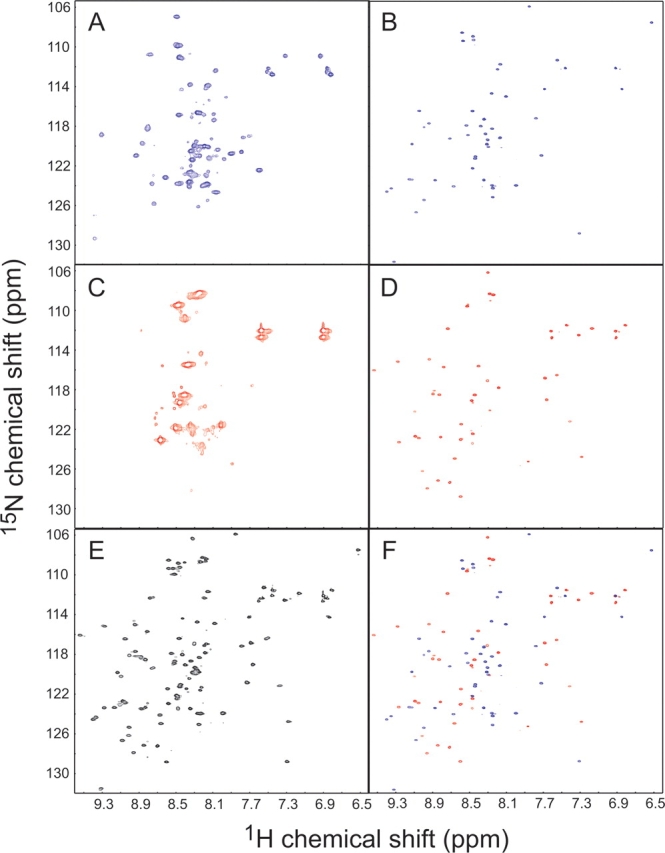
1H-15N HSQC spectra of the 15N-labeled DEFG fragment in isolation (A) and in the presence of an excess amount of unlabeled ABC fragment (B); those of 15N-labeled ABC fragment in isolation (C) and in the presence of an excess amount of unlabeled DEFG fragment (D); and that of the uncut protein (E). The spectrum in F is the composite of those shown in B and D.
Since the HSQC spectrum of the DEFG fragment in isolation showed additional peaks outside the random coil region (Fig. 4A ▶), circular dichroism studies were performed to investigate residual structure. The CD spectra exhibited concentration-dependent changes, indicative of aggregation (data not shown). These results suggest that possible residual structure in the DEFG fragment may be due to aggregation of this fragment, rather than a structured region within the monomeric fragment. Thus, we did not further pursue this line of investigation.
Application of fragment complementation to engineering stable mutants
We examined if the degree of fragment complementation in yeast two-hybrid was correlated with the stability of the uncut protein. We reasoned that mutations that increase the affinity of fragment complementation should also increase the stability of the uncut protein proportionately, provided that such mutations do not change molecular interactions close to the cleavage site. We selected three previously identified mutations that destabilize the protein by varying degrees (Cota et al. 2000) and observed their effect on fragment complementation. We found that the complementation ability of the fragments as detected with the yeast two-hybrid system is well correlated with the stability of the native uncut proteins (Fig. 5A ▶), suggesting the possibility of stabilizing FNfn10 through the identification of a mutation that enhances fragment association.
Figure 5.

(A). Yeast two-hybrid β-galactosidase assays for the interactions of the wild-type ABC fragment and different DEFG fragments (bar graph). Yeast two-hybrid survival assays of the cells containing the indicated fragment pair in minimal media lacking leucine (middle column). The right column shows ΔΔG of the uncut protein, defined as ΔGMutant − ΔGWT. (B) β-Galactosidase assays for the interactions of mutant ABC fragments isolated from yeast two-hybrid screening with the DEFG(I59A) fragment.
To isolate stabilizing mutations using yeast two-hybrid screening, we proceeded to find a fragment pair whose interaction strength is below the detection threshold in our system so that stabilizing mutations in such a pair can be selected. Since only the most destabilized variant I59A showed the LEU− phenotype (Fig. 5A ▶), further screening experiments were done using the DE FG fragment having this mutation so that stabilizing mutations would change the phenotype to LEU+.
As a proof of concept, we generated two small libraries of the ABC fragment and performed screening. We first carried out saturation mutagenesis at Gly37 to make library 1. We chose this position because glycine is not a preferred residue in a β-strand, and the side chain of a newly introduced amino acid residue at this position should be solvent exposed and should minimally disturb the hydrophobic core. Further mutations were added to library 1 through error prone PCR to create library 2. From both libraries we obtained mutant ABC fragments that exhibited significantly stronger interaction with DEFG(I59A) than the wild type (Fig. 5B ▶). Three out of four clones contained mutations at position 37 (the site of saturation mutagenesis) and adjacent position 36. Clone 3 contained additional mutations at positions 23 and 28 (Table 1).
Table 1.
Sequences of selected clones from yeast two hybrid screening of FNfn10 ABC fragment library against C-terminal fragment DEFG(I59A) and their effects on the stability of the wild-type protein and I59A variant
| Position | ||||||
| Clone | 23 | 28 | 36 | 37 | Δ ΔG (WT) (kcal mol−1) | Δ ΔG (I59A) (kcal mol−1) |
| WT | D | T | Y | G | 0 | 0 |
| 1 | D | T | F | R | −0.1 ± 0.2 | 0.7 ± 0.1 |
| 2 | D | T | C | E | −0.5 ± 0.2 | 0.2 ± 0.1 |
| 3 | V | N | C | W | 1.5 ± 0.2 | 2.1 ± 0.5 |
| 4 | D | T | Y | E | −0.3 ± 0.2 | 0.1 ± 0.1 |
Mutated residues are shown in bold. Δ ΔG(WT) is defined as ΔGmutant − ΔGWT and Δ ΔG(I59A) is defined as ΔGmutant − ΔGI59A. The errors associated with ΔG values are the standard deviations determined from independent experiments.
The mutations obtained from the selection were then introduced both into the wild type and the I59A variant of full-length FNfn10. Mutations in three clones that led to stronger interaction between the ABC and the DEF-G(I59A) fragment marginally affected the stability of the full-length proteins (Table 1). Introduction of the mutations present in clone 3, however, stabilized both wild-type FNfn10 and I59A by ~1.5 kcal mol−1 and 2.0 kcal mol−1, respectively.
Discussion
In this work, we discovered complementation of FNfn10 fragments produced by bisection in the BC, CD, and DE loops (Fig. 2 ▶). Earlier studies on complementation of chymotrypsin inhibitor 2 (CI2) and streptococcal protein G B1 domain each found only one bisection site for productive complementation (Ladurner et al. 1997; Kobayashi et al. 1999). It is interesting that a small protein such as FNfn10 tolerates cleavage at multiple loops. These results suggest that the cleavage of these FNfn10 loops minimally affect the stability of the reconstituted complex, consistent with previous results that insertion of glycine residues in all but the EF loop of FNfn10 result in marginal destabilization of the protein (Batori et al. 2002) and NMR studies showing that many FNfn10 loops are quite flexible (Carr et al. 1997).
We also observe strong complementation between fragments containing overlapping sequences (ABCD/DEFG, ABCDE/DEFG and ABCDE/EFG). In these complexes, one of the overlapping segments is presumably unstructured. Complementation of overlapping fragments was also observed for CI2 (Ladurner et al. 1997) and E. coli glycinamide ribonucleotide formyltransferase (PurN) (Ostermeier et al. 1999). Structural studies on CI2 have shown that the thermodynamically most stable complex is formed in the presence of redundant primary sequence (Ladurner et al. 1997). Due to the presence of a single permissible cleavage site in CI2, the point of discontinuity of the polypeptide chain in the reconstituted complex was clearly observed around that site. In contrast, FNfn10 has multiple permissible sites. For complementation of ABCD/DEFG or ABCDE/DEFG fragments the point of discontinuity may lie in either the CD or the DE loop, since cleavage in both of these loops result in successful fragment complementation. Structural studies of these overlapping fragments will be helpful in determining whether there is a preferred point of discontinuity in these complexes.
The ABC and DEFG fragments reconstitute with a Kd of 3.6 nM. A survey of the literature (Table 2) suggests that this is one of the tightest fragment complementation systems reported so far that do not involve metal binding. The high affinity between these fragments is reflected in the high stability of the parent protein and minimal perturbation by the cleavage. The high affinity of this fragment complementation system holds potential as heterodimerization motifs, analogous to the widely used coiled-coil systems (O’Shea et al. 1993). The absence of homodimerizing propensity for the ABC and DEFG fragments is clearly an advantage of our system. The low solubility of the ABC fragment might impose some limitations on the applicability of this system. However, one could improve it by introducing mutations that enhance the solubility.
Table 2.
Comparison of Kdvalues for reconstitution reaction reported in the literature
| Protein | Kd | No. of residues | References |
| FNfn10 | 3.6 nM | 42 + 47 | This work |
| CI2 | 40 nM | 40 + 24 | (Ladurner et al. 1997) |
| Ubiquitin | 38 μM | 35 + 40 | (Jourdan and Searle 2000) |
| Protein G B1 domain | 10 μM | 40 + 15 | (Honda et al. 1999) |
| Barnase | 0.6 μM | 36 + 73 | (Sancho and Fersht 1992) |
| S-protein/S-peptide | 0.6 μM (wild type) 5.4 nM (engineered) |
20 + 104 | (Dwyer et al. 2001) |
| Calbindin D9K | 3 pM (with Ca+2) | 43 + 31 | (Berggard et al. 2001) |
Compared with the ABC/DEFG pair, the Kd value for the reconstitution between the ABCDE and the FG fragments was much higher (1.2 ± 0.28 μM). Our earlier studies indicated that insertion of residues in the EF loop caused significant destabilization of FNfn10 by ~4.4 kcal mol−1 (Batori et al. 2002). The EF loop includes the “tyrosine corner” motif in which a side chain–backbone hydrogen bond is formed between Tyr68 and Lys63 (Hamill et al. 2000). Loss of this interaction in the reconstituted complex may be a major contributing factor to its destabilization. These observations lend further support to the correlation between fragment complementation affinity and the stability of the uncut protein. Previous studies with calbindin D9k have also shown that the affinity of fragment complementation is well correlated to the stability of the uncut protein (Berggard et al. 2001).
We did not find evidence for native-like structures in the ABC and DEFG fragments. Because the bisection in the CD loop disrupts both β-sheets of FNfn10 and also the hydrophobic core (Fig. 1 ▶), it is not unexpected that these fragments lack a native-like structure in isolation. Most of the earlier studies of protein fragment complementation have indicated the presence of residual structure in one of the complementing fragments (Sancho et al. 1992; de Prat Gay and Fersht 1994; Kippen et al. 1994; Kobayashi et al. 1999). The reconstitution of p85α SH2 domain was, however, found to take place from unstructured fragments (Ojennus et al. 2001). The absence of residual structure would lead to stronger affinity between complementary fragments, because most of the free energy difference for the folding of the parental protein is available for binding of the fragments.
Although glycine insertions in the AB or the FG loop do not significantly destabilize FNfn10 (Batori et al. 2002), bisection in these two loops did not result in detectable association between the complementary fragments (A and BCDEFG, and ABCDEF and G, respectively) (Fig. 2 ▶). Furthermore, we were unable to detect any interaction of the ABCDEF and BCDEFG fragments with fragments having overlapping sequences (e.g., FG and AB, respectively). These results suggest that the ABCDEF and BCDEFG fragments may have a native-like residual structure. It is worth mentioning that the NMR structure of the CDEFG fragment of the first FN3 domain of human fibronectin (“Anastellin”) is folded in a conformation similar to the corresponding region of the full-length protein (Briknarova et al. 2003). We cannot, however, exclude the possibility that these fragments aggregate and/or interact with a cellular component so that they are unavailable for complementation in the yeast two-hybrid system.
From the observations above, a guideline for designing tightly associating fragments emerges. A cleavage site should be chosen so that it minimally destabilizes the parent protein. On the other hand, the fragmentations should eliminate the possibility of the formation of the native-like structure in fragments. The ABC–DEFG pair of FNfn10 appears to satisfy this guideline.
The high-affinity complementation of FNfn10 fragments suggests possible roles of fragment complementation in protein evolution. A highly debated question in protein evolution is whether structural units in proteins correspond to exon intron boundaries of their genes (Bertolaet et al. 1995). Gilbert and coworkers have shown that intron positions correlate with module boundaries in ancient proteins (de Souza et al. 1996). Fragment complementation studies with triose phosphate isomerase have shown restoration of catalytic activity with fragments resulting from cleavage in exon–intron boundaries (Bertolaet and Knowles 1995). Interestingly, we found an intron in the human FNfn10 gene at a position corresponding to residue 40, two residues N-terminal to our CD-loop cleavage site (Oldberg and Ruoslahti 1986; Fig. 1B ▶). It is tempting to speculate that early in evolution a functional FN3 domain may have emerged by tight association of two separate peptides.
A number of library screening methods have been successfully employed in engineering proteins with increased stability. Such selection is challenging because it is often difficult to identify a selection criterion that directly reflects protein stability. Direct selection for stable proteins is possible when the function of a protein of interest is related to the survival of its host microorganism (Das et al. 1989). Selection for stably folded proteins based on binding to a target (Kim et al. 1998; Distefano et al. 2002; Kotz et al. 2004) or on their susceptibility to proteolysis (Kristensen and Winter 1998; Sieber et al. 1998; Bai and Feng 2004) have been demonstrated using phage display. Display levels of fusion protein on yeast surface has been shown to be correlated with thermal stability (Shusta et al. 1999), and resistance to decreasing redox potential was successfully employed for selecting proteins that are stable in the absence of native disulphide bonds (Jermutus et al. 2001). Although these examples represent an impressive array of technical innovations and successes, the idiosyncrasy of the target protein often imposes restrictions on the applicability of these methods. In particular, they are not always suitable for improving the stability of an already stable protein (Pedersen et al. 2002).
The fragment complementation screening system represents an alternative to existing approaches of isolating stable variants of a protein. Our yeast two-hybrid results show that the degree of association of complementary fragments is well correlated to the stability of the uncut protein (Fig. 5A ▶). The stabilization of a highly stable protein, FNfn10, by ~1.5 kcal mol−1 shows the potential of our system to improve protein stability.
Our results also illustrate an advantage of our strategy in the choice of the host protein in stability-based selection. Previous studies have shown that the host protein needs to be sufficiently destabilized in order to identify stabilizing mutations (Zhou et al. 1996). However, our host protein for screening, the FNfn10 I59A variant, is modestly stable (ΔΔG ~5.3 kcal mol−1), and one would need to further destabilize it in order to employ a function-based screening. Also, there may not be a convenient functional test for some proteins, as is the case for FNfn10. Our screening system allowed us to isolate stabilizing mutations using a modestly stable host protein without the use of function-based screening. These results suggest that our strategy should be applicable to many other proteins.
Although we successfully identified mutations that improved the FNfn10 stability, not all mutations that restored complementation ability of the ABC fragments with DEFG(I59A) fragment led to enhancement of stability of the I59A variant. It is not straightforward to quantify differences in affinity from yeast two-hybrid data alone (Estojak et al. 1995). A major reason is that it is difficult to compensate for differences in expression levels of interacting proteins in yeast cells. Although we did not observe marked expression differences of different ABC fragments (data not shown), it is still possible that the interaction of DEFG(I59A) fragment with the wild-type ABC fragment in vivo was close to the affinity threshold required to score as positive and small expression differences led to positive scoring in our selection.
Clones 1–3, which had a mutation at position 36, had slightly different effects on the stability of the I59A variant and the wild type (Table 1). Examination of the FNfn10 structure revealed that Tyr36 is spatially close to Ile59, which was mutated in the host protein (I59A), explaining the reason for the different effects of the mutations in these clones. This emphasizes the importance of selecting stabilizing mutations in the context of the wild-type fragments in order to improve the stability of the wild-type protein. A difficulty is to modulate the stringency of selection so that the affinity for the wild-type fragment pair falls below the threshold required to score as positive. Studies are underway in our laboratory to investigate other selection techniques as alternative methods to overcome aforementioned limitations of the method presented here.
Materials and methods
Strains and media
Yeast strains EGY48, MATα his3 trp1 ura3 leu2::6LexAop-LEU2, and RFY206, MATa his3Δ200 leu2–3 lys2Δ201 trp1Δ::hisG ura3–52, have been described (Gyuris et al. 1993; Finley and Brent 1994) and were purchased from Origene. Yeast cells were grown in yeast extract/peptone/dextrose media or yeast complete dropout media following instructions from Invitrogen.
Construction of vectors for yeast two-hybrid
The plasmids encoding different N-terminal fragments of FNfn10 (pFN-X-B42 where X is A, AB, ABC, ABCD, ABCDE, or ABCDEF; see “Nomenclature” in Results for the fragment naming scheme) were constructed by subcloning appropriate PCR fragments in the plasmid pYesTrp2 (Invitrogen) so that the fragments are fused N-terminal to the B42 activation domain. Similarly, plasmids encoding LexA-fusion proteins (pEG-X where X is BCDEFG, CDEFG, DEFG, EFG, FG, or G) were constructed by subcloning appropriate PCR fragments in the plasmid pEG202 (Origene). In addition, the plasmid encoding DEFG fragment having the mutation I59A was made as a LexA fusion (termed pEG-DEFG[I59A]).
Library construction
All mutagenesis experiments were performed using pFN-ABC-B42 as the template. Saturation mutagenesis at position 37 was performed using PCR. The primers 5′-TAATACGACTCAC TATAGGG-3′ (T7P) and 5′-CACCGTTACCACCGGTTTCMNNGTACGTGATACGGTAATAAC-3′, where N denotes a mixture of A, T, G, and C, and M denotes a mixture of A or C, were used to introduce mutations in the ABC fragment at position 37. The primers 5′-GCGTGAATGTAAGCGTGAC-3′ (pYTRev) and 5′-GAAACCGGTGGTAACGGTG-3′ were used to amplify the B-42 segment. These two PCR products were mixed in approximately equimolar concentrations, annealed, extended, and finally amplified with the T7P and pYTRev primers to generate a DNA fragment for creating library1. For creating library 2, essentially the same procedure was followed except that the PCR for the ABC fragment was performed in presence of 0.5 mM MnCl2, 7 mM MgCl2, 0.2 mM of each dATP and dGTP, 1 mM of each dCTP and dTTP, and 5 units of Taq DNA polymerase. To prepare a library in yeast, homologous recombination was used. pFN-ABC-B42 was digested with KpnI, followed by cleaning with the Qiagen gel elution kit to prepare the cut acceptor vector. The yeast strain EGY48 was transformed with 1 μg each of the cut vector and a PCR fragment following the procedure of Geitz and Woods (2001).
Library screening
The yeast strain RFY206 harboring the plasmid pEG-DEFG-(I59A) and the LacZ reporter plasmid pSH18–34 (Origene) was mated with EGY48 containing library 1 or library 2 (Finley and Brent 1994). Diploid cells with the LEU+ phenotype were selected on minimal dropout media (Gal Raf Leu− His−Ura−Trp−) (Sambrook 2001). Selected cells were then tested for galactose dependence of the LEU+ phenotype and β-galactosidase activity. The DNA segment corresponding to the ABC fragment was amplified by PCR and used for DNA sequencing. The phenotypes of the selected clones were confirmed by retransforming yeast cells using these PCR fragments and the cut vector. β-Galactosidase assay was performed with the mated cells as described previously (Koide et al. 2002).
Protein expression and purification
The E. coli expression vector for wild-type FNfn10 has been described (Koide et al. 2001). All mutations were introduced using standard PCR methods and the genes of all the mutants were verified by DNA sequencing. All the mutant proteins were expressed as soluble proteins and subsequently purified using metal affinity chromatography, as previously described for the wild-type protein (Koide et al. 1998).
Fragment purification
FNfn10 mutants containing GGMGG insertion in the CD or the EF loop were dissolved at a concentration of 2 mg/mL in 0.1 M HCl. Two to five milligrams of cyanogen bromide were dissolved and the reaction container was sealed under Argon and incubated at room temperature for 2 h. The reaction mixture was then passed through a SepPak cartridge (Waters) to remove cyanogen bromide, and the bound protein was eluted with 60% acetonitrile and kept on ice. Fragments were separated on a reverse phase column (Resource RPC, Amersham Biosciences) by an acetonitrile gradient from 20% to 65%, and their identity confirmed by mass spectrometry. All procedures involving cyanogen bromide were performed in a fume hood.
The gene for the ubiquitin fusion of the ABC fragment was subcloned into the expression vector pET24a (Novagen) using standard PCR so that the ubiquitin gene (Ohnishi et al. 2001) is fused N-terminal to the ABC fragment. The protein was purified as follows: Cells were suspended in the suspension buffer (20 mM Tris, 100 mM sodium chloride at pH 8). Hen Egg Lysozyme (final concentration 1 mg/mL) and phenyl methyl sulfonyl chloride (final concentration 1 mM) were added to the suspension. The solution was incubated for 30 min at 4°C and sonicated in ice. After centrifugation, the pellet was dissolved in the suspension buffer containing 4 M urea. After a second centrifugation, the supernatant was recovered to which concentrated sodium chloride was added at a final concentration of 0.5 M. The solution was applied to a Hi-Trap chelating column (Amersham) preloaded with nickel and equilibrated with Tris buffer containing 0.5 M sodium chloride and 4 M urea. After washing the column with the same buffer, Ubi-ABC was eluted with the buffer containing 400 mM imidazole. The eluted fractions containing the protein were pooled and dialyzed against 1 mM Glycine HCl buffer at pH 2.6. The dialyzed sample was centrifuged and the protein in the supernatant fraction was used for subsequent studies.
Fluorescence spectroscopy
All fluorescence experiments were performed using a Spectronics AB-2 spectrofluorometer equipped with an automated titrator as described previously (Koide et al. 1998, 1999). To measure the dissociation constant of the reconstitution reaction between ABC and DEFG fragments or ABCDE and FG fragments, increasing amounts of DEFG or FG fragment were added to a fixed concentration (500 nM for ABC and 1 μM for ABCDE, respectively) of N-terminal fragment and, after 2 min incubation, the fluorescence emission at 355 nm with excitation at 295 nm was measured. The sample temperature was kept at 25°C. The dissociation constant of the complex was calculated by fitting to the data the following equation for association with a 1:1 stoichiometry:
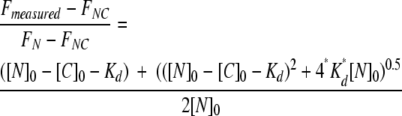 |
where [N]0 is the total concentration of the N-terminal fragment, [C]0 is the total concentration of the C-terminal fragment, Kd is the dissociation constant, FN is the fluorescence of the free N-terminal fragment, FNC is the fluorescence of the complex, and Fmeasured is the observed value of fluorescence. The fitting was performed using the program Igor Pro 3.0 (Wavemetrics).
For chemical denaturation measurements, proteins were dissolved at a final concentration of 1 μM in 20 mM sodium citrate buffer at pH 6 containing 100 mM sodium chloride. Increasing concentrations of guanidine hydrochloride (GuHCl) were added and the change in fluorescence from the single tryptophan residue in FNfn10 was monitored at 30°C as described (Koide et al. 1998). The GuHCl concentrations before and after a titration experiment were determined using an Abbe refractometer (Spectronic Instruments) as described (Pace and Sholtz 1997). Data were fitted to the standard two-state unfolding model (Koide et al. 1998) using the program Igor Pro 3.0.
NMR spectroscopy
NMR experiments were performed at 30°C on a Varian INOVA 600 spectrometer. The 1H, 15N-heteronuclear single-quantum correlation (HSQC) spectrum (Kay et al. 1992) of the DEFG fragment (0.5 mM) in isolation was recorded in sodium phosphate buffer at pH 6 containing 100 mM sodium chloride and 5% (v/v) deuterium oxide as described previously. The spectrum of the ABC fragment (0.25 mM) was recorded in the above buffer containing 5% glycerol to prevent aggregation. To prepare a NMR sample for a reconstituted complex, a 15N-labeled fragment was mixed with its unlabeled complementary fragment in 20 mM sodium phosphate at pH 6, containing 4 M GuHCl. The molar ratio of the labeled and unlabeled fragments during the reconstitution reaction was 1:3. GuHCl was removed by dialysis and the samples were concentrated. NMR data were processed using the NMR Pipe Package (Kay et al. 1992) and analyzed using the NMRView software (Johnson and Blevins 1994).
Acknowledgments
We thank Gregory Darnell and Dr. Scott Kennedy for technical assistance. This work was supported in part by NIH grants GM55042 and DK62316 to S.K.
Abbreviations
CI2, chymotrypsin inhibitor 2
FN3, fibronectin type III domain
FNfn10, tenth FN3 of human fibronectin
GuHCl, guanidine hydrochloride
HSQC, heteronuclear single-quantum correlation
NMR, nuclear magnetic resonance
PCR, polymerase chain reaction.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051603005.
References
- Bai, Y. and Feng, H. 2004. Selection of stably folded proteins by phage-display with proteolysis. Eur. J. Biochem. 271 1609–1614. [DOI] [PubMed] [Google Scholar]
- Batori, V., Koide, A., and Koide, S. 2002. Exploring the potential of the monobody scaffold: Effects of loop elongation on the stability of a fibronectin type III domain. Protein Eng. 15 1015–1020. [DOI] [PubMed] [Google Scholar]
- Berggard, T., Thulin, E., Akerfeldt, K.S., and Linse, S. 2000. Fragment complementation of calbindin D28k. Protein Sci. 9 2094–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berggard, T., Julenius, K., Ogard, A., Drakenberg, T., and Linse, S. 2001. Fragment complementation studies of protein stabilization by hydro-phobic core residues. Biochemistry 40 1257–1264. [DOI] [PubMed] [Google Scholar]
- Bertolaet, B.L. and Knowles, J.R. 1995. Complementation of fragments of triosephosphate isomerase defined by exon boundaries. Biochemistry 34 5736–5743. [DOI] [PubMed] [Google Scholar]
- Bertolaet, B.L., Seidel, H.M., and Knowles, J.R. 1995. Introns and the origin of protein-coding genes. Science 268 1367 [author reply 1367– 1369]. [DOI] [PubMed] [Google Scholar]
- Briknarova, K., Akerman, M.E., Hoyt, D.W., Ruoslahti, E., and Ely, K.R. 2003. Anastellin, an FN3 fragment with fibronectin polymerization activity, resembles amyloid fibril precursors. J. Mol. Biol. 332 205–215. [DOI] [PubMed] [Google Scholar]
- Carr, P.A., Erickson, H.P., and Palmer 3rd, A.G. 1997. Backbone dynamics of homologous fibronectin type III cell adhesion domains fromfibronectin and tenascin. Structure 5 949–959. [DOI] [PubMed] [Google Scholar]
- Cota, E., Hamill, S.J., Fowler, S.B., and Clarke, J. 2000. Two proteins with the same structure respond very differently to mutation: The role of plasticity in protein stability. J. Mol. Biol. 302 713–725. [DOI] [PubMed] [Google Scholar]
- Das, G., Hickey, D.R., McLendon, D., McLendon, G., and Sherman, F. 1989. Dramatic thermostabilization of yeast iso-1-cytochrome c by an asparagine—Isoleucine replacement at position 57. Proc. Natl. Acad. Sci. 86 496–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Prat-Gay, G. 1996. Association of complementary fragments and the elucidation of protein folding path-ways. Protein Eng. 9 843–847. [DOI] [PubMed] [Google Scholar]
- de Prat Gay, G. and Fersht, A.R. 1994. Generation of a family of protein fragments for structure-folding studies. 1. Folding complementation of two fragments of chymotrypsin inhibitor-2 formed by cleavage at its unique methionine residue. Biochemistry 33 7957–7963. [DOI] [PubMed] [Google Scholar]
- de Prat Gay, G., Ruiz-Sanz, J., Neira, J.L., Corrales, F.J., Otzen, D.E., Ladurner, A.G., and Fersht, A.R. 1995a. Conformational pathway of the polypeptide chain of chymotrypsin inhibitor-2 growing from its N terminus in vitro. Parallels with the protein folding pathway. J. Mol. Biol. 254 968–979. [DOI] [PubMed] [Google Scholar]
- de Prat Gay, G., Ruiz-Sanz, J., Neira, J.L., Itzhaki, L.S., and Fersht, A.R. 1995b. Folding of a nascent polypeptide chain in vitro: Cooperative formation of structure in a protein module. Proc. Natl. Acad. Sci. 92 3683–3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza, S.J., Long, M., Schoenbach, L., Roy, S.W., and Gilbert, W. 1996. Intron positions correlate with module boundaries in ancient proteins. Proc. Natl. Acad. Sci. 93 14632–14636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson, C.D., Veerapandian, B., Dai, X.P., Hamlin, R.C., Xuong, N.H., Ruoslahti, E., and Ely, K.R. 1994. Crystal structure of the tenth type III cell adhesion module of human fibronectin. J. Mol. Biol. 236 1079–1092. [DOI] [PubMed] [Google Scholar]
- Distefano, M.D., Zhong, A., and Cochran, A.G. 2002. Quantifying β-sheet stability by phage display. J. Mol. Biol. 322 179–188. [DOI] [PubMed] [Google Scholar]
- Dwyer, J.J., Dwyer, M.A., and Kossiakoff, A.A. 2001. High affinity RNase S-peptide variants obtained by phage display have a novel “hot-spot” of binding energy. Biochemistry 40 13491–13500. [DOI] [PubMed] [Google Scholar]
- Dyson, H.J., Merutka, G., Waltho, J.P., Lerner, R.A., and Wright, P.E. 1992a. Folding of peptide fragments comprising the complete sequence of proteins. Models for initiation of protein folding. I. Myohemerythrin. J. Mol. Biol. 226 795–817. [DOI] [PubMed] [Google Scholar]
- Dyson, H.J., Sayre, J.R., Merutka, G., Shin, H.C., Lerner, R.A., and Wright, P.E. 1992b. Folding of peptide fragments comprising the complete sequence of proteins. Models for initiation of protein folding. II. Plastocyanin. J. Mol. Biol. 226 819–835. [DOI] [PubMed] [Google Scholar]
- Estojak, J., Brent, R., and Golemis, E.A. 1995. Correlation of two-hybrid affinity data with in vitro measurements. Mol. Cell. Biol. 15 5820–5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley Jr., R.L. and Brent, R. 1994. Interaction mating reveals binary and ternary connections between Drosophila cell cycle regulators. Proc. Natl. Acad. Sci. 91 12980–12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz, R.D. and Woods, R.A. 2001. Genetic transformation of yeast. Biotechniques 30 816–820. [DOI] [PubMed] [Google Scholar]
- Gyuris, J., Golemis, E., Chertkov, H., and Brent, R. 1993. Cdi1, a human G1 and S phase protein phosphatase that associates with Cdk2. Cell 75 791–803. [DOI] [PubMed] [Google Scholar]
- Hamill, S.J., Cota, E., Chothia, C., and Clarke, J. 2000. Conservation of folding and stability within a protein family: The tyrosine corner as an evolutionary cul-de-sac. J. Mol. Biol. 295 641–649. [DOI] [PubMed] [Google Scholar]
- Honda, S., Kobayashi, N., Munekata, E., and Uedaira, H. 1999. Fragment reconstitution of a small protein: Folding energetics of the reconstituted immunoglobulin binding domain B1 of streptococcal protein G. Biochemistry 38 1203–1213. [DOI] [PubMed] [Google Scholar]
- Jermutus, L., Honegger, A., Schwesinger, F., Hanes, J., and Pluckthun, A. 2001. Tailoring in vitro evolution for protein affinity or stability. Proc. Natl. Acad. Sci. 98 75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, B.A. and Blevins, R.A. 1994. NMR View—A computer-program for the visualization and analysis of NMR data. J. Biomol. NMR 4 603–614. [DOI] [PubMed] [Google Scholar]
- Johnsson, N. and Varshavsky, A. 1994. Split ubiquitin as a sensor of protein interactions in vivo. Proc. Natl. Acad. Sci. 91 10340–10344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdan, M. and Searle, M.S. 2000. Cooperative assembly of a nativelike ubiquitin structure through peptide fragment complexation: Energetics of peptide association and folding. Biochemistry 39 12355–12364. [DOI] [PubMed] [Google Scholar]
- Karimova, G., Pidoux, J., Ullmann, A., and Ladant, D. 1998. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc. Natl. Acad. Sci. 95 5752–5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato, I. and Anfinsen, C.B. 1969. On the stabilization of ribonuclease S-protein by ribonuclease S-peptide. J. Biol. Chem. 244 1004–1007. [PubMed] [Google Scholar]
- Kay, L.E., Keifer, P., and Saarinen, T. 1992. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J. Am. Chem. Soc. 114 10663–10665. [Google Scholar]
- Kim, D.E., Gu, H., and Baker, D. 1998. The sequences of small proteins are not extensively optimized for rapid folding by natural selection. Proc. Natl. Acad. Sci. 95 4982–4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kippen, A.D., Sancho, J., and Fersht, A.R. 1994. Folding of barnase in parts. Biochemistry 33 3778–3786. [DOI] [PubMed] [Google Scholar]
- Kobayashi, N., Honda, S., Yoshii, H., Uedaira, H., and Munekata, E. 1995. Complement assembly of two fragments of the streptococcal protein G B1 domain in aqueous solution. FEBS Lett. 366 99–103. [DOI] [PubMed] [Google Scholar]
- Kobayashi, N., Honda, S., and Munekata, E. 1999. Fragment reconstitution of a small protein: Disulfide mutant of a short C-terminal fragment derived from streptococcal protein G. Biochemistry 38 3228–3234. [DOI] [PubMed] [Google Scholar]
- Koide, A., Bailey, C.W., Huang, X., and Koide, S. 1998. The fibronectin type III domain as a scaffold for novel binding proteins. J. Mol. Biol. 284 1141–1151. [DOI] [PubMed] [Google Scholar]
- Koide, S., Bu, Z., Risal, D., Pham, T.N., Nakagawa, T., Tamura, A., and Engelman, D.M. 1999. Multistep denaturation of Borrelia burgdorferi OspA, a protein containing a single-layer β-sheet. Biochemistry 38 4757–4767. [DOI] [PubMed] [Google Scholar]
- Koide, A., Jordan, M.R., Horner, S.R., Batori, V., and Koide, S. 2001. Stabilization of a fibronectin type III domain by the removal of unfavorable electrostatic interactions on the protein surface. Biochemistry 40 10326–10333. [DOI] [PubMed] [Google Scholar]
- Koide, A., Abbatiello, S., Rothgery, L., and Koide, S. 2002. Probing protein conformational changes in living cells by using designer binding proteins: Application to the estrogen receptor. Proc. Natl. Acad. Sci. 99 1253–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotz, J.D., Bond, C.J., and Cochran, A.G. 2004. Phage-display as a tool for quantifying protein stability determinants. Eur. J. Biochem. 271 1623–1629. [DOI] [PubMed] [Google Scholar]
- Kristensen, P. and Winter, G. 1998. Proteolytic selection for protein folding using filamentous bacteriophages. Fold. Des. 3 321–328. [DOI] [PubMed] [Google Scholar]
- Ladurner, A.G., Itzhaki, L.S., de Prat Gay, G., and Fersht, A.R. 1997. Complementation of peptide fragments of the single domain protein chymotrypsin inhibitor 2. J. Mol. Biol. 273 317–329. [DOI] [PubMed] [Google Scholar]
- Magliery, T.J., Wilson, C.G., Pan, W., Mishler, D., Ghosh, I., Hamilton, A.D., and Regan, L. 2005. Detecting protein–protein interactions with a green fluorescent protein fragment reassembly trap: Scope and mechanism. J. Am. Chem. Soc. 127 146–157. [DOI] [PubMed] [Google Scholar]
- Neira, J.L., Davis, B., Ladurner, A.G., Buckle, A.M., Gay Gde, P., and Fersht, A.R. 1996. Towards the complete structural characterization of a protein folding pathway: The structures of the denatured, transition and native states for the association/folding of two complementary fragments of cleaved chymotrypsin inhibitor 2. Direct evidence for a nucleation-condensation mechanism. Fold. Des. 1 189–208. [DOI] [PubMed] [Google Scholar]
- Ohnishi, S., Koide, A., and Koide, S. 2001. The roles of turn formation and cross-strand interactions in fibrillization of peptides derived from the OspA single-layer β-sheet. Protein Sci. 10 2083–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojennus, D.D., Fleissner, M.R., and Wuttke, D.S. 2001. Reconstitution of a native-like SH2 domain from disordered peptide fragments examined by multidimensional heteronuclear NMR. Protein Sci. 10 2162–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldberg, A. and Ruoslahti, E. 1986. Evolution of the fibronectin gene. Exon structure of cell attachment domain. J. Biol. Chem. 261 2113– 2116. [PubMed] [Google Scholar]
- O’Shea, E.K., Lumb, K.J., and Kim, P.S. 1993. Peptide “Velcro”: Design of a heterodimeric coiled coil. Curr. Biol. 3 658–667. [DOI] [PubMed] [Google Scholar]
- Ostermeier, M., Nixon, A.E., Shim, J.H., and Benkovic, S.J. 1999. Combinatorial protein engineering by incremental truncation. Proc. Natl. Acad. Sci. 96 3562–3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace, C.N. and Sholtz, J.M. 1997. Measuring the conformational stability of a protein. In Protein structure. A practical approach (ed. T.E. Creighton), 2nd ed., pp. 299–321. IRL Press, Oxford, UK.
- Pedersen, J.S., Otzen, D.E., and Kristensen, P. 2002. Directed evolution of barnase stability using proteolytic selection. J. Mol. Biol. 323 115–123. [DOI] [PubMed] [Google Scholar]
- Pelletier, J.N., Campbell-Valois, F.X., and Michnick, S.W. 1998. Oligomerization domain-directed reassembly of active dihydrofolate reductase from rationally designed fragments. Proc. Natl. Acad. Sci. 95 12141–12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi, F., Charlton, C.A., and Blau, H.M. 1997. Monitoring protein– protein interactions in intact eukaryotic cells by β-galactosidase complementation. Proc. Natl. Acad. Sci. 94 8405–8410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook, J. 2001. Molecular cloning: A laboratory manual, 3rd ed., p. A2.9. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Sancho, J. and Fersht, A.R. 1992. Dissection of an enzyme by protein engineering. The N- and C-terminal fragments of barnase form a native-like complex with restored enzymic activity. J. Mol. Biol. 224 741–747. [DOI] [PubMed] [Google Scholar]
- Sancho, J., Neira, J.L., and Fersht, A.R. 1992. An N-terminal fragment of barnase has residual helical structure similar to that in a refolding intermediate. J. Mol. Biol. 224 749–758. [DOI] [PubMed] [Google Scholar]
- Shiba, K. and Schimmel, P. 1992. Functional assembly of a randomly cleaved protein. Proc. Natl. Acad. Sci. 89 1880–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shusta, E.V., Kieke, M.C., Parke, E., Kranz, D.M., and Wittrup, K.D. 1999. Yeast polypeptide fusion surface display levels predict thermal stability and soluble secretion efficiency. J. Mol. Biol. 292 949–956. [DOI] [PubMed] [Google Scholar]
- Sieber, V., Pluckthun, A., and Schmid, F.X. 1998. Selecting proteins with improved stability by a phage-based method. Nat. Biotechnol. 16 955–960. [DOI] [PubMed] [Google Scholar]
- Ullmann, A., Jacob, F., and Monod, J. 1967. Characterization by in vitro complementation of a peptide corresponding to an operator-proximal segment of the β-galactosidase structural gene of Escherichia coli. J. Mol. Biol. 24 339–343. [DOI] [PubMed] [Google Scholar]
- Yu, W.F., Tung, C.S., Wang, H., and Tasayco, M.L. 2000. NMR analysis of cleaved Escherichia coli thioredoxin (1–73/74–108) and its P76A variant: cis/trans peptide isomerization. Protein Sci. 9 20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, H.X., Hoess, R.H., and DeGrado, W.F. 1996. In vitro evolution of thermodynamically stable turns. Nat. Struct. Biol. 3 446–451. [DOI] [PubMed] [Google Scholar]