

Abstract
Methionine sulfoxide reductases B (MsrBs) catalyze the reduction of methionine-R-sulfoxide via a three-step chemical mechanism including a reductase step, formation of an intradisulfide bond followed by a thioredoxin recycling process. Fifty percent of the MsrBs, including the Escherichia coli enzyme, possess a metal binding site composed of two CXXC motifs of unknown function. It is located on the opposite side of the active site. The overexpressed E. coli MsrB tightly binds one atom of zinc/iron. Substitution of the cysteines of E. coli MsrB results in complete loss of bound metal and reductase activity, and leads to a low-structured conformation of the protein as shown by CD, fluorescence, and DSC experiments. Introduction of the two CXXC motifs in Neisseria meningitidis MsrB domain leads to a MsrB that tightly binds one atom of zinc/iron, shows a strongly increased thermal stability and displays a reductase activity similar to that of the wild-type but lacking thioredoxin recycling activity. These results demonstrate the stabilizing effect of the metal and the existence of a preformed metal binding site in the nonbound metal MsrB. The data also indicate that metal binding to N. meningitidis MsrB induces subtle structural modifications, which prevent formation of a competent binary complex between oxidized MsrB and reduced thioredoxin but not between reduced MsrB and substrate. The fact that the E. coli and the N. meningitidis MsrBs exhibit a similar thermal stability suggests the existence of other structural factors in the nonbound metal MsrBs that compensate the metal bound stabilizing effect.
Keywords: methionine sulfoxide reductase B, metal binding site, zinc, iron, apoenzyme, thioredoxin
Methionine residues in proteins are susceptible to oxidation to methionine sulfoxide (MetSO) by reactive oxygen species (ROS). This post-translation modification can lead to loss of protein function (Vogt 1995). Therefore, organisms have developed various defense strategies against the action of ROS. Among these strategies, one possibility includes restoration of the biological function of the modified protein targets. This is the role of the methionine sulfoxide reductases (Msr), which reduces protein-bound MetSO back to methionine. Two classes of monomeric Msrs called MsrA and MsrB have been recently characterized, which are specific for the S and the R epimers, respectively, of the sulfoxide of MetSO (Sharov et al. 1999; Moskovitz et al. 2000; Grimaud et al. 2001; Olry et al. 2002). Although they show unrelated three-dimensional structures (Lowther et al. 2000, 2002; Tete-Favier et al. 2000; Taylor et al. 2003), the MsrAs and MsrBs characterized so far share a similar catalytic mechanism (Fig. 1 ▶). It consists of three steps that include a reductase step resulting in formation of a sulfenic acid intermediate on the catalytic cysteine and release of 1 mol of methionine per mol of Msr followed by a second step in which an intramolecular disulfide bond is formed between the catalytic and the recycling cysteines. Finally, in the third step, called the recycling step, thioredoxin (Trx), which interacts specifically with the oxidized form of the Msr, reduces the Msr disulfide bond leading to regeneration of the reduced form of the Msr (Boschi-Muller et al. 2000, 2005; Olry et al. 2002). The kinetics of the three steps have also been extensively studied. The overall rate-limiting step is associated with the recycling process, but more precisely with dissociation of reduced Msr and oxidized Trx, while the rate-determining step in the process leading to disulfide formation is associated with the sulfenic acid formation (Antoine et al. 2003; Olry et al. 2004; Boschi-Muller et al. 2005).
Figure 1.

Proposed catalytic mechanism of MsrA and MsrB from N. meningitidis. Nucleophilic attack of Cys-X on the sulfur atom of the sulfoxide function of MetSO leads to formation of a tetrahedral intermediate or a transition state (step IA), which rearranges to form a sulfenic acid intermediate with release of 1 mol of Met per mol of enzyme (step IB). Step IA is favored by protonation of the sulfoxide via acid catalysis. In step II, nucleophilic attack of Cys-Y on the sulfur atom of the sulfenic acid leads to formation of a Cys-X/CysY disulfide bond and a release of a water molecule, which is also favored by acid catalysis. In step III, return of the active site to a fully reduced state proceeds via reduction of the Msr disulfide bond by reduced Trx. Cys-X represents Cys-117 and Cys-51 in MsrB and MsrA, respectively, and Cys-Y represents Cys-63 and Cys-198 in MsrB and MsrA, respectively. The numbering of amino acid residues indicated is based on the numbering of the E. coli MsrA and MsrB sequence.
Alignment of the primary structures of MsrBs revealed that in addition to the catalytic C117— which is always present—and the recycling C63 or C31 residues—which is present in at least 80% of the MsrB putative sequences (Neiers et al. 2004; Boschi-Muller et al. 2005)—at least 50% of the available MsrB putative sequences possesses four other cysteines included in two CXXC signatures at positions 45, 48, 94, and 97 (numbering of Escherichia coli MsrB) (Fig. 2 ▶). In contrast, Neisseria gonorrhoeae MsrB domain contains no CXXC signatures as revealed by its crystal structure, and represents the only published report of MsrB 3D structure so far (Lowther et al. 2002). It shows a core domain composed of two anti-parallel β-sheets surrounded by three α-helices and five 310-helices. One of the two active sites in the asymmetric unit is occupied by a cacodylate molecule that likely mimicks the binding of the substrate with one of the two methyl groups representing the ɛ-methyl group of the MetSO and which points to W65. The site resides in a pocket exposed on the surface of the protein. The catalytic C117 is located on a β-strand, while the recycling C63 and the fluorescent probe W65 (Olry et al. 2004) are both situated within a loop. The respective position of the two cysteines is compatible with formation of a disulfide bond between C117 and C63. The three-dimensional structure also reveals a putative metal binding site, at a distance of 20 Å from the active site, the fold of which is structurally related to that of the exchange factor Mss4 in which a Zn2+ atom is strongly bound via two CXXC signatures (Yu and Schreiber 1995a,b).
Figure 2.

Sequence alignment of E. coli, N. meningitidis, N. gonorrhoeae, Rhodobacter capsulatus, X. campestris, and Drosophila melanogaster MsrBs. The sequences of MsrB from E. coli (E. coli), N. meningitidis (N. menin.) (the C-terminal domain of the PilB protein), N. gonorrhoeae (N. gono.) (the C-terminal domain of the PilB protein), R. capsulatus (R. caps.), X. campestris (X. camp.), and Drosophila melanogaster (D. mela.) were aligned with Bioedit software. The cysteines residues are shown in white on black boxes. The consensus sequences of metal-bound and -free MsrBs (determined from the alignment of 41 putative metal-bound and 31 metal-free MsrBs, respectively, extracted from complete sequencing genome from PEDANT database) are indicated below the alignment (only conserved residues at ≥90% are reported). Black points indicate the positions of the two catalytic residues of E. coli and N. meningitidis MsrBs, i.e., C63 (at least 50% invariant in all putative Msr sequences [72]) and C117 (at least 95% invariant), and black stars indicate the positions of the cysteines residues involved in binding of zinc/iron (C45, C48, C94, and C97). The numbering of amino acid residues indicated is based on the numbering of the E. coli MsrB sequence. α-helices and β-strands of the N. gonorrhoeae MsrB (Lowther et al. 2002) are indicated by gray lines and black arrows, respectively.
Recent studies done by Gladyshev et al. (Kumar et al. 2002) have shown that C45, C48, C94. and C97 of the two signatures CXXC are responsible for tight binding of a zinc atom in Drosophila MsrB. The fact that the zinc atom was postulated to stabilize the Mss4 structure supports a similar role in the metallo MsrBs.
In the present study, substitutions of C45, C48, C94, and C97 of the E. coli MsrB (eMsrB) with the corresponding amino acids of the consensus sequence D45, S48, S94, S97, or A97 of the MsrBs devoid of the CXXC signatures have been performed (Fig. 2 ▶). Substitution of cysteines for D45, S48, S94, and A97 of the Neisseria meningitidis MsrB (nmMsrB) has also been carried out in reverse. The results show that (1) the absence of bound metal in the eMsrB provokes drastic structural modifications that explains why the reductase activity is lost; (2) D45C/S48C/S94C/A97C nmMsrB tightly binds the metal that is indicative of a metal binding site preformed in MsrBs which does not possess the two CXXC signatures; and (3) metal binding strongly increases the thermal stability of the D45C/S48C/S94C/A97C nmMsrB, although the eMsrB and nmMsrB, which contain bound metal or not, respectively, display similar thermal stabilization as shown by differential scanning calorimetry (DSC) analysis. These results are discussed in the context of the knowledge of the three-dimensional structure of the Neisseria MsrBs and in terms of evolution.
Results
Properties of the eMsrB
Nature of the metal bound
When overexpressed, the purified wild-type eMsrB appeared pink-colored. The UV/visible spectrum showed an absorbance maximum at 372 and 485 nm in accord with the presence of oxidized iron (curve not shown). Increasing the concentration of FeSO4 up to 1 mM in the culture medium led to an increase in intensity of the pink color. Metal analyses of the purified enzyme by mass spectrometry under nondenaturating conditions and by atomic emission spectrometry showed the presence of zinc/iron in a stoichiometry of 1 per mol of enzyme. The ratio zinc:iron decreased from 8:2 to 6:4, upon increasing concentration of FeSO4 in the culture medium (Table 1). It is a plausible assumption that zinc is the metal relevant from a physiological point of view and the presence of iron is likely due to the shortage of zinc when eMsrB is overexpressed in E. coli. To support this experimentally, it would be necessary to isolate the eMsrB from a nontransformed E. coli strain.
Table 1.
Metal content of wild-type eMsrB
| Electrospray mass spectrometry analyses | Atomic absorption spectrometry analyses | ||||
| FeSO4 (mM) added | Under denaturating conditions (Da) | Under nondenaturating conditions (Da) | Difference (Da)a | mol Zinc/mol enzyme | mol Iron/mol enzyme |
| 0 | 15,319 ± 1b | 15,381 ± 1 (100%) | 62 | 0.80 | 0.08 |
| 0.1 | 15,319 ± 1b | 15,371 ± 0.2 (32%) | 52 | 0.64 | 0.22 |
| 15,381 ± 0.2 (68%) | 62 | ||||
| 1 | 15,319 ± 1b | 15,371 ± 0.2 (40%) | 52 | 0.55 | 0.38 |
| 15,381 ± 0.2 (60%) | 62 | ||||
The values of the metal content represent the average of three independent measurements (SD range 10%). When two masses were observed, an estimation of the quantity of each population is indicated. Molecular masses and differences in mass are expressed in daltons. The values of 62 and 52 Da of mass differences correspond to the presence of one atom of zinc and of iron, respectively.
a Mass differences measured by electrospray mass spectrometry correspond to the differences calculated between the mass obtained in nondenaturating conditions and the mass obtained under denaturating conditions.
b The mass of 15,319 Da correspond to that expected from the protein sequence.
Addition of chelatants like EDTA, pyridine-2,6-dicarboxylic acid, or 1,10 o-phenanthroline did not change the metal content and the Msr activity. Such data suggest a high association constant for the metal as postulated for the MsrB from Drosophila (Kumar et al. 2002). Precipitation with TCA followed by urea treatment and refolding in Tris buffer, permitted isolation of eMsrB form with no significant amount of bound metal (Table 2).
Table 2.
Metal content and catalytic properties of different forms of E. coli and N. meningitidis MsrBs
| Enzyme | mol Zinc/mol enzyme | mol Iron/mol enzyme | kcat with Trx (sec−1) | kobs with DTT (sec−1) |
| Wild-type eMsrBa | 0.80 | 0.08 | 0.18 | 0.010 |
| Wild-type eMsrB apoform | 0.02 | 0.02 | 2.10−2 | 1.7.10−3 |
| C45D/C48S/C94S/C97S eMsrBb | 0.02 | 0.02 | <3.10−3 | <1.10−3 |
| Wild-type nmMsrB | 0.01 | 0.01 | 0.20c | 0.050 |
| D45C/S48C/S94C/A97C nmMsrB | 0.60 | 0.17 | <3.10−3 | 0.045 |
For determination of the metal content of wild-type and mutant MsrBs, the values represent the average of three independent measurements (SD range 10%). The activities of wild-type and mutant enzymes were assayed with 150 mM D,L-Met-R,S-SO, 10 mM DTT, and 50–100 μM enzyme (SD range 10%) or with 20–150 mM D,L-Met-R,S-SO, 100–800 μM Trx, 1.28 μM Trx reductase, and 0.5–50 μM enzyme (SD range 10%) as described in Materials and Methods. For wild-type eMsrB, KM values for L-Met-R,S-SO and Trx were 2.2 ± 0.6 mM and 140 ± 22 μM, respectively.
a The kinetic parameters of the MsrBs with a bound metal were determined with a form having a zinc:iron ratio of 8:2.
b Similar results were obtained with the C45D/C48S/C94S/C97A eMsrB.
c From Olry et al. (2002).
When the four cysteines of the two CXXC motifs were substituted with the corresponding amino acids of the consensus sequence D45, S48, S94, S/A97 present in MsrBs lacking CXXC signatures, the metal content was not significant (Table 2).
Catalytic properties of the wild type and of the C45D/C48S/C94S/C97S eMsrB
In the presence of either saturating concentration of Trx or 10 mM DTT, a kcat value of 0.18 sec−1 and a kobs value of 1.10−2 sec−1, respectively, were determined for the wild-type eMsrB, similar to those of the nmMsrB (Table 2; Olry et al. 2002). MsrBs with different ratios of zinc and iron displayed similar kinetic parameters, suggesting that the nature of the metal bound has no effect on the conformation of the protein particularly in relation to the active site (data not shown). When C45, C48, C94, and C97 were altogether substituted, no activity was detected for the quadruple-mutated eMsrB in the presence of either Trx or DTT (Table 2).
The reductase step of the wild type was studied under single turnover quenched-flow conditions, at pH 8, in the absence of reductant, at a saturating concentration of Ac-L-Met-R,S-SO-NHMe as already described by Olry et al. (2004). The results showed formation of 1 mol Ac-Met-NHMe per mol of wild-type eMsrB within the aging time of the stopped-flow apparatus adapted for quenched-flow use, which is ~30 msec. This indicated a rate of the reductase step of at least 33 sec−1, in the range of that observed for wild-type nmMsrB (Olry et al. 2004). For the C45D/C48S/C94S/C97S eMsrB, no Ac-Met-NHMe was formed after an overnight incubation with saturating concentration of Ac-L-Met-R,S-SO-NHMe, which confirms the absence of activity with DTT.
Biochemical properties of the MsrBs with no bound metal, and comparison with those of the wild-type eMsrB
As shown in Figure 3A ▶, the CD spectrum of the wild-type eMsrB and of the apo wild-type and C45D/C48S/C94S/C97S eMsrBs were quite different. In particular, the negative band at 208 nm observed for the wild-type eMsrB, which is likely representative of the content of α-helices, shifted <204 nm. These results suggested that eMsrB in the nonmetal-bound state has either a significant proportion of random coil structures or regions of high flexibility, which probably reflect a less ordered conformation of the eMsrBs. The fact that the same perturbed spectrum was observed with the apo wild-type and the C45D/C48S/C94S/C97S eMsrBs showed that the structural perturbation observed is likely caused by the absence of the metal and not by the amino acids, which have been introduced in place of the cysteines. This is confirmed by the fact that adding zinc to the apo form led to an active enzyme with kinetic properties and CD spectrum similar to those of the wild type (data and spectrum not shown).
Figure 3.
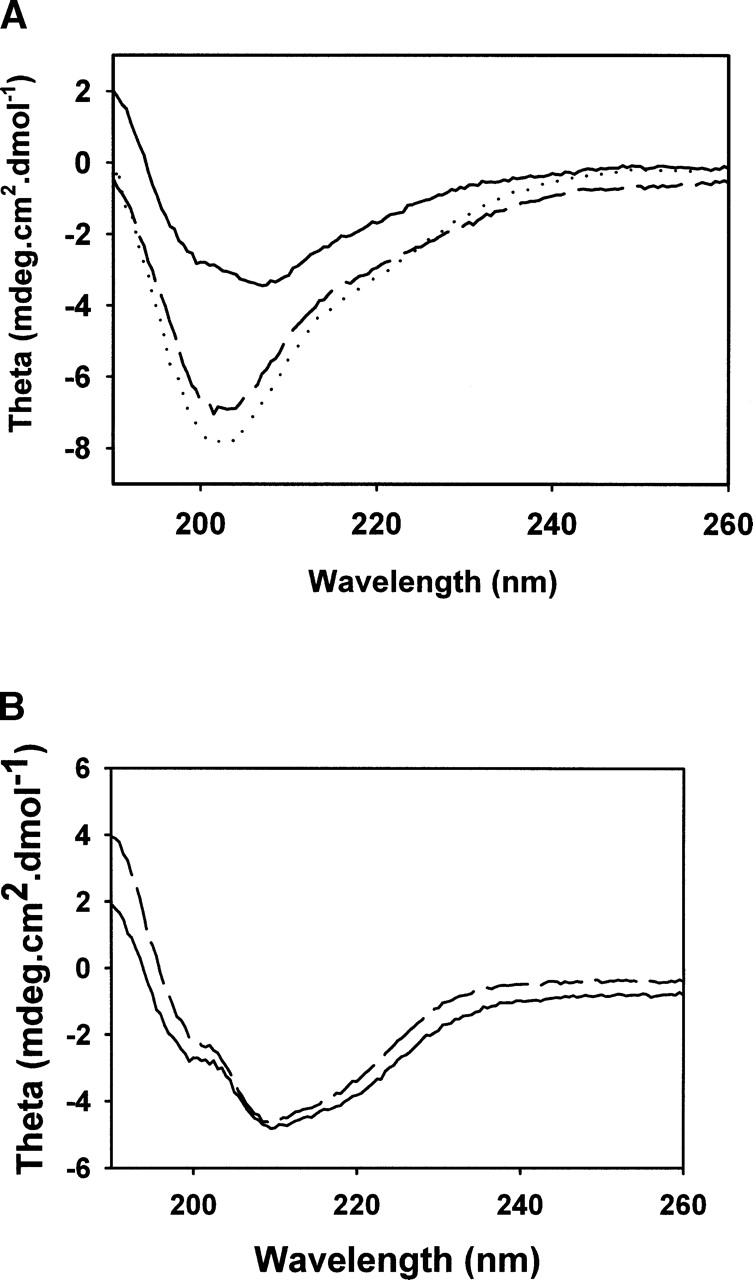
CD spectra of different forms of eMsrBs (A) and nmMsrBs (B). (A) Wild-type (solid line), C45D/C48S/C94S/C97S (dashed line) and wild-type apoform (dotted line) eMsrBs. (B) Wild-type (solid line) and D45C/S48C/S94C/A97C (dashed line) nmMsrBs. CD spectra were scanned from 260 to 190 nm in potassium phosphate buffer 10 mM (pH 7.1) at 25°C, and at an enzyme concentration of 5 μM.
To get more structural information on the effect of the absence of the metal, the fluorescence emission spectrum of W65, a residue which is located within the active site and is known to be a good probe of the conformation of the active site, was done (Olry et al. 2004). For the wild-type eMsrB, the λmax of the W65 fluorescence emission was centered at 334 nm under native conditions. The λmax was shifted to 353 nm in the case of the apo wild-type and C45D/C48S/C94S/C97S eMsrBs (Fig. 4A ▶), similarly to what is observed with the wild type under denaturating conditions (in the presence of urea 4 M; curve not shown).
Figure 4.
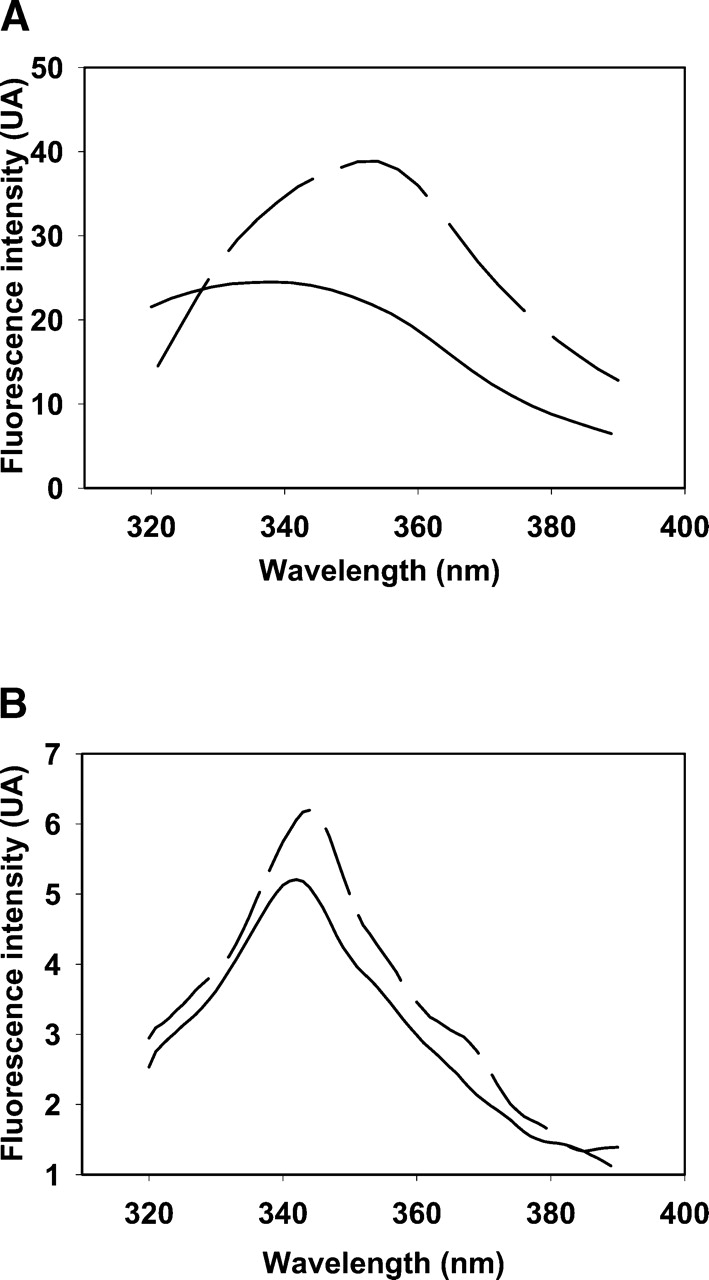
Emission fluorescence spectra of different forms of eMsrBs (A) and nmMsrBs (B). (A) Wild-type (solid line) and C45D/C48S/C94S/C97S (dashed line) eMsrBs. Fluorescence spectra of 10 μM eMsrB were recorded at 25°C in buffer A on excitation at maximum wavelength (291 nm). The emission fluorescence spectrum of the apo wild-type eMsrB was superimposable with that of the C45D/C48S/C94S/C97S eMsrB and was not represented for reason of clarity. (B) Wild-type (solid line) and D45C/S48C/S94C/A97C (dashed line) nmMsrBs. Fluorescence spectra of 10 μM nmMsrB were recorded at 25°C in buffer A on excitation at 310 nm.
DSC experiments showed an endotherm with a Tm of 40°C for the wild-type enzyme (Fig. 5 ▶). In contrast, no thermogram was observed for the apo wild type and the C45D/C48S/C94S/C97S eMsrB that reflects the absence of a significant ΔH contribution in the thermal unfolding.
Figure 5.

Heat denaturation curves of wild-type eMsrB (short-dashed line) and wild-type (solid line), D45C/S48C/S94C/A97C (long-dashed line), and D45C/S48C/S94C/A97C apoform (dotted line) nmMsrBs. Differential scanning spectra were obtained between 25°C and 105°C at a rate of 1° K/min in potassium phosphate buffer 10 mM (pH 7.1) at an enzyme concentration of 1 mg.mL−1. The corresponding melting temperatures are 40.2°C, 42.3°C, 91.4°C, and 38.7°C for wild-type eMsrB and wild-type, D45C/S48C/S94C/S97C and D45C/S48C/S94C/S97C apoform nmMsrBs, respectively.
No fluorescence intensity increase and no difference in the λmax of the fluorescence emission was observed in the presence of hydrophobic fluorescent probe such as 8-anilino-1-naphtalenesulfonic acid (ANS) for wild-type, apo wild-type, and C45D/C48S/C94S/C97S eMsrBs under native conditions. On the contrary, an increase and a shift of λmax from 480 to 520 nm were observed for all the three forms under conditions compatible with the presence of a molten globule intermediate (in the presence of 0.1% SDS) (curves not shown). This suggested that apo and C45D/C48S/C94S/C97S eMsrBs have acquired a fold, although different from that of the wild type, with their hydrophobic residues buried within the protein interior.
Biochemical and catalytic properties of the D45C/S48C/S94C/A97C nmMsrB and comparison with those of the wild-type nmMsrB
nmMsrB belongs to a larger three-domain protein called PilB. PilB is composed of an N-terminal domain, which displays a disulfide reductase activity (Wu et al. 2005), a central domain, which bears an MsrA activity, while the C-terminal domain shows an MsrB activity (Olry et al. 2002). In the present study, the nmMsrB corresponds to the C-terminal domain of PilB, as described by Orly et al. (2002).
When overexpressed D45C/S48C/S94C/A97C nmMsrB was isolated, the concentrated protein solution was also pink colored. Metal analysis by atomic emission spectrometry showed a binding of zinc:iron with a ratio 8:2 and a global stoichiometryof 1atom of metal permol of protein in contrast to the wild type, in which no zinc/iron is bound (Table 2). Moreover, chelating reagents had no effect on the metal content and on the activity, suggesting again a tight binding of the metal (data not shown).
No Trx recycling activity was observed, whereas activity measured with DTT was in the range of that of the wild-type nmMsrB (Table 2). This showed that although the Trx recycling process is not operative, the reductase step was likely as efficient as that of the wild type. To validate this assumption, the rate of the reductase step of the D45C/S48C/S94C/A97C nmMsrB was determined by following the rate associated with a change of the W65 fluorescence intensity upon going from the reduced to the sulfenic acid form as already described by Olry et al. (2004). From the curve of kobs versus substrate concentration, a KS value of 170 ± 30 mM for Ac-L-Met-R,S-SO-NH-Me and a kobs max value of 65 ± 4 sec−1 were determined at pH 8, similar to those described for the wild-type nmMsrB (Fig. 6 ▶; Olry et al. 2004). The fact that an efficient Trx recycling activity was observed for the apo D45C/S48C/S94C/A97C nmMsrB with a kcat value of 0.06 sec−1 similar to that of the wild-type nmMsrB showed that the loss of the Trx recycling activity is likely due to the presence of the bound metal.
Figure 6.

Determination of the catalytic parameters of the reductase step of D45C/S48C/S94C/S97C nmMsrB measured by fluorescence stopped-flow under single turnover kinetics. The nmMsrB fluorescence decrease was recorded on a stopped-flow apparatus at 25°C in buffer A. Final concentration of nmMsrB was 10 μM. Excitation wavelength was set at 291 nm, and emitted light was collected above 320 nm using a cutoff filter. Symbols (•) represent experimental points. Data were fit to Equation 1 by least-squares regression, which gave kobs max and KS values of 65 ± 4 sec−1 and 170 ± 30 mM, respectively.
To obtain more structural information on the effect of the presence of the metal in the nmMsrB, the CD (Fig. 3B ▶) and the emission fluorescence (Fig. 4B ▶) spectra of the wild-type and D45C/S48C/S94C/A97C nmMsrBs were recorded. No significant difference was observed, which indicates the absence of major structural modifications caused by the metal binding.
DSC experiments carried out on the nmMsrB showed that the Tm value is shifted from 39°–42°C for the apo D45C/S48C/S94C/A97C and the wild-type nmMsrBs to 91°C for the D45C/S48C/S94C/A97C nmMsrB in which a metal is bound (Fig. 5 ▶).
Discussion
As indicated in the introduction section, inspection of the alignment of the primary structure shows that >50% of the MsrBs contains two CXXC signatures at positions 45, 48, 94, and 97, e.g., the eMsrB, the selenoprotein R, and the Drosophila MsrB. Recently, Gladyshev et al. have shown that changing C45, C48, C94, and C97 in the Drosophila MsrB resulted not only in loss of zinc binding but also of DTT and Trx recycling activities (Kumar et al. 2002). The absence of activity with DTT, a reagent which can reduce the sulfenic acid intermediate formed in the first step of the catalytic mechanism (Olry et al. 2002), suggested at least a conformational change of the active site that prevented formation of a productive binary complex MsrB-MetSO. That is the case for the eMsrB. Substituting the cysteines of the two CXXC signatures with the corresponding amino acids in MsrBs in which no CXXC signature is present led as expected to loss of metal but, more importantly, to an active site more exposed to the solvent and to a less-structured folding of the protein. These two latter conclusions are based on (1) the red shift, from 343 to 353 nm, of the fluorescent emission peak of W65, which indicates a position of W65 in the active site totally exposed to the solvent; (2) the UV shift accompanied by an increase of the CD message in the 195–210 nm region, which supports a less ordered conformation of the eMsrBs devoid of metal; (3) the absence of a significant ΔH contribution in the thermal unfolding as revealed by DSC analysis in contrast to the wild-type eMsrB, which presents an endotherm with a Tm of 40.2°C.
Therefore, the role of the metal in eMsrB is likely to stabilize the core structure, which renders the conformation of the active site competent for the reductase activity. This is in apparent contradiction with the fact that the nmMsrB, lacking the two CXXC signatures and therefore has no bound metal, is as active as the eMsrB and thus has a tertiary structure competent for revealing activity. Indeed, the kobs max value for the reductase step, the kcat value obtained with the Trx/Trx reductase recycling system, and the KM values for the MetSO substrate and Trx are similar to those of the eMsrB values. Inspection of the three-dimensional structure of nmMsrB (B. Kauffmann, unpubl.), shows a putative metal binding site at a distance of 20 Å from the active site similar to that observed in the structure of the N. gonorrhoeae MsrB (Lowther et al. 2002). A water molecule, hydrogen bonded to the invariant residues D45, S48, and S94, replaces the bound metal. Substituting cysteines for D45, S48, S94, and A97 resulted in a nmMsrB that tightly binds one metal atom per mol of enzyme. Such a result confirms the existence of a preformed metal binding site in the subclass of MsrBs in which C45, C48, C94, and C97 are not present. Moreover, the rate and the affinity constant of the quadruple mutated nmMsrB for the MetSO in the reductase step remained unchanged but the recycling activity with Trx was lost. These results suggest that metal binding induces subtle structural modifications transmitted to the active site that prevents formation of a competent binary complex between oxidized MsrB and reduced Trx but not between the reduced MsrB and MetSO substrate. These hypotheses are supported by the fact that no significant change was observed in the CD spectrum and in the λmax of the W65 fluorescence emission peak of the reduced quadruple mutated nmMsrB. However, what is particularly striking is the strong thermal stabilization brought by the metal binding as shown by an increase in Tm from 42° to 91°C of the endotherm in DSC. The relationship between the drastic increase of the thermal stability and the fact that a competent complex between oxidized MsrB and reduced Trx is not formed remains to be understood at the structural level.
From an evolutionary point of view, it is probable that, as suggested by Gladyshev et al. (Kumar et al. 2002), the zinc-containing MsrB is the prototypical enzyme that has lost the two CXXC signatures later in evolution. This assumption is supported by the present study which shows that MsrBs, that either possess or lack the two signatures CXXC, exhibit a similar DSC thermal stability. When the two CXXC signatures are present, the metal binding should, however, participate in the thermal stabilization of the MsrB. Thus, the loss of the two CXXC signatures should provoke a destabilization of the MsrB due to the absence of bound metal. Therefore, the destabilizing effect has to be compensated probably by other interactions acquired along the evolutionary cycle that stabilize the core structure of MsrB with no bound metal. Inspection of the X-ray structures of the MsrBs from N. meningitidis and gonorrhoeae does not show the presence of evident stabilizing interactions within the core structure that would be absent in MsrBs with bound metal except the presence of an ion pair between D57 and R70. Positions 57 and 70 are usually occupied by Asp/Glu and Arg/Lys residues, respectively, in MsrB with no bound metal (Fig. 2 ▶). Substituting D57 of the nmMsrB by Ala has been recently carried out, while Asp and Lys at positions 57 and 70, respectively, were introduced in the C45D/C48S/C94S/C97S eMsrB. In fact, the biochemical properties of the two mutant MsrBs were shown to be unchanged (A. Olry, unpubl.). Clearly, the stabilizing elements involved in MsrBs with no bound metal remain to be characterized at the structural level. In that context, knowledge of the three-dimensional structure of MsrBs with bound metal and comparison to that of MsrBs with no bound metal could be very informative.
Materials and methods
Plasmid constructions, site-directed mutageneses, production, and purification of wild-type and mutants eMsrBs and nmMsrBs
Plasmid pETYeaA, designed for eMsrB production, was obtained by cloning the yeaA open reading frame, synthesized by PCR using E. coli K12 genomic DNA, into the plasmid pET30a(+) between the NdeI and HindIII sites. Plasmid pSKPILBMsrB, containing the N. meningitidis MsrB coding sequence under the lac promoter, was used for nmMsrB production as previously described by Olry et al. (2002). Site-directed mutageneses were performed using the QuikChange Site-Directed Mutagenesis Kit (Stratagene).
The E. coli strain used for all eMsrB productions was BL21(DE3) transformed with a pETYeaA plasmid containing the wild-type or mutated eMsrB coding sequence under the control of the T7 promoter. The overexpression of eMsrBs was performed by the addition of 1 mM IPTG in the culture medium at 0.6 A600. After 5 h of induction, cells were harvested by centrifugation. The E. coli strain used for all nmMsrB productions was HB101 transformed with a pSKPILBMsrB plasmid, as previously described by Olry et al. (2002).
All wild-type and mutant MsrBs were produced in a soluble form except for C45D/C48S/C94S/C97S eMsrB. For nmMsrBs, the protocol of purification was carried out as previously described (Olry et al. 2002), except that buffer used was devoid of EDTA. For wild-type eMsrB purification, cells were harvested by centrifugation, resuspended in buffer A (Tris-HCl 50 mM at pH 8) containing 20 mM dithiothreitol (DTT), and sonicated. The MsrB was then precipitated at 50% (NH4)2SO4 saturation. The contaminating proteins were removed by applying the enzymatic solutions onto exclusion size chromatography on ACA 54 resin (IBF) at pH 8 (buffer A). Purified fractions were then pooled and applied onto a Q-Sepharose column equilibrated with buffer A, followed by a linear gradient of KCl (0–0.4 M) using a fast protein liquid chromatography system (Amersham Biosciences). The eMsrB was eluted at 250 mM KCl. Finally, 100 mg/L of culture of the homogeneous eMsrB protein was obtained.
For C45D/C48S/C94S/C97S eMsrB, the protocol was different due to its production under a nonsoluble form. Pellets obtained after sonication were resuspended in buffer A containing 6 M urea. The contaminating proteins and nucleic acids were removed by applying the enzymatic solution diluted to 2 M urea final concentration onto a Q-Sepharose column equilibrated with buffer A containing 2 M urea and 1 mM DTT using a fast protein liquid chromatography system (Amersham Biosciences). Elution was performed in two steps: first by a linear inverse gradient of urea (2–0 M), and then by a linear gradient of KCl (0–0.5 M). The C45D/C48S/C94S/C97S eMsrB was eluted at 250 mM KCl. Finally, 80 mg/L of culture of the homogeneous eMsrB protein was obtained.
Purity of wild-type and mutated MsrBs was checked by electrophoresis on 15% SDS-polyacrylamide gel and by electrospray ionization (ESI) mass spectrometry analyses. Purified enzymes were stored at −20°C in the presence of 50 mM DTT and 50% (NH4)2SO4. Under these conditions, the enzymes were stable for several weeks. Their molar concentrations were determined spectrophotometrically, using theoretical extinction coefficients at 280 nm deduced from the method of Scopes (1974), i.e., 13,730 M−1 • cm−1 for wild-type and C45D/C48S/C94S/C97S eMsrBs, and 17,330 M−1 • cm−1 for wild-type and D45C/S48C/S94C/S97C nmMsrBs.
Determination of metal content
The zinc and iron contents of wild-type and mutant MsrBs from E. coli and N. meningitidis were analyzed using inductively coupled plasma-atomic emission spectrometry (ICP-AES). In parallel, analyses of control buffer samples showed that zinc and iron concentrations were <50 and 10 μg • L−1, respectively.
The metal content of wild-type eMsrB was also determined by ESI mass spectrometry analyses. Before molecular mass analyses, all samples were desalted by applying the enzymatic solution onto an exclusion size column (HIPREP 26/10 Desalting, Amersham Biosciences) equilibrated in 50 mM ammonium bicarbonate (pH 7.6) buffer. ESI mass spectrometry measurements were performed on an ESI time-of-flight mass spectrometer (LCT, Micromass) fitted with a standard Z-spray source. Measurements were performed in nondenaturing conditions by diluting enzymes to 10 pmol/μL in ammonium acetate 50 mM (pH 6.8) buffer. For mass analysis in denaturing conditions, MsrB samples were diluted to 10 μM in a 1:1 water–acetonitrile mixture (v/v) containing 1% formic acid. Samples were continuously infused into the ion source at a flow rate of 5 μL/min. The accelerating voltage applied on the sample was set to 80 V. Mass spectra were recorded in the positive ion mode on the mass range 1000–4000 m/z, after calibration with a separate injection of horse heart myoglobin diluted to 2 pmol/μL in a 1:1 water–acetonitrile mixture (v/v) acidified with 1% formic acid.
Preparation of metal-free MsrBs
To prepare metal-free wild-type eMsrB and D45C/S48C/S94C/A97C nmMsrB, enzymes were precipitated at room temperature with trichloroacetic acid (TCA) (10% [w/v]). The precipitate was centrifuged and the pellet was dissolved in Tris-HCl buffer 0.5 M (pH 8) containing 0.1 M DTT. The solution was then incubated for 3 h and the MsrB was then again precipitated with TCA (10% [w/v]). The pellet was dissolved in Tris-HCl buffer 0.5 M (pH 8) containing 8 M urea, and diluted with Tris-HCl buffer 0.5 M (pH 8) to a final urea concentration of 1 M. Urea and DTT were finally eliminated with a small Econopac EP 10DG column (Biorad) equilibrated with Tris-HCl buffer 50 mM (pH 8). The absence of bound metal was checked by plasma atomic emission spectrometry.
Determination of MsrBs activity
Kinetic studies were carried out with D,L-Met-R,S-SO as a substrate and E. coli Trx1 as a reductant in the presence of a Trx-regenerating system (1.28 μM E. coli Trx reductase and 0.3 mM NADPH) as previously described by Olry et al. (2002). The initial rate data were fit to the Michaelis-Menten relationship using least-squares analysis to determine kcat and KM with the program Sigmaplot (Jandel Scientific Software). All KM values were determined at saturating concentrations of the other substrate.
With the DTT regenerating system, the reaction mixture contained 10 mM DTT and 20–100 μM wild-type or mutated MsrBs in buffer A. Inital rate measurements were carried out at 25°C by following the appearance of free Met measured by high pressure liquid chromatography as previously described by Boschi-Muller et al. (2000).
Kinetics of the reductase activity by single turnover experiments
Kinetics of the emission fluorescence intensity decrease associated with formation of the sulfenic acid intermediate in the D45C/S48C/S94C/A97C nmMsrB were measured with Ac-L-Met-R,S-SO-NHMe as a substrate at 25°C on a SX18MV-R stopped-flow apparatus (Applied PhotoPhysics) adapted for fluorescence measurements as previously described (Olry et al. 2004). Data were fit to Equation 1 using least-squares analysis to determine kobs max and KS for Ac-L-Met-R,S-SO-NHMe. S represents the Ac-L-Met-R,S-SO-NHMe concentration, and K represents the KS value for the substrate.
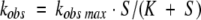 |
(1) |
For eMsrB, the formation of Ac-L-Met-NHMe was followed by single turnover quenched-flow experiments at 25°C on a SX18MV-R stopped-flow apparatus (Applied PhotoPhysics) fitted for the double-mixing mode and adapted to recover the quenched samples as previously described (Olry et al. 2004). Briefly, the mixture containing 300 μM MsrB and 350 mM Ac-L-Met-R,S-SO-NHMe in buffer A was allowed to react for 30 msec before being mixed with an equal volume of a quenching aqueous solution containing 2% of trifluoroacetic acid. Ac-L-Met-NHMe quantification in the quenched samples was carried out by reverse-phase chromatography as previously described (Olry et al. 2004).
Fluorescence properties of wild-type and mutated MsrBs
The fluorescence emission spectra of the wild-type and mutated MsrBs (10 μM) in buffer A were recorded on a spectrofluorometer (flx SAFAS) thermostated at 25°C at 291-nm and 310-nm excitation wavelength for eMsrBs and nmMsrBs, respectively. For ANS binding experiments, fluorescence emission was measured by excitation at 380-nm and emission was recorded from 400 to 600 nm in buffer A in the presence or the absence of 0.1% SDS. ANS concentration was 1 mM and protein concentration was 10 μM.
Differential scanning calorimetry
Heat denaturation measurements were carried out on a Micro-Cal VP-DSC instrument in 0.51-mL cells at a scan rate of 60°K • h−1. Samples were extensively dialyzed against potassium phosphate buffer 10 mM (pH 7.1) at 4°C, degassed for 7 min twice before the experiment. Protein concentration was 1 mg • mL−1. The final dialysis buffer was used for the reference DSC cell and to record the buffer baseline. Thermograms were analyzed using Microcal ORIGIN software.
Circular dichroism
Circular dichroism (CD) spectra of the enzymes (5 μM in phosphate buffer 10 mM at pH 7.1) were obtained using a Jobin-Yvon CD6 spectrometer. The spectra were scanned at 25°C with 1-nm steps from 260 to 190 nm and averaged over six scans.
Acknowledgments
This research was supported by the CNRS (Programme “Protéomique et Génie des Protéines CNRS 2001”), the Ministère délégué à la Recherche (ACI BCMS047), the University Henry Poincaré Nancy I, the Association pour la Recherche sur le Cancer (Grant ARC no. 4393), and the Institut Fédératif de Recherches 111 Bioingénierie. H.Y. and A.O. were supported by a fellowship from the French Ministère de la Recherche and the Association pour la Recherche sur le Cancer, respectively. We are very grateful to B. Thouvenot for the gift of the pETYeaA plasmid, to Drs. A. Van Dorsselaer and S. Sanglier-Cianferani for the mass spectrometry analysis, and to Dr. S. Sonkaria for careful reading of the manuscript. We also thank C. Gauthier for his efficient technical help.
Abbreviations
MetSO, methionine sulfoxide
ROS, reactive oxygen species
Msr, MsrA, and MsrB, methionine sulfoxide reductase, methionine sulfoxide reductase A, methionine sulfoxide reductase B, respectively
eMsrB and nmMsrB, Escherichia coli and Neisseria meningitidis MsrB, respectively
IPTG, isopropyl-β-D-thiogalactopyranoside
DTT, dithiothreitol
Trx, thioredoxin
TCA, trichloroacetic acid
CD, circular dichroism
ESI, electrospray ionization
DSC, differential scanning calorimetry
ANS, 8-anilino-1-naphtalenesulfonic acid.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051711105.
References
- Antoine, M., Boschi-Muller, S., and Branlant, G. 2003. Kinetic characterization of the chemical steps involved in the catalytic mechanism of methionine sulfoxide reductase A from Neisseria meningitidis. J. Biol. Chem. 278 45352–45357. [DOI] [PubMed] [Google Scholar]
- Boschi-Muller, S., Azza, S., Sanglier-Cianferani, S., Talfournier, F., Van Dorsselear, A., and Branlant, G. 2000. A sulfenic acid enzyme intermediate is involved in the catalytic mechanism of peptide methionine sulfoxide reductase from Escherichia coli. J. Biol. Chem. 275 35908–35913. [DOI] [PubMed] [Google Scholar]
- Boschi-Muller, S., Olry, A., Antoine, M., and Branlant, G. 2005. The enzymology and biochemistry of methionine sulfoxide reductases. Biochim. Biophys. Acta 1703 231–238. [DOI] [PubMed] [Google Scholar]
- Grimaud, R., Ezraty, B., Mitchell, J.K., Lafitte, D., Briand, C., Derrick, P.J., and Barras, F. 2001. Repair of oxidized proteins. Identification of a new methionine sulfoxide reductase. J. Biol. Chem. 276 48915–48920. [DOI] [PubMed] [Google Scholar]
- Kumar, R.A., Koc, A., Cerny, R.L., and Gladyshev, V.N. 2002. Reaction mechanism, evolutionary analysis, and role of zinc in Drosophila methionine-R-sulfoxide reductase. J. Biol. Chem. 277 37527–37535. [DOI] [PubMed] [Google Scholar]
- Lowther, W.T., Brot, N., Weissbach, H., Honek, J.F., and Matthews, B.W. 2000. Thioldisulfide exchange is involved in the catalytic mechanism of peptide methionine sulfoxide reductase. Proc. Natl. Acad. Sci. 97 6463–6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowther, W.T., Weissbach, H., Etienne, F., Brot, N., and Matthews, B.W. 2002. The mirrored methionine sulfoxide reductases of Neisseria gonorrhoeae pilB. Nat. Struct. Biol. 9 348–352. [DOI] [PubMed] [Google Scholar]
- Moskovitz, J., Poston, J.M., Berlett, B.S., Nosworthy, N.J., Szczepanowski, R., and Stadtman, E.R. 2000. Identification and characterization of a putative active site for peptide methionine sulfoxide reductase (MsrA) and its substrate stereospecificity. J. Biol. Chem. 275 14167–14172. [DOI] [PubMed] [Google Scholar]
- Neiers, F., Kriznik, A., Boschi-Muller, S., and Branlant, G. 2004. Evidence for a new sub-class of methionine sulfoxide reductases B with an alternative thioredoxin recognition signature. J. Biol. Chem. 279 42462–42468. [DOI] [PubMed] [Google Scholar]
- Olry, A., Boschi-Muller, S., Marraud, M., Sanglier-Cianferani, S., Van Dorsselear, A., and Branlant, G. 2002. Characterization of the methionine sulfoxide reductase activities of PILB, a probable virulence factor from Neisseria meningitidis. J. Biol. Chem. 277 12016–12022. [DOI] [PubMed] [Google Scholar]
- Olry, A., Boschi-Muller, S., and Branlant, G. 2004. Kinetic characterization of the catalytic mechanism of methionine sulfoxide reductase B from Neisseria meningitidis. Biochemistry 43 11616–11622. [DOI] [PubMed] [Google Scholar]
- Scopes, R.K. 1974. Measurement of protein by spectrophotometry at 205 nm. Anal. Biochem. 59 277–282. [DOI] [PubMed] [Google Scholar]
- Taylor, A.B., Benglis Jr., D.M., Dhandayuthapani, S., and Hart, P.J. 2003. Structure of Mycobacterium tuberculosis methionine sulfoxide reductase A in complex with protein-bound methionine. J. Bacteriol. 185 4119–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tete-Favier, F., Cobessi, D., Boschi-Muller, S., Azza, S., Branlant, G., and Aubry, A. 2000. Crystal structure of the Escherichia coli peptide methionine sulphoxide reductase at 1.9 Å resolution. Struct. Fold. Des. 8 1167–1178. [DOI] [PubMed] [Google Scholar]
- Vogt, W. 1995. Oxidation of methionyl residues in proteins: Tools, targets, and reversal. Free Radic. Biol. Med. 18 93–105. [DOI] [PubMed] [Google Scholar]
- Wu, J., Neiers, F., Boschi-Muller, S., and Branlant, G. 2005. The N-terminal domain of PilB from Neisseria meningitidis is a disulfide reductase that can recycle methionine sulfoxide. J. Biol. Chem. 280 12344–12350. [DOI] [PubMed] [Google Scholar]
- Yu, H. and Schreiber, S.L. 1995a. Cloning, Zn2+ binding, and structural characterization of the guanine nucleotide exchange factor human Mss4. Biochemistry 34 9103–9110. [DOI] [PubMed] [Google Scholar]
- ———. 1995b. Structure of guanine-nucleotide-exchange factor human Mss4 and identification of its Rab-interacting surface. Nature 376 788–791. [DOI] [PubMed] [Google Scholar]