

Abstract
We have determined the critical concentrations of a set of 18 variants of Alzheimer’s Aβ(1–40) peptide, each carrying a different residue at position 18. We find that the critical concentrations depend on the hydrophobicity and β-sheet propensity of residue 18, and therefore on properties that we identified previously to affect also the kinetics by which these peptides aggregate. Since the critical concentrations can be related to the Gibbs free energy of aggregation (ΔG), these data imply a link between the thermodynamics and the kinetics of aggregation in that sequences that form very stable aggregates are also those that form such aggregates very rapidly.
Keywords: amyloidosis, conformational disease, neurodegeneration, protein folding, prion
Amyloid fibrils represent a specific and β-sheet-rich structural form of the polypeptide chain that differs from the β-sheet structure present in native, globular proteins (Dobson 2001; Zandomeneghi et al. 2004). The effect of mutation on the kinetics of aggregation and amyloid formation has been subject to many investigations (Chiti et al. 2003; DuBay et al. 2004; Tartaglia et al. 2004; Christopeit et al. 2005), while comparatively little is known about sequence effects on the thermodynamics of aggregation. For example, the kinetics of aggregation is known to be affected by intrinsic properties of the unfolded polypeptide chain, such as β-sheet propensity and hydrophobicity, the presence and the stability of competing globular protein structures, and also by environmental parameters. Here, we have determined the critical concentrations (cc) of a set of 18 variants of the Alzheimer’s Aβ(1–40) peptide where residue 18 has been replaced by all other standard and non-sulphur-containing amino acids. cc represents the maximum concentration of full solubility above which the soluble peptide fraction does not increase (Fig. 1A ▶). The value of cc can be determined conveniently by centrifugation and relates to the Gibbs free energy of aggregation ΔG (Oosama and Asakura 1975). This method has been used previously to explore the thermodynamics of the formation of native protein fibrils, such as flagella (Oosama and Asakura 1975), and amyloid fibrils (Harper and Lansbury 1997; Williams et al. 2004).
Figure 1.
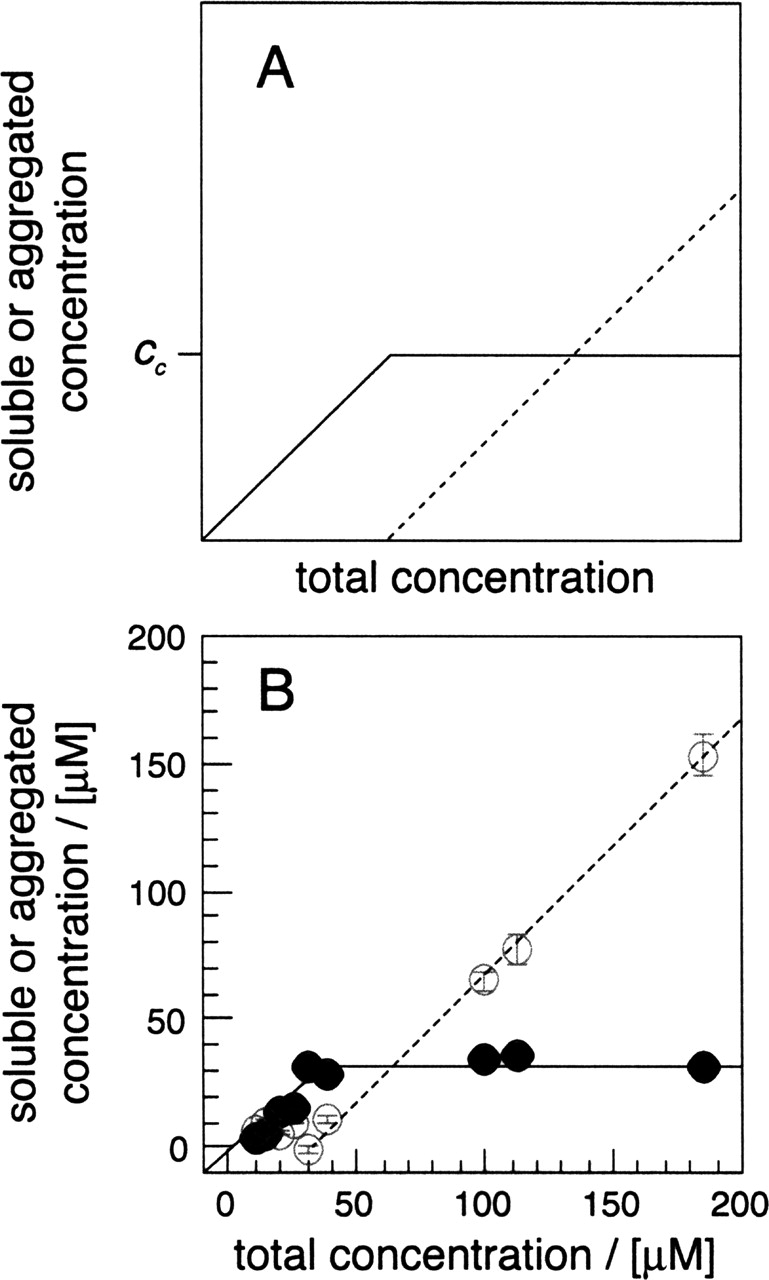
Measurement of cc. (A) Schematic representation of the critical concentration (cc). (Continuous line) Soluble peptide concentration; (dotted line) aggregated peptide concentration. (B) Soluble peptide (filled symbols) and insoluble peptide (open symbols) after 28 d of incubation. The soluble fraction was obtained from two samples per concentration that were incubated separately. Data points represent the averages, while error bars show the standard deviations (mostly smaller than the symbol size).
To demonstrate that Aβ(1–40) complies with critical concentration theory, solutions of wild-type Aβ peptide were incubated at different concentration. After 28 d, aggregation was quantified by ultracentrifugation. The resulting data set correlates well with expectation and the peptide is fully soluble at concentrations up to 32 ± 2 μM (Fig. 1B ▶). Above this concentration, the amount of soluble peptide does not increase further. Although a cc value of 32 ± 2 μM is well within the broad range reported for the reduced peptide (0.9–40 μM) (Harper and Lansbury 1997; Williams et al. 2004), numeric comparisons are hampered by different experimental setups, solution conditions, and redox states. Furthermore, a plot similar to the one shown in Figure 1B ▶ is obtained when incubation was terminated after 14 d.
Next, we have measured and compared the cc values of the 18 mutants. The resulting data show considerable differences among them (Table 1): While Val18Tyr is least soluble, Val18Lys, Val18Asp, Val18Ser, Val18Pro, and Val18Glu are associated with the highest cc values (Table 1). The high solubility of Val18Pro is consistent with previous data (Morimoto et al. 2004; Williams et al. 2004; Christopeit et al. 2005), and can be attributed to an interference of proline with the hydrogen-bonded network of β-sheets (Moriarty and Raleigh 1999). Charges, on the other hand, can impair aggregation by electrostatic repulsion (Fändrich and Dobson 2002). We have compared the cc values after 28 d with those measured after 14 d of incubation. Both data sets are very similar and a plot of the two sets of cc values fits with a straight line that has a slope of 1.03 and a correlation coefficient of 0.98 (data not presented). We conclude that incubation for 28 d is sufficient to ensure that aggregation is close to its end point. Moreover, cc does not change significantly when we change the centrifugation time between 10 min and 2 h.
Table 1.
Results of the ccanalysis
| Residue 18 | cc (μM) | ΔΔG (kcal/mol) |
| A | 66 ± 7 | 0.41 |
| D | 247 ± 7 | 1.23 |
| E | 95 ± 7 | 0.64 |
| F | 24 ± 1 | −0.21 |
| G | 33 ± 2 | 0.00 |
| H | 74 ± 5 | 0.49 |
| I | 11 ± 2 | −0.68 |
| K | 264 ± 28 | 1.27 |
| L | 29 ± 1 | −0.09 |
| N | 62 ± 10 | 0.38 |
| P | 176 ± 12 | 1.02 |
| Q | 31 ± 3 | −0.05 |
| R | 48 ± 5 | 0.22 |
| S | 187 ± 8 | 1.06 |
| T | 40 ± 6 | 0.11 |
| V | 32 ± 2 | −0.03 |
| W | 26 ± 3 | −0.17 |
| Y | 10 ± 1 | −0.75 |
Errors on cc are the standard deviation from four measurements. ΔΔG represents the difference ΔG(residue X) – ΔG(Gly18).
From the cc values obtained with this method we calculated ΔG and compared the thermodynamic values with several intrinsic properties of residue 18. This shows that ΔG fits well with β-sheet propensity (R value 0.70) (Street and Mayo 1999; Fig. 2A ▶) and hydrophobicity of residue 18 (R value 0.64) (Creighton 1993). By contrast, there was no discernible correlation with the van der Waals volume or the α-helix stabilizing effect (Creighton 1993; Fersht 1998). The correlation coefficient with these properties is always worse than 0.36 (data not shown). Recently, we have examined also the kinetics by which these mutants aggregate. We extracted two kinetic parameters, the lag time, which relates to the nucleation propensity, and the rate of aggregation, which relates to the polymerization propensity (Christopeit et al. 2005). Both kinetic parameters were found to show the same dependences on residue 18 as are revealed here for ΔG; i.e., β-sheet propensity and hydrophobicity correlate well with the aggregation kinetics, while van der Waals volume and α-helix stabilizing show no good correlation (Christopeit et al. 2005). Consistent with this, ΔG is directly proportional to the lag time (Fig. 2B ▶) and to the rate of aggregation, although this dependence is less clear (R value 0.64; data not shown). Taken together, these data imply that there is a link between the thermodynamics and the kinetics of aggregation in that mutants that form very stable aggregates are also those that form such aggregates very readily. Of course, this relationship may only be valid at certain positions of the polypeptide chain, i.e., the ones located within the β-sheet core of the aggregates. And indeed, residue 18 was chosen initially because several different techniques had suggested consistently that it occurs within the core of the aggregated β-sheet structure of the peptide (Christopeit et al. 2005).
Figure 2.
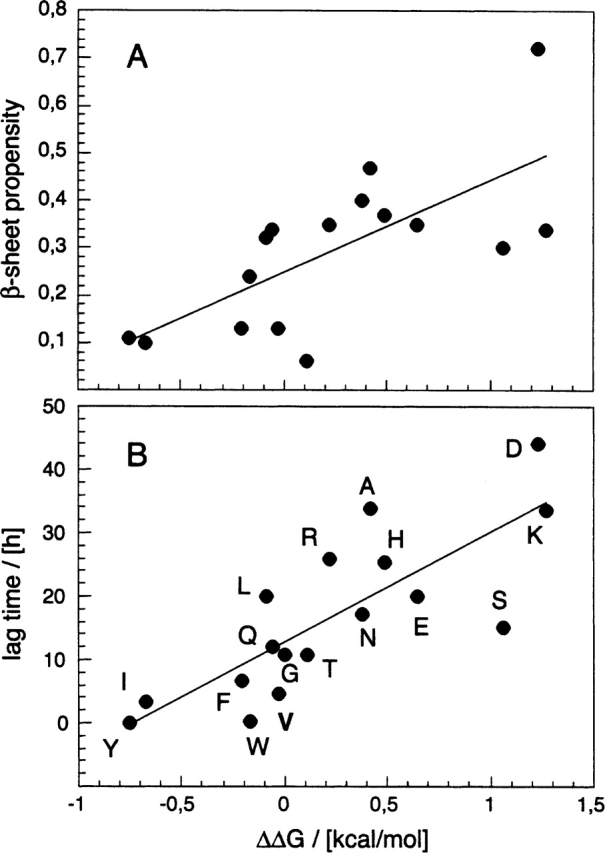
Correlation between the thermodynamics and the kinetics of aggregation. (A) Correlation between ΔΔG and the β-sheet propensity scale published by Street and Mayo (1999), which represents an average from several independent measurements (R value 0.70). Note that for Pro or Gly no propensity values have been reported. (B) Correlation between ΔΔG and the lag time as a measure of the aggregation kinetics (R value 0.80). Values of ΔΔG are taken from Table 1. The lag time data are taken from Christopeit et al. (2005).
However, we noted an apparent discrepancy between the present thermodynamic and our previous kinetic data in that the cc values of Val18Asp, Val18Lys, and Val18Ser measured here exceed the 120 μM concentration under which these mutants formed aggregates previously. Although there have been small differences in the experimental solution conditions (the previous analysis was carried out in 96-well plates, and samples contained, besides peptide and buffer, 20 μM thioflavine-T dye and 10 mM sodium azide), aggregates were evidenced before with thioflavine-T, which is a very sensitive method and may detect aggregates that are small and that cannot be spun down, such as soluble oligomers. Support for this notion is provided by the fact that thioflavine-T dye can detect small quantities of aggregates in the supernatant of all three mutants after centrifugation (data not presented). An alternative but much less likely possibility to explain this finding is the mutagenic stabilization of an alternative conformation that would hinder aggregate formation. Since wild-type Aβ(1–40) was shown to approach a random-coil structure under the aqueous conditions used here (Hortschansky et al. 2005), one should not expect that mutation would induce the formation of a sufficiently stable conformation that prevents aggregation during the very long incubation times of the present experiment. Therefore, the present data enable conclusions only on the formation of larger aggregates but not on the formation of very small aggregates that are traceable by other techniques.
Given these limitations, we observe a correlation between the kinetic and the thermodynamic data sets. A possible explanation for this finding might come from the mechanism of aggregation. Consider equimolar solutions of the different monomeric Aβ(1–40) variants; one should then expect that all peptide solutions possess approximately the same probability of bimolecular collisions between the fully dissolved polypeptide chains. However, if the resulting dimers are held together by intermolecular interactions of differing strength, this will result, inevitably, in different dissociation constants and different effective lifetimes of these species. The same considerations hold, of course, when larger oligomers (trimers, tetramers, etc.) are included in this scenario. Increasing the thermodynamic strength of the peptide–peptide interactions therefore increases the probability by which the peptide forms aggregation nuclei or larger aggregated states, which is observed by experiment. In vivo, of course, the nucleation-dependent aggregation reaction is modified by further parameters and interactions with other cellular components, for example, lipid rafts (Gellermann et al. 2005). These may modulate the mechanism and properties revealed here for the aggregation of pure polypeptide chains in vitro.
Materials and methods
Peptide incubation and ultracentrifugation
All experiments were carried out using the oxidized peptide; i.e., Met35 is present as sulfoxide. This form is more redoxstable than the reduced peptide (Watson et al. 1998). Potential interferences of redox reactions with our measurements are minimized. Peptides were produced recombinantly, oxidized, and prepared for analysis as described previously (Christopeit et al. 2005; Hortschansky et al. 2005). The resulting peptide solution (1.0–1.5 mg/mL in 50 mM sodium phosphate buffer at pH 7.4) was UV-sterilized by irradiation at 254 nm for 1.5 h. Incubation at 37° C was carried out in Oil-Free tubes (MoBi-Tec) and terminated by spinning down the sample for 30 min at 120,000 rpm (513,000g) and 4° C in a Beckman TLA-100 table-top centrifuge using a TLA-120.2 fixed angle rotor. After centrifugation the supernatant was taken off and the soluble peptide fraction was determined using a Micro-BC-Assay (Uptima) calibrated with a standard of wild-type Aβ peptide (Christopeit et al. 2005). The reported insoluble peptide fraction represents the difference between the soluble fraction and the total peptide concentration.
Data analysis
The Gibbs free energy of aggregation relates to the critical concentration as described by the simple equation ΔG = −RT ln(1/cc) in which R represents the gas constant and T represents the absolute temperature.
Acknowledgments
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (DFG) and a BioFuture grant from the Bundesministerium für Bildung und Forschung (BMBF). We thank Sylke Fricke for technical assistance.
Competing interests We claim a conflict of interest arising from a commercial partner that precludes free distribution of any DNA constructs described here. Basis vectors are available from commercial vendors.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051585905.
References
- Chiti, F., Stefani, M., Taddei, N., Ramponi, G., and Dobson, C.M. 2003. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature 424 805–808. [DOI] [PubMed] [Google Scholar]
- Christopeit, T., Hortschansky, P., Schroeckh, V., Gührs, K.H., Zandomeneghi, G., and Fändrich, M. 2005. Mutagenic analysis of the nucleation propensity of oxidized Alzheimer’s β-amyloid peptide. Protein Sci. 14 2125–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creighton, T. 1993. Proteins, structure and molecular properties, 2nd ed. W.H. Freeman, New York.
- Dobson, C.M. 2001. The structural basis of protein folding and its links with human disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 356 133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuBay, K.F., Pawar, A.P., Chiti, F., Zurdo, J., Dobson, C.M., and Vendruscolo, M. 2004. Prediction of the absolute aggregation rates of amyloidogenic polypeptide chains. J. Mol. Biol. 341 1317–1326. [DOI] [PubMed] [Google Scholar]
- Fändrich, M. and Dobson, C.M. 2002. The behaviour of polyamino acids reveals an inverse side chain effect in amyloid structure formation. EMBO J. 21 5682–5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fersht, A. 1998. Structure and mechanism in protein sciences: A guide to enzyme catalysis and protein folding. H.W. Freeman, New York.
- Gellermann, G.P., Appel, T.R., Tannert, A., Radestock, A., Hortschansky, P., Leisner, C., Lütkepohl, T., Shtrasburg, S., Röcken, C., Pras, M., et al. 2005. Raft lipids as novel and common components of human, extracellular amyloid fibrils. Proc. Natl. Acad. Sci. 102 6297–6302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper, J.D. and Lansbury Jr., P.T. 1997. Models of amyloid seeding in Alzheimer’s disease and scrapie: Mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu. Rev. Biochem. 66 385–407. [DOI] [PubMed] [Google Scholar]
- Hortschansky, P., Schroeckh, V., Christopeit, T., Zandomeneghi, G., and Fändrich, M. 2005. The aggregation kinetics of Alzheimer’s β-amyloid peptide is controlled by stochastic nucleation. Protein Sci. 14 1753–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriarty, D.F. and Raleigh, D.P. 1999. Effects of sequential proline substitutions on amyloid formation by human amylin 20–29. Biochemistry 38 1811–1818. [DOI] [PubMed] [Google Scholar]
- Morimoto, A., Irie, K., Murakami, K., Masuda, Y., Ohigashi, H., Nagao, M., Fukuda, H., Shimizu, T., and Shirasawa, T. 2004. Analysis of the secondary structure of β-amyloid (Aβ42) fibrils by systematic proline replacement. J. Biol. Chem. 279 52781–52788. [DOI] [PubMed] [Google Scholar]
- Oosama, F. and Asakura, S. 1975. Thermodynamics of polymerisation of protein. Academic Press, London, UK.
- Street, A.G. and Mayo, S.L. 1999. Intrinsic β-sheet propensities result from van der Waals interactions between side chains and the local backbone. Proc. Natl. Acad. Sci. 96 9074–9076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia, G.G., Cavalli, A., Pellarin, R., and Caflisch, A. 2004. The role of aromaticity, exposed surface, and dipole moment in determining protein aggregation rates. Protein Sci. 13 1939–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson, A.A., Fairlie, D.P., and Craik, D.J. 1998. Solution structure of methionine-oxidized amyloid β-peptide (1–40). Does oxidation affect conformational switching? Biochemistry 37 12700–12706. [DOI] [PubMed] [Google Scholar]
- Williams, A.D., Portelius, E., Kheterpal, I., Guo, J.T., Cook, K.D., Xu, Y., and Wetzel, R. 2004. Mapping Aβ amyloid fibril secondary structure using scanning proline mutagenesis. J. Mol. Biol. 335 833–842. [DOI] [PubMed] [Google Scholar]
- Zandomeneghi, G., Krebs, M.R., McCammon, M.G., and Fändrich, M. 2004. FTIR reveals structural differences between native β-sheet proteins and amyloid fibrils. Protein Sci. 13 3314–3321. [DOI] [PMC free article] [PubMed] [Google Scholar]