

Abstract
The solution structure of protein AF2095 from the thermophilic archaea Archaeglobus fulgidis, a 123-residue (13.6-kDa) protein, has been determined by NMR methods. The structure of AF2095 is comprised of four α-helices and a mixed β-sheet consisting of four parallel and anti-parallel β-strands, where the α-helices sandwich the β-sheet. Sequence and structural comparison of AF2095 with proteins from Homo sapiens, Methanocaldococcus jannaschii, and Sulfolobus solfataricus reveals that AF2095 is a peptidyl-tRNA hydrolase (Pth2). This structural comparison also identifies putative catalytic residues and a tRNA interaction region for AF2095. The structure of AF2095 is also similar to the structure of protein TA0108 from archaea Thermoplasma acidophilum, which is deposited in the Protein Data Bank but not functionally annotated. The NMR structure of AF2095 has been further leveraged to obtain good-quality structural models for 55 other proteins. Although earlier studies have proposed that the Pth2 protein family is restricted to archeal and eukaryotic organisms, the similarity of the AF2095 structure to human Pth2, the conservation of key active-site residues, and the good quality of the resulting homology models demonstrate a large family of homologous Pth2 proteins that are conserved in eukaryotic, archaeal, and bacterial organisms, providing novel insights in the evolution of the Pth and Pth2 enzyme families.
Keywords: NMR, Archaeglobus fulgidis, protein AF2095, solution structure, peptidyl-tRNA hydrolase Pth2, Pth2 evolution
Peptidyl-tRNA hydrolase (Pth) is a crucial bacterial enzyme that was first identified in Escherichia coli (Koessel 1970; Menninger 1979; Refugio Garcia-Villegas et al. 1991; Heurgue-Hamard et al. 1996) and subsequently classified in eukaryotes (Brun et al. 1971). Pth cleaves the peptide from peptidyl-tRNA molecules that have prematurely dissociated from the ribosome during protein translation (Meinnel et al. 1993; Schmitt et al. 1996). The freed tRNA can then be recycled back into the protein synthesis process. Aminoacyl tRNAfMet, the initiator of the translation process, is a relatively poor substrate for E. coli Pth and is correspondingly protected from hydrolysis. This permits free aminoacyl tRNAfMet to readily participate in the formation of the relatively slow ribosomal initiation complex without being prematurely hydrolyzed. The low hydrolase activity of Pth toward tRNAfMet has been attributed to the lack of base pairing for residues 1–72 in tRNAfMet and the resulting incorrect orientation of the 5′-phosphate that is necessary for Pth activity (Schulman and Pelka 1975; Dutka et al. 1993). The accumulation of tRNALys as peptidyl-tRNA in mutant E. coli cell lines has been identified as the cause of slow growth and cell death (Menninger 1979; Heurgue-Hamard et al. 1996). Presumably, this arises from the high propensity of tRNALys to disassociate from the translation complex during protein synthesis. The 1.2 Å X-ray structure of E. coli Pth indicates the protein adopts an α/β fold with a twisted mixed β-sheet (Schmitt et al. 1997). This E. coli Pth structure was determined to be similar to an amino-peptidase, which permitted the mapping of the enzyme’s active site to a catalytic triad composed of residues His 20, Asp 93, and His 113 (Goodall et al. 2004).
Although Pth is essential for viability of bacteria (Menez et al. 2002) and present in most eukaryotes, a number of archaea genomes contain no recognizable Pth homologs (Rosas-Sandoval et al. 2002; Fromant et al. 2003). Recently, unique Pth (Pth2) activities have been identified in archaea Methanocaldococcus jannaschii and the thermophilic archaea Sulfolobus solfataricus. These enzymes, MJ0051 from M. jannaschii (Y051_METJA) and SSO0175 from S. solfataricus (Y175_SULSO), lack any sequence similarity with the well-characterized bacterial Pth enzyme class (Schmitt et al. 1997; de Pereda et al. 2004). The recently determined X-ray crystal structure of human Pth2 (Protein Data Bank [PDB] ID 1q7s; de Pereda et al. 2004), the first 3D structure of a Pth2 enzyme, reveals that in addition to the lack of sequence similarity between Pth and Pth2, these enzymes also exhibit completely different folds. The Pth2 structure resembles the thioredoxin family fold, but exhibits E. coli tRNALys hydrolase activity. The active site of human Pth2 was assigned to residues in the β1–α1 loop and β3–β4 loop based on the close spatial proximity of conserved residues of Pth2 enzymes. Despite this lack of sequence and structural similarity between Pth and Pth2, the biological activities of the two enzyme classes are complimentary. Indeed, E. coli strains lacking endogenous Pth are rescued by the expression of Pth2 (Rosas-Sandoval et al. 2002; Fromant et al. 2003). Eukaryotes contain both Pth and Pth2 enzymes, where Pth2 enzymes are thought to be involved in mitochondrial protein synthesis (Rosas-Sandoval et al. 2002; Jan et al. 2004; Stupack and Cheresh 2004). In addition, a Pth/ Pth2 double mutant in Saccharomyces cerevisiae is viable, suggesting functional redundancy of Pth enzymes with some other eukaryotic enzyme(s).
Previously reported sequence analysis based on the Pth2 enzymes from human, archaea M. jannaschii, and thermophilic archaea S. solfataricus indicated that the Pth2 enzyme class was identified in numerous archaea and eukaryote organisms but was not present in bacteria (Rosas-Sandoval et al. 2002; Fromant et al. 2003; de Pereda et al. 2004). In addition, these enzymes were identified as members of the unannotated UPF0099 Pfam family, which only contained archaeal and eukaryotic proteins at the time of this analysis.
The human Pth2 enzyme is also known as Bcl-2 inhibitor of transcription 1 (Bit1). Bit1 is a mitochondrial protein that regulates apoptosis in a caspase-independent mechanism (Jan et al. 2004; Stupack and Cheresh 2004). The enzyme is located in the outer mitochondrial membrane, where, in the absence of integrin-mediated cell attachment, it migrates to the cytosol. Apoptosis is then initiated when Bit1 forms a complex with N-terminal enhancer of split (AES), which is a member of the Groucho family of transcriptional co-repressors. The exact details of the Bit1-mediated apoptosis pathway and the role of its Pth activity, are not well understood.
The thermophilic archaea Archaeglobus fulgidis AF2095 protein is an example of a protein of unknown biological function targeted for structural analysis by the Northeast Structural Genomics Consortium (NESG; http://www.nesg.org) (Liu et al. 2004; Wunderlich et al. 2004), which also belongs to the previously unannotated Pfam family UPF0099. In this article, we describe the 3D solution NMR structure of AF2095, together with the structural and functional insights provided by this structure. Comparison of the 3D NMR structure of A. fulgidis AF2095 and the 1.95 Å X-ray crystal structure of the functionally unannotated thermophilic archaea T. acidophilum protein TA0108 that has recently been deposited in the PDB (ID 1RLK) with the human Pth2 X-ray structure (de Pereda et al. 2004) suggests that these proteins are also Pth2 enzymes. Although it had been proposed that the Pth2 family has no bacterial members (Rosas-Sandoval et al. 2002; Fromant et al. 2003; de Pereda et al. 2004), assessments of homology models based on the 3D structure of AF2095 reported here demonstrate a wide phylogenetic distribution of the Pth2 enzyme class, including many bacterial proteins, consistent with the recent inclusion of bacterial members in Pfam family UPF0099. The structural and functional analysis also provide some insight in the evolution of the Pth and Pth2 enzyme families.
Results and Discussion
AF2095 NMR structure
The AF2095 structure is well defined by the NMR data where a total of 3201 constraints were used to refine the structure (Table 1). This is evident by a best-fit superposition of the backbone atoms shown in Figure 1 ▶, where the atomic root mean square distribution (RMSD) of the 30 simulated annealing structures about the mean coordinate positions for residues 4–81 and 90–111 is 0.41 ±0.06 Å for the backbone atoms. The quality of the AF2095 NMR structure is also evident by the results of the PROCHECK analysis, where an overall G-factor of −0.16 and 9.8 bad contacts per 100 residues is consistent with a good-quality structure (Laskowski et al. 1996). In addition, all of the backbone torsion angles for nonglycine residues lie within expected regions of the Ramachandran plot, where 82.2% of the residues lie within the most favored region of the Ramachandran φ,ψ plot, 15.9% lie in the additionally allowed region, and 1.9% lie in the generously allowed region.
Table 1.
Structural statistics and atomic RMS differences
| A. Structural statistics | <SA> | (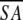 )r )r
|
| RMS deviations from experimental distance restraints (Å) | ||
| All (2750) | 0.041 ± 0.005 | 0.040 |
| Interresidue sequential (|i–j| = 1) (719) | 0.038 ± 0.008 | 0.046 |
| Interresidue short range (1 <|i–j| ≤5) (825) | 0.043 ± 0.005 | 0.035 |
| Interresidue long range (|i–j| > 5) (651) | 0.045 ± 0.009 | 0.040 |
| Intraresidue (483) | 0.008 ± 0.004 | 0.004 |
| H-bonds (72)a | 0.096 ± 0.013 | 0.104 |
| RMS deviation from exptl dihedral restraints (deg) (220)b,c | 1.433 ± 0.794 | 0.642 |
| RMS deviation from exptl Cα restraints (ppm) (94) | 0.89 ± 0.03 | 0.91 |
| RMS deviation from exptl Cβ restraints (ppm) (84) | 0.91 ± 0.02 | 0.94 |
| RMS deviation from 3JNHα restraints (Hz) (53) | 0.61 ± 0.02 | 0.54 |
| FNOE (kcal/mol)c | 234 ± 57 | 219 |
| Ftor (kcal/mol)c | 36 ± 45 | 5.5 |
| Frepel (kcal/mol)d | 108 ± 23 | 28.2 |
| FL-J (kcal/mol)e | −374 ± 21 | −362 |
| Deviations from idealized covalent geometry | ||
| Bonds (Å) (2268) | 0.005 ± 0 | 0.004 |
| Angles (deg) (4109) | 0.621 ± 0.030 | 0.608 |
| Impropers (deg) (1163)f | 0.519 ± 0.063 | 0.483 |
| PROCHECKg | ||
| Overall G-factor | −0.11 ± 0.03 | −0.16 |
| % Residues in most favorable region of Ramachandran plot | 83.5 ± 2.5 | 82.2 |
| H-bond energy | 1.0 ± 0.06 | 0.90 |
| No. of bad contacts/100 residues | 14.2 ± 2.1 | 9.8 |
| B. Atomic RMS differences (Å) | ||||
| Residues 6–142 | Secondary structureh | |||
| Backbone atoms | All atoms | Backbone atoms | All atoms | |
The notation of the structures is as follows: <SA> are the final 30 simulated annealing structures; 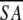 is the mean structure obtained by averaging the coordinates of the individual is the mean structure obtained by averaging the coordinates of the individual 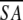 structures best fit to each other (excluding residues 1–3, 82–89, and 112–123); and ( structures best fit to each other (excluding residues 1–3, 82–89, and 112–123); and (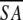 )r is the restrained minimized mean structure obtained by restrained minimization of the mean structure SA (Nilges et al. 1988a). The number of terms for the various restraints is given in parentheses. )r is the restrained minimized mean structure obtained by restrained minimization of the mean structure SA (Nilges et al. 1988a). The number of terms for the various restraints is given in parentheses. | ||||
| a For backbone NH-CO hydrogen bond there are two restraints: rNH-O = 1.5–2.3 Å and rN-O = 2.5–3.3 Å. All hydrogen bonds involve slowly exchanging NH protons. | ||||
| b The torsion angle restraints comprise 110 φ and 110 ψ. | ||||
| c The values of the square-well NOE (FNOE) and torsion angle (Ftor) potentials (cf. Equations 2 and 3 in Clore et al. 1986) are calculated with force constants of 50 kcal mol−1 Å −2 and 200 kcal mol−1 rad−2, respectively. | ||||
| d The value of the quartic van der Waals repulsion term (Frep) (cf. Equation 5 in Nilges et al. 1988c) is calculated with a force constant of 4 kcal mol−1 Å −4 with the hard-sphere van der Waals radius set to 0.8 times the standard values used in the CHARMM (Brooks et al. 1983) empirical energy function (Nilges et al. 1988a,b,c). | ||||
| e EL-J is the Lennard-Jones-van der Waals energy calculated with the CHARMM empirical energy function and is not included in the target function for simulated annealing or restrained minimization. | ||||
| f The improper torsion restraints serve to maintain planarity and chirality. | ||||
| g These were calculated by using the PROCHECK program. | ||||
| h The residues in the regular secondary structure are 17–36(α1), 39–45(α2), 58–72(α3), 101–108(α4), 4–10(β1), 49–54(β2), 75–78(β3) and 92–98(β4). | ||||
<SA>vs (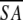 ) ) |
0.41 ± 0.06 | 1.00 ± 0.07 | 0.33 ± 0.04 | 0.94 ± 0.08 |
<SA>vs (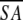 )r )r
|
0.49 ± 0.07 | 1.17 ± 0.11 | 0.41 ± 0.05 | 1.09 ± 0.15 |
(SA)r vs 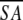
|
0.27 | 0.61 | 0.25 | 0.57 |
Figure 1.

(A) Ribbon diagram of the NMR structure of AF2095 for residues 1–112 colored by secondary structure. Disordered residues 113–123 were removed for clarity. (B) Superposition of the backbone (N,C,C′) atoms for the 30 best structures determined for AF2095 for residues 1–112. The disorder for residues Q79–I91 in the loop that connects β-strands β3 and β4 is evident by the large RMSD spread. The figures were generated with MOLSCRIPT (Kraulis 1991) and rendered with Raster3D (Merritt and Bacon 1997).
The AF2095 protein adopts an α/β fold composed of a twisted, mixed four-stranded β-sheet (II-I-IV-III) with two α-helices packed against each surface of the β-sheet. In one case, the two α-helices may be viewed as one continuous helix with an <90° bend that separates the two helical regions (Fig. 1 ▶). Looking along the edge of the β-sheet, the three primary α-helices form a triangular arrangement with the β-strands sandwiched between the helices. The four stranded β-sheet corresponds to residues 4–10 (β1), 49–54 (β2), 75–78 (β3), and 92–98 (β4) and the four helical regions correspond to residues 17–36 (α1), 39–45 (α2), 58–72 (α3), and 101–108 (α4).
There are two distinct regions of the AF2095 structure that are suggestive of being dynamic and disordered as evident by a lack of chemical shift assignments (Powers et al. 2004). The C-terminal region of AF2095 that includes residues P112–H123 appears to be disordered. This region of the protein also includes the LEHHHHHH purification tag. It is also interesting to note that this region of the protein includes four closely spaced leucine residues (L111, L113, L114, L116). Early on in the data collection and resonance assignments for AF2095, the NMR data quality was unexpectedly low for a protein the size of AF2095. A significant improvement in spectral quality was obtained with elevated temperatures, and the subsequent observation of disorder in the partially hydrophobic C-terminal tail appears to suggest a weak aggregation of the protein through an association of the C-terminal region of the protein. The improvement in the quality of the NMR data with a higher temperature is also consistent with the thermophilic nature of A. fulgidis and is probably closer to the functional conditions for which this protein has evolved.
The loop region consisting of residues Q79–I91, which connects β-strands β3 and β4, also appears to be dynamic and disordered in nature (Fig. 1 ▶). Residues L83–V86 have limited or a complete lack of NMR resonance assignments and, as a result, an absence of structural information. The lack of NMR assignments for residues L83–V86 arises because of a complete absence of any peaks that could be attributed to these residues in any NMR experiment, including the 2D 1H-15N HSQC spectra. There is no evidence to suggest that the lack of assignments could be attributed to peak overlap (Powers et al. 2004). The residues neighboring L83–V86 also exhibit generally weaker peak intensity and minimal structural NOEs.
Analysis of the surface structure of AF2095 using GRASP reveals a distinct cavity with a positive electrostatic potential suggestive of a potential binding site (Fig. 2 ▶). The AF2095 cavity is bordered by helix α1 and β-strand β2, and the base of the cavity is primarily formed by residues I91–V95 and residues L7–R10. These residues correspond to β-strands β4 and β1 and the intervening loop that connects the two β-strands.
Figure 2.
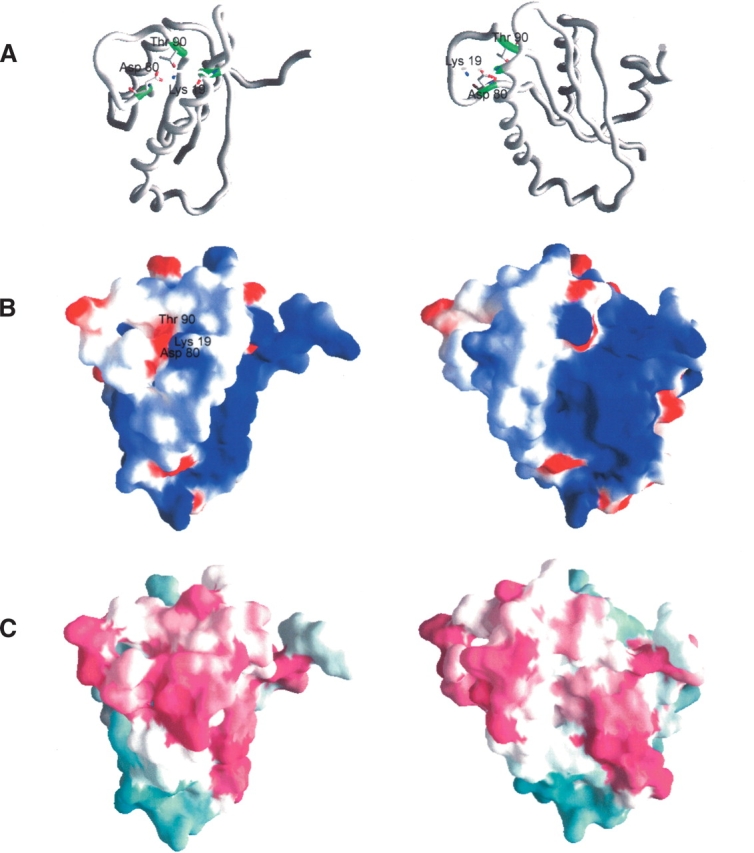
Structures of A. fulgidis AF2095. (A) Carbon-α backbone of AF2095. Residues proposed to form the catalytic triad (Lys 19, Asp 80, Thr 90) are shown in sticks in green. (B) Electrostatic surface of AF2095, in the same orientation as in A. The electrostatic surface was calculated by using a salt concentration of 0.1 M, and the color scale is −5 kt (negative, red) to 5 kt (positive, blue). (C) Phylogenetic analysis of AF2095 performed with the program ConSurf (Glaser et al. 2003). Maroon indicates conserved; cyan, variable. Sequences used in the ConSurf analysis were obtained from a PSIBLAST search against the nonredundant database using an e-value <0.001 after four iterations. Conserved residues cluster to the β3–β4 loop and the N terminus of α1, as well as to the region containing a surface patch of positive electrostatic potential. The right column is rotated −45° about the Y-axis, relative to the left column.
Function analysis and leverage of AF2095
Sequence homologs of AF2095
At the time of structure determination, AF2095 was annotated as a “conserved hypothetical protein” in major protein databases: SWISS-PROT (YK95_ARCFU, O28185), PIR (G69511), GenBank (AAB89160.1, NP_07-0920.1), TIGRFAM (TIGR00283), and PFAM (PF01981). AF2095 shares 40% and 36% identity with archaeal proteins MJ0051 from M. jannaschii (Y051_METJA) and SSO0175 from S. solfataricus (Y175_SULSO), respectively. These two archaea proteins have recently been experimentally identified as Pth2 enzymes (Rosas-Sandoval et al. 2002; Fromant et al. 2003). The sequences of both these enzymes also coincide with the previously unannotated Pfam family UPF0099, where AF2095 is also a member. This sequence analysis strongly suggests that AF2095 belongs to this Pth2 family of peptidyl-tRNA hydrolases (E.C.3.1.1.29).
Structural homologs of AF2095
The recent X-ray crystal structure of human Pth2 (YCE7_HUMAN; PDB ID 1q7s), also called Bcl-2 inhibitor of transcription 1 (Bit1), indicates that Pth2 enzymes adopt a novel protein fold (de Pereda et al. 2004). Even though eukaryotic/archaeal Pth2 and bacterial Pth (Schmitt et al. 1997) protein structures have very different folds, it has been proposed that Pth and Pth2 have essentially the same catalytic mechanism as do other α/β-fold hydrolases, a catalytic triad composed of a nucleophile, a basic, and an acidic amino acid. This is supported by the observation that human Pth2 exhibits catalytic activity in cleaving E. coli purified diacetyllysyl-tRNALys to produce free diacetyl-lysine and tRNA. A proposed tRNA interaction region of human Pth2 was inferred from a nonuniform distribution of a positive electrostatic surface potential comprising the β1–α1 loop, the β3–β4 loop, and α1. Residues involved in the catalytic activity of human Pth2 were inferred from conserved and spatially proximal surface residues that form a potential active site.
AF2095 shares 36% sequence identity with human Pth2 and also shares 44% sequence identity to conserved hypothetical protein TA0108 from T. acidophilum (Y108_THEAC; PDB ID 1rlk). Both the human Pth2 and TA0108 sequences were identified in the PSI-BLAST searches seeded with the AF2095 sequence. The 1.95 Å X-ray structure for TA0108 has been determined by the Midwest Center for Structural Genomics Research and is currently unpublished. The reported 2.0 Å X-ray structure for human Pth2 corresponds to residues 66–179 of the complete protein sequence, where residues 1–65 have been tentatively assigned to a localization signal in eukaryotes. Structural alignment of AF2095 with both TA0108 and human Pth2 in PrISM (Yang and Honig 1999) yields 3.8 Å and 3.7 Å RMSD, respectively (Fig. 3 ▶). The largest deviations in tertiary structure between AF2095 and human Pth2 occur in the β1–α1 loop, the β3–β4 loop, and the N terminus of α1, which have different relative orientations in these structures.
Figure 3.

Ribbon diagrams of the aligned views of (A) human Pth2 (PDB ID 1q7s), (B) T. acidophilum TA0108 (PDB ID 1rlk), and (C) A. fulgidis AF2095 (GR4; PDB ID 1rzw).
While the disordered β3–β4 loop in AF2095 lacks any experimental data to define its relative conformation, the distinct orientation and packing of the α1-α2 region is defined by 156 long-range NOEs and 363 short-range NOEs. These NOEs are equally distributed over the length of the helices and are all satisfied in the AF2095 NMR structure. Similarly, the α1–α2 region is also consistent with φ/ψ torsion angles based on 13Cα/Cβ chemical shifts as defined by TALOS (Cornilescu et al. 1999). Specifically, the carbon chemical shifts and TALOS predict a modest deviation from typical φ/ψ α-helical values between residues K34 and D36. Additional validation for the proper packing of the AF2095 NMR structure is provided by MolProbity analysis of bad contacts (Lovell et al. 2003). The packing violations observed for AF2095 are consistent with good-quality NMR structures. In addition, the violations are distributed throughout the 3D structure and are not concentrated in the β1–α1 loop, the β3–β4 loop, or the N terminus of α1, which differ from those of the human Pth2 and TA0108 structures. Presumably, if there was a packing error in these regions of the AF2095 structure, there would be a concentration of bad contacts identified by the MolProbity analysis (see Supplemental Material).
The sequence-based alignment between AF2095 and human Pth2 indicates a three-residue deletion in the region of α1 and α2. The shorter length of this helical region in AF2095 may explain the different orientation of the N terminus of α1 compared with that of human Pth2. The N terminus of α1 in AF2095 may be displaced to compensate for this three-residue deletion and to allow for the proper alignments of β1 and β4 with the remainder of the β-sheet. Also, residues that comprise the disordered β3–β4 loop in AF2095 are proximal to the N terminus of α1. This interaction may contribute to the displacement of α1, especially since the average conformation of the disordered loop in the AF2095 NMR structure is distinct from the human Pth2 X-ray structure. Further contributing to these observed structural differences, may be the fact that the AF2095 NMR structure was determined at an elevated temperature (40°C).
The proposed active site residues of human Pth2 are conserved in AF2095; however, due to the structural differences in the two proteins, particularly the dynamic loop conformations, these residues are not well aligned in the structures. Superposition of the AF2095 NMR structure with human Pth2 and TA0108 only aligns D80 with the corresponding acid residues, where T90 from AF2095 actually aligns with the basic catalytic residues. Again, these putative functional residues correspond to a dynamic and disordered loop region (Q79–I91) in the AF2095 structure, so a low alignment with the Pth2 X-ray structures is not surprising. Based on the conservation of the putative functional residues of human Pth2 in these three proteins and in the AF2095 family, the active-site residues for AF2095 and TA0108 can be inferred (Table 2).
Table 2.
Identification of active site residues
| Protein structure | Base residue | Acid residue | Nucleophile residue |
| AF2095 | K19 | D80 | T90 |
| human Pth2 | K81 | D145 | S155 |
| TA0108 | K19 | D83 | T93 |
The dynamic property of the β3–β4 loop and the poor alignment of the catalytic triad may imply a conformational adjustment or stabilization of the AF2095 structure when the protein binds a peptidyl-tRNA. A conformational change or an induced fit upon the binding of a ligand in the enzyme’s active site is a common occurrence (Burkhard et al. 1999; Hynson et al. 2004; Romanowski et al. 2004; Venkitakrishnan et al. 2004). Enzymes that bind tRNA exhibit similar ligand-induced conformational changes upon binding tRNA (Crepin et al. 2003; Zhang et al. 2003; Phannachet and Huang 2004). In fact, the binding of tRNA to aminoacyl-tRNA synthetase is required to properly arrange the active site for the catalytic activity of these enzymes (Francklyn et al. 2002; Sherlin and Perona 2003). A similar requirement may also exist for AF2095 and other Pth2 enzymes. Thus, the differences observed between the alignment of the NMR structure of AF2095 with the X-ray structures of TA0108 and human Pth2 may simply reflect changes in the dynamic behavior of the proteins in the solution and solid state. Again, significant differences in the dynamic behavior of proteins have been observed when comparing NMR and X-ray structures (Powers et al. 1993; Moy et al. 1997, 1998).
Similar to TA0108 and human Pth2, the surface of AF2095 exhibits a positive electrostatic potential, consistent with a nucleic acid binding function. In addition, all three proteins have a negatively charged cluster in the vicinity of the proposed active site surface (Fig. 2B ▶). The sequence and structure homology between human Pth2 and AF2095 implies that AF2095 functions as a Pth2 enzyme. Furthermore, it also implies that protein TA0108 from T. acidophilum, which is currently defined as a conserved hypothetical protein, is also a Pth2 enzyme.
Leverage analysis of AF2095
NESG prioritizes protein targets for structure elucidation by targeting sequence clusters (Liu et al. 2004; Wunderlich et al. 2004). In this manner, experimental structural information that is obtained for any representative protein in a given cluster can be leveraged to predict a quality structural model for the remaining protein members of the cluster by means of homology modeling. AF2095 has been assigned to NESG Cluster ID 17431, which contains a total of 14 proteins from human, Drosophila, Caenorhabditis elegans, Arabidopsis, yeast, archaeal, and eubacteria. When the structural analysis of AF2095 was initiated, no experimental structures were available for any members of this NESG cluster.
PSI-BLAST searches with AF2095 as the initial query against the nonredundant (NR) database were performed, which identified 67 significant hits. A 68th protein (hypothetical protein Mbur208901) was identified by inspecting the multiple sequence alignment of MJ0051 homologs (Rosas-Sandoval et al. 2002). Essentially, all the sequences identified to be homologous to AF2095 are annotated as proteins of unknown function (see Supplemental Table 1S). Thus, the NMR structure for AF2095 reported herein may be leveraged to obtain both structural and functional information for an additional 68 proteins.
An effort was made to validate homologs identified by PSI-BLAST by evaluating the quality of the resulting homology models (see Supplemental Table 1S). After removal of human Pth2, TA0108, and two proteins with >95% sequence identity with AF2095 from the leverage list, MODELLER (Sanchez and Sali 1998a) yielded good models based on the normalized ProsaII Z-scores (pG > 0.7) (Sippl 1993) for 63 sequences. Removing likely isoforms or polymorphisms yielded a revised leverage list of 53 high-quality and sequence-unique models for the AF2095 NMR structure.
For two sequences (EAA17753 and T05298) that are significantly similar to AF2095 (e-values of 1e−28 and 8e−18 after the last round of PSI-BLAST, respectively), initial models obtained were unreliable with pG scores of 0.55 and 0.00, respectively. In such cases, the alignment most likely contains errors. As an example, T05298 has a 42-residue insertion (region 144–185) that is not covered by the AF2095 template. Removal of the 42-residue insertion region and modeling of the remainder of the sequence provided a relatively reliable model for T05298 (pG score, 0.66). For EAA17753, a reliable model (pG score 0.78) could be made by using 1q7s and 1rlk as additional templates to the AF2095 NMR structure. Therefore, we estimate that the leverage for the AF2095 NMR structure is at least 55 sequences when EAA17753 and T05298 are included. The total increases to 65 with the inclusion of highly similar sequences.
Previous sequence alignments, which did not include the analysis of the quality of predicted homology models, have been reported by using human Pth2 (de Pereda et al. 2004) or archaeal Pth2 proteins from M. jannaschii (Rosas-Sandoval et al. 2002) and S. solfataricus (Fromant et al. 2003). A significant observation reported from these prior alignment efforts indicated that Pth2 homologs were not present in bacteria. Conversely, the AF2095 leverage list contains proteins from archaea, eukaryotes, and bacteria proteins from Corynebacterium diptheriae, Corynebacterium efficiens, Corynebacterium glutamicum, Streptomyces avermitilis, and Streptomyces coelicolor. These bacterial proteins have 26%–29% sequence similarity to AF2095, with pG scores ranging from 0.81 to 0.98 for the homology models. Our analysis of sequence and structure homologs of AF2095 has clearly indicated that Pth2 enzymes have a wider phylogenetic distribution than previously thought. Recently, the Pfam family UPF0099 has been updated to include these bacterial proteins.
Phylogenic analysis and evolution of Pth and Pth2 enzymes
The complete lack of sequence and structural similarity between the Pth and Pth2 enzyme families, while maintaining comparable hydrolase activity, is suggestive of convergent evolution. Our structural and functional analysis of AF2095 allows us to propose a potential evolutionary pathway for the Pth and Pth2 enzymes. It appears that eukaryotes inherited Pth2 enzymes from the archaea lineage during the formation of the mitochondria from the endosymbiosis of two prokaryotes (Brown and Doolittle 1997). Similarly, bacteria appear to have inherited Pth2 from archaea by lateral gene transfer (Hao and Golding 2004) after the emergence of the three separate domains. A phylogenic tree (Fig. 4 ▶) based on sequence similarity of at least <30% with AF2095 supports this proposed Pth2 evolutionary pathway.
Figure 4.
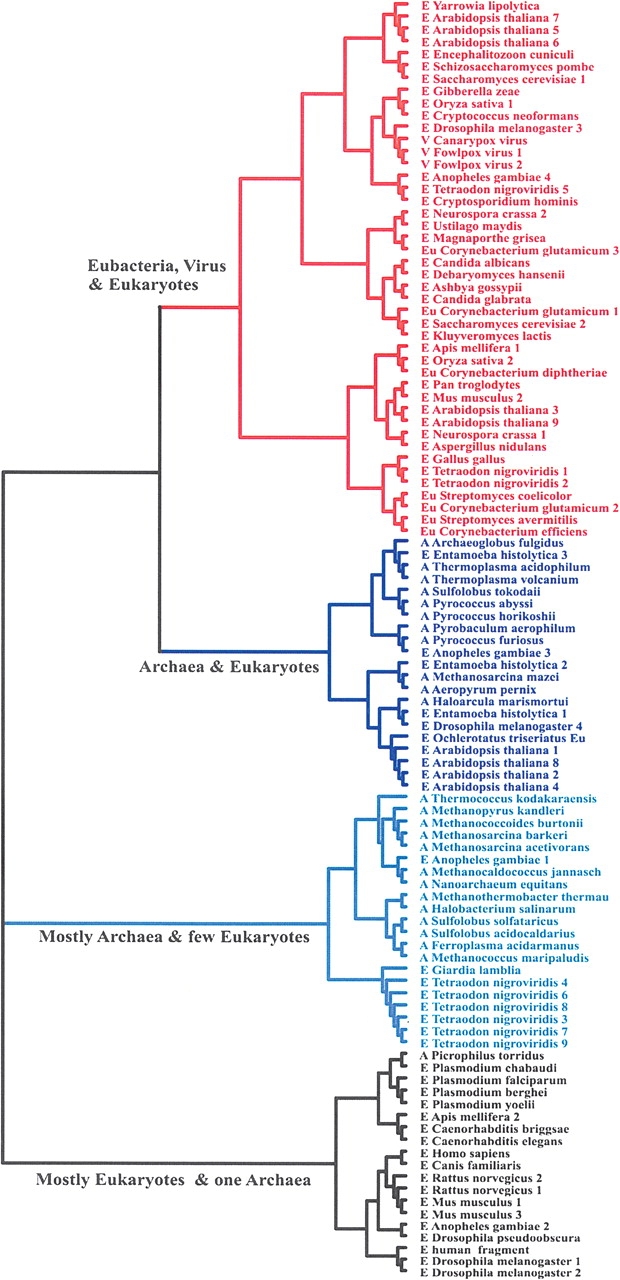
Phylogenic tree of Pth2 enzymes with <≥30% sequence similarity to AF2095, including homologs. For clarity, each major branch is colored separately, each organism is labeled archaea (A), eukaryote (E), and eubacteria (Eu), and the homologs are numbered sequentially.
Pth enzymes are essential for bacteria viability, where Pth2 enzymes are known to be conserved in all archaea. Conversely, Pth and Pth2 enzymes are nonessential in yeast (Menez et al. 2002; Rosas-Sandoval et al. 2002). Archaea contain only Pth2 enzymes, which have been implicated as being involved in mitochondrial protein synthesis in eukaryotes (Rosas-Sandoval et al. 2002; Jan et al. 2004; Stupack and Cheresh 2004). These results, combined with the observation that the primary branch (colored blue) in the AF2095 phylogenic tree contains only archaea and eukaryotic proteins, are consistent with eukaryotes inheriting Pth2 from archaea as part of the formation of the mitochondria. Furthermore, the closest Pth2 enzyme to AF2095 in this branch of the phylogenic tree comes from Entamoeba histoltica, an evolutionarily distant organism (Woese 2000) that contains a number of proteins, in addition to AF2095, that are common to A. fulgidis. This result strongly suggests a common ancestor (Field et al. 2000) that is consistent with the proposed evolutionary pathway that would relate A. fulgidis and E. histoltica through the formation of the mitochondria in eukaryotes.
The next closely related branch in the AF2095 phylogenic tree (colored red) contains no archaea Pth2 sequences, but it contains all the bacteria and virus genomes with an identified Pth2 enzyme. Again, this would be consistent with lateral gene transfer of Pth2 to bacteria from archaea after the separation of these domains. All the bacteria genomes that were identified to contain a Pth2 enzyme also contain Pth enzymes. Since Pth activity is crucial for bacteria viability, this observation would be consistent with bacteria inheriting the redundant Pth2 gene from archaea as opposed to the simultaneous evolution within the same organism of functionally redundant enzymes that are sequence and structurally distinct. This analysis would also imply that the Pth gene evolved separately through the bacteria lineage and was inherited by eukaryotes when eukaryotes and eubacteria diverged (Rosas-Sandoval et al. 2002). Again, this is consistent with eukaryotes containing both Pth and Pth2 enzymes, where Pth2 is a mitochondrial protein. The two remaining branches are almost exclusively either archaea or eukaryotic proteins, where the eukaryotes are primarily higher organisms—such as human, mouse, rat, and fly—representing distant evolution of the Pth2 enzyme.
Conclusion
The NMR solution structure for AF2095 has provided insight into the function of AF2095. The sequence and structure similarity between AF2095 and human Pth2 suggests that AF2095 is a peptidyl-tRNA hydrolase, specifically a Pth2 enzyme. Structural comparison between the Pth2 structures and the prediction that the hydrolase contains a catalytic triad composed of a nucleophile, a basic, and an acidic amino acid (Sanishvili et al. 2003; de Pereda et al. 2004) enables the prediction of residues in AF2095 that may comprise a catalytic site, specifically K19 (located at the beginning of α1), D80, and T90 (located at the β3–β4 loop), which are conserved in AF2095 homologs. These residues are proximal to the proposed tRNA interaction region, which has a strong electrostatic positive field. However, the distances between these putative catalytic residues in AF2095 would likely preclude them from forming a triad in the conformation observed in the AF2095 structure. Thus, the proper assembly of the active site may first require binding the peptidyl-tRNA as previously observed with aminoacyl-tRNA synthetase. The properties of the electrostatic surface of AF2095 are consistent with a nucleic acid binding function as well as with the previous analysis of Pth2 structures. The AF2095 NMR structure provides structural and functional leverage for 55 protein sequences from a variety of organisms, including five bacterial proteins. The AF2095 leverage analysis implies that Pth2 is an extensive family present in archaea, bacteria, and eukaryotes, which is contrary to prior predictions but consistent with the recent addition of bacterial proteins to Pfamfamily UPF0099. Phylogenic analysis of the Pth2 enzymes homologous to AF2095 supports convergent evolution of the Pth and Pth2 enzymes, suggesting that eukaryotes inherited Pth2 as part of the mitochondria organelle.
Materials and methods
Sample preparation and data collection
Uniformly (>95%) 15N- and 15N/13C-enriched recombinant AF2095 (13.6 kDa, 123 amino acids, PI 9.4) was expressed in E. coli and purified as described elsewhere (Acton et al. 2005). The NMR samples contained 1 mM AF2095 in 95% H2O/5% D2O solution containing 20 mM MES, 100 mM NaCl, 10 mM DTT, 5 mM CaCl2, and 0.02% NaN3 at pH 6.5. All spectra were recorded at 40°C on Bruker and Varian 500-, 600-, 750-, and 800-MHz NMR spectrometer using a gradient enhanced triple-resonance 1H/13C/15N probe.
1H, 15N, 13C, and 13CO assignments and secondary structure determination of AF2095 were reported previously (Powers et al. 2004). In addition to the NMR experiments used for the AF2095 resonance assignments, the present structure is based on the following series of spectra: HNHA (Vuister and Bax 1993), 1H-15N HSQC, and 3D 15N- (Marion et al. 1989; Zuiderweg and Fesik 1989) and 13C-edited NOESY (Ikura et al. 1990; Zuiderweg et al. 1990) experiments. The 15N-edited NOESY and 13C-edited NOESY experiments were collected with 100-msec and 80-msec mixing times, respectively. Spectra were processed by using the NMRPipe software package (Delaglio et al. 1995) and analyzed with PIPP (Garrett et al. 1991) on a Linux workstation.
Interproton distance restraints
The NOEs assigned from 3D 13C-edited NOESY and 3D 15N-edited NOESY experiments were classified into strong, medium, weak, and very weak corresponding to interproton distance restraints of 1.8–2.7 Å (1.8–2.9 Å for NOEs involving NH protons), 1.8–3.3 Å (1.8–3.5 Å for NOEs involving NH protons), 1.8–5.0 Å, and 3.0–6.0 Å, respectively (Williamson et al. 1985; Clore et al. 1986). Upper distance limits for distances involving methyl protons and nonstereospecifically assigned methylene protons were corrected appropriately for center averaging (Wüthrich et al. 1983). Hydrogen bond restraints were deduced on the basis of slowly exchanging NH protons, which were identified by recording a 1H-15N-HSQC spectrum after exchanging an AF2095 sample from H2O to D2O. Two distance restraints were used for each hydrogen bond (rNH-O = 1.5–2.3 Å, rN-O = 2.4–3.3 Å).
Torsion angle restraints
The φ and ψ torsion angle restraints were obtained from chemical shift analysis by using the TALOS program (Cornilescu et al. 1999) and from consistency with distance restraints for intraresidue and sequential NOEs involving NH, CαH, and CβH protons, and 3J(HN-Hα) coupling constants measured from the relative intensity of Hα cross-peaks to the HN diagonal in the HNHA experiment (Vuister and Bax 1993). The minimum ranges employed for the φ, and ψ torsion angle restraints were ±30° and ±50°, respectively.
Structure calculations
An initial structural fold for AF2095 was determined by automated analysis of NMR data using the programs AutoStructure (Huang 2001; Huang et al. 2003; Zheng et al. 2003; Huang et al. 2005) and DYANA (Guntert et al. 1997) along with slow amide exchange and 3J(HN-Hα). The best DYANA structure was then used in further refinement by using XPLOR-NIH software. The structures were further refined using the hybrid distance geometry dynamical-simulated annealing method of Nilges et al. (1988c) with minor modifications (Clore et al. 1990), using the program XPLOR-NIH (Schwieters et al. 2003), adapted to incorporate pseudopotentials for 3J(HN-Hα) coupling constants (Garrett et al. 1994), secondary 13Cα/13Cβ chemical shift restraints (Kuszewski et al. 1995), radius of gyration (Kuszewski et al. 1999), and a conformational database potential (Kuszewski et al. 1996, 1997; Kuszewski and Clore 2000). The target function that is minimized during restrained minimization and simulated annealing comprises only quadratic harmonic terms for covalent geometry, 3J(HN-Hα) coupling constants, and secondary 13Cα/13Cβ chemical shift restraints, square-well quadratic potentials for the experimental distance, radius of gyration and torsion angle restraints, and a quartic van der Waals term for nonbonded contacts. The radius of gyration can be predicted with reasonable accuracy on the basis of the number of residues using a relationship determined empirically from the analysis of high-resolution X-ray structures (Kuszewski et al. 1999). The force constant for the conformational database and radius of gyration potentials were kept relatively low throughout the simulation to allow the experimental distance and torsion angle restraints to predominately influence the resulting structures. The force constant for the NOE and dihedral restraints was 30 times and 10 times stronger then was the force constants used for the conformational database and radius of gyration potentials, respectively. All peptide bonds were constrained to be planar and trans. There were no hydrogen-bonding, electrostatic, or 6–12 Lennard-Jones empirical potential energy terms in the target function.
Leverage analysis
Determination of the number of protein sequences for which quality homology models can be built with the structure of AF2095 as a modeling template was performed in several automated steps. First, a PSI-BLAST (Altschul et al. 1997) search was performed against the National Center for Biotechnology Information (NCBI) NR protein database with AF2095 as the query sequence. The threshold for inclusion of a hit in the PSI-BLAST position-specific scoring matrix (or PSSM) was 0.0005, and 10 rounds of iteration were allowed. Significant hits were defined as those hits with e-values in the final round of <0.001. Next, the sequences of proteins of known structure and redundant sequences from the same species were removed from the hit list. For the purpose of our analysis, two sequences from the same species were considered redundant if their regions aligned to AF2095 are of the same length and >95% identical to each other. The remaining significant hits comprise our “homolog list.”
Protein structure models were built for all sequences in the homolog list as well as for nonsignificant PSI-BLAST hits (i.e., e-value > 0.001) if these had >50 residues aligned to AF2095. Including nonsignificant hits allowed us to test for the existence of possible structural homologs whose sequences have diverged from AF2095 to an extent too great to be detected with confidence by PSI-BLAST. Ten models were calculated for each PSI-BLAST alignment using comparative modeling, as implemented in program MODELLER (Sali and Blundell 1993). Subsequently, each model’s quality was assessed by ProsaII (Sippl 1993). ProsaII Z-scores describe the difference in free energy of a model being evaluated and the average free energy of an ensemble of models obtained by threading the same sequence through unrelated folds. For each set of 10 alignments, only the model with the most significant (lowest) ProsaII Z-score was retained. The probability that a model is reliable, given its Z-score value and length, was calculated by the pG server (Sanchez and Sali 1998b; http://sanchezlab.org/servers/pg). pG values provide a way to compare statistical significance among models of different lengths. Based on past analyses (Sanchez and Sali 1998b), models with pG scores between 0.7 and 1 are considered reliable and, thereby, likely to have the same fold as AF2095.
Models, generated as described above, that were assessed as unreliable (i.e., pG < 0.7) were considered manually. Generally, it was possible to derive significantly improved models by manually editing the sequence alignment between homolog and template or by using alternative templates, i.e., 1rlk or 1q7s, that are structurally homologous to AF2095.
Comparison of AF2095 with homologs of known structure
AF2095 structure (PDB ID 1rzw) and the structures of its homologs, hypothetical protein TA0108 from T. acidophilu (PDB ID 1rlk) and human Pth2, also called Bcl-2 inhibitor of transcription 1 (Bit1) (PDB ID 1q7s; de Pereda et al. 2004), were superimposed by MODELLER and PRISM (Yang and Honig 1999). Two residues were considered structurally equivalent if their Cα atoms are <3.5 Å apart upon rigid body superposition. The sets of homologs for 1rlk and 1q7s were obtained as described for AF2095 above.
Pth2 family multiple sequence alignment
The multiple sequence alignment for AF2095 and its homologs incorporated those sequence fragments aligned to AF2095 by PSI-BLAST. The alignment was performed by ClustalW (Thompson et al. 1994) and refined manually by matching secondary structure information about AF2095 and its homologs of known structure (TA0108 and human Pth2) to secondary structure predictions (Kelley et al. 2000) for the homologs of unknown structure. Similarly, ClustalW was used to generate the phylogenic tree based on sequence alignment to AF2095.
Electrostatic calculations
Electrostatic potentials of structures and models were calculated and visualized in GRASP (Nicholls et al. 1991). In all calculations, the monovalent salt concentration in the continuum solvent was set to 0.1 M. The surface-mapped potential is graded from −5 kT/e (red) to 5 kT/e (blue).
Electronic supplemental material
Electronic supplemental material contains images summarizing the experimental data and quality assessment of the AF2095 NMR structure that supports the differences observed between the Pth2 structures illustrated in Figure 3 ▶, as well as a table listing the AF2095 homologs identified from PSI-BLAST, with corresponding information about structural models.
Acknowledgments
This work was supported by grants from the Protein Structure Initiative of the National Institutes of Health (P50 GM62413), the National Science Foundation (DBI-9904841), and Nebraska Tobacco Settlement Biomedical Research Development Funds. NMR spectra were acquired at both the Center for Advanced Biotechnology and Medicine NMR facility at Rutgers University and the Environmental Molecular Sciences Laboratory (a national scientific user facility sponsored by the U.S. Department of Energy Office of Biological and Environmental Research) located at Pacific Northwest National Laboratory and operated for DOE by Battelle (contract KP130103). Atomic coordinates and the NMR chemical shift assignments for the restrained minimized mean structure of AF2095 have been deposited in the RCSB PDB (ID 1rzw).
Abbreviations
3D, three dimensional
AES, N-terminal enhancer of split
Bit1, Bcl-2 inhibitor of transcription 1
DTT, dithiothreitol
HNHA, amide proton to nitrogen to CαH proton
HSQC, hetero-nuclear single quantum coherence spectroscopy
MES, 2-[N-morpholino]ethanesulfonic acid
NCBI, National Center for Biotechnology Information
NMR, nuclear magnetic resonance
NR, nonredundant NOE, nuclear Overhauser effect
NOESY, nuclear Overhauser enhancement spectroscopy
Pth, peptidyl tRNA hydrolase
PSSM, position-specific scoring matrix
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051666705.
Supplemental material: see www.proteinscience.org
References
- Acton, T.B., Gunsalus, K.C., Xiao, R., Ma, L.C., Aramini, J., Baran, M.C., Chiang, Y.-W., Climent, T., Cooper, B., Denissova, N.G., et al. 2005. Robotic cloning and protein production platform of the northeast structural genomics consortium. Methods Enzymol. 394 210–243. [DOI] [PubMed] [Google Scholar]
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks, B.R., Bruccoleri, R.E., Olafson, B.D., States, D.J., Swaminathan, S., and Karplus, M. 1983. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4 187–217. [Google Scholar]
- Brown, J.R. and Doolittle, W.F. 1997. Archaea and the prokaryote-to-eukaryote transition. Microbiol. Mol. Biol. Rev. 61 456–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brun, G., Paulin, D., Yot, P., and Chapeville, F. 1971. Peptidyl-tRNA hydrolase: Characterization in some organisms. Enzymic activity in the presence of ribosomes. Biochimie 53 225–231. [DOI] [PubMed] [Google Scholar]
- Burkhard, P., Tai, C.-H., Ristroph, C.M., Cook, P.F., and Jansonius, J.N. 1999. Ligand binding induces a large conformational change in O-acetylserine sulfhydrylase from Salmonella typhimurium. J. Mol. Biol. 291 941–953. [DOI] [PubMed] [Google Scholar]
- Clore, G.M., Nilges, M., Sukumaran, D.K., Brünger, A.T., Karplus, M., and Gronenborn, A.M. 1986. The three-dimensional structure of α1- purothionin in solution: Combined use of nuclear magnetic resonance, distance geometry and restrained molecular dynamics. EMBO J. 5 2729–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clore, G.M., Appella, E., Yamada, M., Matsushima, K., and Gronenborn, A.M. 1990. Three-dimensional structure of interleukin 8 in solution. Biochemistry 29 1689–1696. [DOI] [PubMed] [Google Scholar]
- Cornilescu, G., Delaglio, F., and Bax, A. 1999. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 13 289–302. [DOI] [PubMed] [Google Scholar]
- Crepin, T., Schmitt, E., Mechulam, Y., Sampson, P.B., Vaughan, M.D., Honek, J.F., and Blanquet, S. 2003. Use of analogues of methionine and methionyl adenylate to sample conformational changes during catalysis in Escherichia coli methionyl-tRNA synthetase. J. Mol. Biol. 332 59–72. [DOI] [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6 277–293. [DOI] [PubMed] [Google Scholar]
- de Pereda, J.M., Waas, W.F., Jan, Y., Ruoslahti, E., Schimmel, P., and Pascual, J. 2004. Crystal structure of a human peptidyl-tRNA hydrolase reveals a new fold and suggests basis for a bifunctional activity. J. Biol. Chem. 279 8111–8115. [DOI] [PubMed] [Google Scholar]
- Dutka, S., Meinnel, T., Lazennec, C., Mechulam, Y., and Blanquet, S. 1993. Role of the 1–72 base pair in tRNAs for the activity of Escherichia coli peptidyl-tRNA hydrolase. Nucleic Acids Res. 21 4025–4030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field, J., Rosenthal, B., and Samuelson, J. 2000. Early lateral transfer of genes encoding malic enzyme, acetyl-CoA synthetase and alcohol dehydrogenases from anaerobic prokaryotes to Entamoeba histolytica. Mol. Microbiol. 38 446–455. [DOI] [PubMed] [Google Scholar]
- Francklyn, C., Perona, J.J., Puetz, J., and Hou, Y.-M. 2002. Aminoacyl-tRNA synthetases: Versatile players in the changing theater of translation. RNA 8 1363–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromant, M., Ferri-Fioni, M.-L., Plateau, P., and Blanquet, S. 2003. Peptidyl-tRNA hydrolase from Sulfolobus solfataricus. Nucleic Acids Res. 31 3227–3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett, D.S., Powers, R., Gronenborn, A.M., and Clore, G.M. 1991. A common sense approach to peak picking in two-, three-, and four-dimensional spectra using automatic computer analysis of contour diagrams. J. Magn. Reson. 95 214–220. [DOI] [PubMed] [Google Scholar]
- Garrett, D.S., Kuszewski, J., Hancock, T.J., Lodi, P.J., Vuister, G.W., Gronenborn, A.M., and Clore, G.M. 1994. The impact of direct refinement against three-bond HN-CαH coupling constants on protein structure determination by NMR. J. Magn. Reson. B 104 99–103. [DOI] [PubMed] [Google Scholar]
- Glaser, F., Pupko, T., Paz, I., Bell, R.E., Bechor-Shental, D., Martz, E., and Ben-Tal, N. 2003. ConSurf: Identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 19 163–164. [DOI] [PubMed] [Google Scholar]
- Goodall, J.J., Chen, G.J., and Page, M.G.P. 2004. Essential role of histidine 20 in the catalytic mechanism of Escherichia coli peptidyl-tRNA hydrolase. Biochemistry 43 4583–4591. [DOI] [PubMed] [Google Scholar]
- Guntert, P., Mumenthaler, C., and Wüthrich, K. 1997. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 273 283–298. [DOI] [PubMed] [Google Scholar]
- Hao, W. and Golding, G.B. 2004. Patterns of bacterial gene movement. Mol. Biol. Evol. 21 1294–1307. [DOI] [PubMed] [Google Scholar]
- Heurgue-Hamard, V., Mora, L., Guarneros, G., and Buckingham, R.H. 1996. The growth defect in Escherichia coli deficient in peptidyl-tRNA hydrolase is due to starvation for Lys-tRNALys. EMBO J. 15 2826– 2833. [PMC free article] [PubMed] [Google Scholar]
- Huang, Y.J. 2001. “Automated determination of protein structures from NMR data by iterative analysis of self-consistent contact patterns.” Ph.D. thesis, Rutgers University, New Brunswick, NJ.
- Huang, Y.J., Swapna, G.V.T., Rajan, P.K., Ke, H., Xia, B., Shukla, K., Inouye, M., and Montelione, G.T. 2003. Solution NMR structure of ribosome-binding factor A (RbfA), a cold-shock adaptation protein from Escherichia coli. J. Mol. Biol. 327 521–536. [DOI] [PubMed] [Google Scholar]
- Huang, Y.J., Moseley, H.N., Baran, M.C., Arrowsmith, C., Powers, R., Tejero, R., Szyperski, T., and Montelione, G.T. 2005. An integrated platform for automated analysis of protein NMR structures. Methods Enzymol. 394 111–141. [DOI] [PubMed] [Google Scholar]
- Hynson, R.M.G., Kelly, S.M., Price, N.C., and Ramsay, R.R. 2004. Conformational changes in monoamine oxidase A in response to ligand binding or reduction. Biochim. Biophys. Acta 1672 60–66. [DOI] [PubMed] [Google Scholar]
- Ikura, M., Kay, L.E., Tschudin, R., and Bax, A. 1990. Three-dimensional NOESY-HMQC spectroscopy of a carbon-13–labeled protein. J. Magn. Reson 86 204–209. [Google Scholar]
- Jan, Y., Matter, M., Pai, J.-t., Chen, Y.-L., Pilch, J., Komatsu, M., Ong, E., Fukuda, M., and Ruoslahti, E. 2004. A mitochondrial protein, Bit1, mediates apoptosis regulated by integrins and Groucho/TLE corepressors. Cell 116 751–762. [DOI] [PubMed] [Google Scholar]
- Kelley, L.A., MacCallum, R.M., and Sternberg, M.J.E. 2000. Enhanced genome annotation using structural profiles in the program 3D-PSSM. J. Mol. Biol. 299 499–520. [DOI] [PubMed] [Google Scholar]
- Koessel, H. 1970. Purification and properties of peptidyl-tRNA hydrolase from Escherichia coli. Biochim. Biophys. Acta 204 191–202. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 945–949. [Google Scholar]
- Kuszewski, J. and Clore, G.M. 2000. Sources of and solutions to problems in the refinement of protein NMR structures against torsion angle potentials of mean force. J. Magn. Reson. 146 249–254. [DOI] [PubMed] [Google Scholar]
- Kuszewski, J., Qin, J., Gronenborn, A.M., and Clore, G.M. 1995. The impact of direct refinement against 13Cα and 13Cβ chemical shifts on protein structure determination by NMR. J. Magn. Reson. B 106 92–96. [DOI] [PubMed] [Google Scholar]
- Kuszewski, J., Gronenborn, A.M., and Clore, G.M. 1996. Improving the quality of NMR and crystallographic protein structures by means of a conformational database potential derived from structure databases. Protein Sci. 5 1067–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1997. Improvements and extensions in the conformational database potential for the refinement of NMR and x-ray structures of proteins and nucleic acids. J. Magn. Reson. 125 171–177. [DOI] [PubMed] [Google Scholar]
- ———. 1999. Improving the packing and accuracy of NMR structures with a pseudopotential for the radius of gyration. J. Am. Chem. Soc. 121 2337–2338. [Google Scholar]
- Laskowski, R.A., Rullmann, J.A., MacArthur, M.W., Kaptein, R., and Thornton, J.M. 1996. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. Biomol. NMR 8 477–486. [DOI] [PubMed] [Google Scholar]
- Liu, J., Hegyi, H., Acton, T.B., Montelione, G.T., and Rost, B. 2004. Automatic target selection for structural genomics on eukaryotes. Proteins 56 188–200. [DOI] [PubMed] [Google Scholar]
- Lovell, S.C., Davis, I.W., Arendall III, W.B., de Bakker, P.I.W., Word, J.M., Prisant, M.G., Richardson, J.S., and Richardson, D.C. 2003. Structure validation by Cα geometry: φ, ψ and Cβ deviation. Proteins 50 437–450. [DOI] [PubMed] [Google Scholar]
- Marion, D., Driscoll, P.C., Kay, L.E., Wingfield, P.T., Bax, A., Gronenborn, A.M., and Clore, G.M. 1989. Overcoming the overlap problem in the assignment of proton NMR spectra of larger proteins by use of three-dimensional heteronuclear proton-nitrogen-15 Hartmann-Hahn-multiple quantum coherence and nuclear Overhauser-multiple quantum coherence spectroscopy: Application to interleukin 1β. Biochemistry 28 6150–6156. [DOI] [PubMed] [Google Scholar]
- Meinnel, T., Mechulam, Y., and Blanquet, S. 1993. Methionine as translation start signal: A review of the enzymes of the pathway in Escherichia coli. Biochimie 75 1061–1075. [DOI] [PubMed] [Google Scholar]
- Menez, J., Buckingham Richard, H., de Zamaroczy, M., and Campelli Celine, K. 2002. Peptidyl-tRNA hydrolase in Bacillus subtilis, encoded by spoVC, is essential to vegetative growth, whereas the homologous enzyme in Saccharomyces cerevisiae is dispensable. Mol. Microbiol. 45 123–129. [DOI] [PubMed] [Google Scholar]
- Menninger, J.R. 1979. Studies on the metabolic role of peptidyl-tRNA hydrolase, 5: Accumulation of peptidyl tRNA is lethal to Escherichia coli. J. Bacteriol. 137 694–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt, E.A. and Bacon, D.J. 1997. Raster3D: Photorealistic molecular graphics. Methods Enzymol. 277 505–524. [DOI] [PubMed] [Google Scholar]
- Moy, F.J., Pisano, M.R., Chanda, P.K., Urbano, C., Killar, L.M., Sung, M.-L., and Powers, R. 1997. Assignments, secondary structure and dynamics of the inhibitor-free catalytic fragment of human fibroblast collagenase. J. Biomol. NMR 10 9–19. [DOI] [PubMed] [Google Scholar]
- Moy, F.J., Chanda, P.K., Cosmi, S., Pisano, M.R., Urbano, C., Wilhelm, J., and Powers, R. 1998. High-resolution solution structure of the inhibitor-free catalytic fragment of human fibroblast collagenase determined by multidimensional NMR. Biochemistry 37 1495–1504. [DOI] [PubMed] [Google Scholar]
- Nicholls, A., Sharp, K., and Honig, B. 1991. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11 281–296. [DOI] [PubMed] [Google Scholar]
- Nilges, M., Clore, G.M., and Gronenborn, A.M. 1988a. Determination of three-dimensional structures of proteins from interproton distance data by dynamical simulated annealing from a randomarray of atoms:Circumventing problems associated with folding. FEBS Lett. 239 129–136. [DOI] [PubMed] [Google Scholar]
- ———. 1988b. Determination of three-dimensional structures of proteins from interproton distance data by hybrid distance geometry-dynamical stimulated annealing calculations. FEBS Lett. 229 317–324. [DOI] [PubMed] [Google Scholar]
- Nilges, M., Gronenborn, A.M., Bruenger, A.T., and Clore, G.M. 1988c. Determination of three-dimensional structures of proteins by simulated annealing with interproton distance restraints: Application to crambin, potato carboxypeptidase inhibitor and barley serine proteinase inhibitor 2. Protein Eng. 2 27–38. [DOI] [PubMed] [Google Scholar]
- Phannachet, K. and Huang, R.H. 2004. Conformational change of pseudouridine 55 synthase upon its association with RNA substrate. Nucleic Acids Res. 32 1422–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers, R., Clore, G.M., Garrett, D.S., and Gronenborn, A.M. 1993. Relationships between the precision of high-resolution protein NMR structures, solution-order parameters, and crystallographic B factors. J. Magn. Reson. B 101 325–327. [Google Scholar]
- Powers, R., Acton, T.B., Chiang, Y., Rajan, P.K., Cort, J.R., Kennedy, M.A., Liu, J., Ma, L., Rost, B., and Montelione, G.T. 2004. Letter to the editor: 1H, 13C and 15N assignments for the Archaeglobus fulgidis protein AF2095. J. Biomol. NMR 30 107–108. [DOI] [PubMed] [Google Scholar]
- Refugio Garcia-Villegas, M., De la Vega, F.M., Galindo, J.M., Segura, M., Buckingham, R.H., and Guarneros, G. 1991. Peptidyl-tRNA hydrolase is involved in l inhibition of host protein synthesis. EMBO J. 10 3549–3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanowski, M.J., Scheer, J.M., O’Brien, T., and McDowell, R.S. 2004. Crystal structures of a ligand-free and malonate-bound human caspase- 1: Implications for the mechanism of substrate binding. Structure 12 1361–1371. [DOI] [PubMed] [Google Scholar]
- Rosas-Sandoval, G., Ambrogelly, A., Rinehart, J., Wei, D., Cruz-Vera, L.R., Graham, D.E., Stetter, K.O., Guarneros, G., and Soll, D. 2002. Orthologs of a novel archaeal and of the bacterial peptidyl-tRNA hydrolase are nonessential in yeast. Proc. Natl. Acad. Sci. 99 16707–16712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali, A. and Blundell, T.L. 1993. Comparative protein modeling by satisfaction of spatial restraints. J. Mol. Biol. 234 779–815. [DOI] [PubMed] [Google Scholar]
- Sanchez, R. and Sali, A. 1998a. Evaluation of comparative protein structure modeling by MODELLER-3. Proteins Suppl. 1: 50–58. [DOI] [PubMed]
- ———. 1998b. Large-scale protein structure modeling of the Saccharomyces cerevisiae genome. Proc. Natl. Acad. Sci. 95 13597–13602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanishvili, R., Yakunin, A.F., Laskowski, R.A., Skarina, T., Evdokimova, E., Doherty-Kirby, A., Lajoie, G.A., Thornton, J.M., Arrowsmith, C.H., Savchenko, A., et al. 2003. Integrating structure, bioinformatics, and enzymology to discover function: BioH, a new carboxylesterase from Escherichia coli. J. Biol. Chem. 278 26039–26045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt, E., Guillon, J.M., Meinnel, T., Mechulam, Y., Dardel, F., and Blanquet, S. 1996. Molecular recognition governing the initiation of translation in Escherichia coli: A review. Biochimie 78 543–554. [DOI] [PubMed] [Google Scholar]
- Schmitt, E., Mechulam, Y., Fromant, M., Plateau, P., and Blanquet, S. 1997. Crystal structure at 1.2 A¢ª resolution and active site mapping of Escherichia coli peptidyl-tRNA hydrolase. EMBO J. 16 4760–4769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulman, L.H. and Pelka, H. 1975. Structural basis for the resistance of Escherichia coli formylmethionyl transfer ribonucleic acid to cleavage by Escherichia coli peptidyl transfer ribonucleic acid hydrolase. J. Biol. Chem. 250 542–547. [PubMed] [Google Scholar]
- Schwieters, C.D., Kuszewski, J.J., Tjandra, N., and Marius Clore, G. 2003. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 160 65–73. [DOI] [PubMed] [Google Scholar]
- Sherlin, L.D. and Perona, J.J. 2003. tRNA-dependent active site assembly in a class I aminoacyl-tRNA synthetase. Structure 11 591–603. [DOI] [PubMed] [Google Scholar]
- Sippl, M.J. 1993. Recognition of errors in three-dimensional structures of proteins. Proteins 17 355–362. [DOI] [PubMed] [Google Scholar]
- Stupack, D.G. and Cheresh, D.A. 2004. A Bit-role for integrins in apoptosis. Nat. Cell Biol. 6 388–389. [DOI] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkitakrishnan, R.P., Zaborowski, E., McElheny, D., Benkovic, S.J., Dyson, H.J., and Wright, P.E. 2004. Conformational changes in the active site loops of dihydrofolate reductase during the catalytic cycle. Biochemistry 43 16046–16055. [DOI] [PubMed] [Google Scholar]
- Vuister, G.W. and Bax, A. 1993. Quantitative J correlation: A new approach for measuring homonuclear three-bond J(HNHα) coupling constants in 15N-enriched proteins. J. Am. Chem. Soc. 115 7772–7777. [Google Scholar]
- Williamson, M.P., Havel, T.F., and Wüthrich, K. 1985. Solution conformation of proteinase inhibitor IIA from bull seminal plasma by proton nuclear magnetic resonance and distance geometry. J. Mol. Biol. 182 295–315. [DOI] [PubMed] [Google Scholar]
- Woese, C.R. 2000. Interpreting the universal phylogenetic tree. Proc. Natl. Acad. Sci. 97 8392–8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunderlich, Z., Acton Thomas, B., Liu, J., Kornhaber, G., Everett, J., Carter, P., Lan, N., Echols, N., Gerstein, M., Rost, B., et al. 2004. The protein target list of the Northeast Structural Genomics Consortium. Proteins 56 181–187. [DOI] [PubMed] [Google Scholar]
- Wüthrich, K., Billeter, M., and Braun, W. 1983. Pseudo-structures for the 20 common amino acids for use in studies of protein conformation by measurements of intramolecular proton–proton distance constraints with nuclear magnetic resonance. J. Mol. Biol. 169 949–961. [DOI] [PubMed] [Google Scholar]
- Yang, A.-S. and Honig, B. 1999. Sequence to structure alignment in comparative modeling using PrISM. Proteins Suppl. 3: 66–72. [DOI] [PubMed]
- Zhang, C.-M., Perona, J.J., and Hou, Y.-M. 2003. Amino acid discrimination by a highly differentiated metal center of an aminoacyl-tRNA synthetase. Biochemistry 42 10931–10937. [DOI] [PubMed] [Google Scholar]
- Zheng, D., Huang, Y.J., Moseley, H.N.B., Xiao, R., Aramini, J., Swapna, G.V.T., and Montelione, G.T. 2003. Automated protein fold determination using a minimal NMR constraint strategy. Protein Sci. 12 1232–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuiderweg, E.R.P. and Fesik, S.W. 1989. Heteronuclear three-dimensional NMR spectroscopy of the inflammatory protein C5a. Biochemistry 28 2387–2391. [DOI] [PubMed] [Google Scholar]
- Zuiderweg, E.R.P., McIntosh, L.P., Dahlquist, F.W., and Fesik, S.W. 1990. Three-dimensional carbon-13–resolved proton NOE spectroscopy of uniformly carbon-13–labeled proteins for the NMR assignment and structure determination of larger molecules. J. Magn. Reson. 86 210–216. [Google Scholar]