

Abstract
Ketol-acid reductoisomerase (KARI; EC 1.1.1.86) catalyzes two steps in the biosynthesis of branched-chain amino acids. Amino acid sequence comparisons across species reveal that there are two types of this enzyme: a short form (Class I) found in fungi and most bacteria, and a long form (Class II) typical of plants. Crystal structures of each have been reported previously. However, some bacteria such as Escherichia coli possess a long form, where the amino acid sequence differs appreciably from that found in plants. Here, we report the crystal structure of the E. coli enzyme at 2.6 Å resolution, the first three-dimensional structure of any bacterial Class II KARI. The enzyme consists of two domains, one with mixed α/β structure, which is similar to that found in other pyridine nucleotide-dependent dehydrogenases. The second domain is mainly α-helical and shows strong evidence of internal duplication. Comparison of the active sites between KARI of E. coli, Pseudomonas aeruginosa, and spinach shows that most residues occupy conserved positions in the active site. E. coli KARI was crystallized as a tetramer, the likely biologically active unit. This contrasts with P. aeruginosa KARI, which forms a dodecamer, and spinach KARI, a dimer. In the E. coli KARI tetramer, a novel subunit-to-subunit interacting surface is formed by a symmetrical pair of bulbous protrusions.
Keywords: active site, domain duplication, enzyme structure, NADPH, X-ray crystallography
It is over 10 years since Holm and Sander (1994) wrote “More and more frequently, a newly determined [protein] structure is similar in fold to a known one, even when no sequence similarity is detectable.” It is implicit in this statement that when proteins show sequence similarity, the fold will be the same, and a host of examples have verified this belief. Nevertheless, some interesting variations have emerged. One example is the structures of the first pair of thiamine diphosphate-dependent enzymes to be solved: transketolase (Lindqvist et al. 1992) and pyruvate oxidase (Muller and Schulz 1993). While the overall structure is very similar, consisting of three domains each of ~180 residues, the order of these domains along the primary structure differs; that is, one protein is a circular permutation of the other. This is but one of many examples where individual structural domains are conserved but may be shuffled into new combinations.
An unusual example of this phenomenon is in the enzyme ketol-acid reductoisomerase (KARI; EC 1.1.1.86) (Dumas et al. 2001). The first reported structure was that of the spinach enzyme (Biou et al. 1997; Thomazeau et al. 2000), which consists of a 225-residue N-terminal domain and a 287-residue C-terminal domain. The N-terminal domain, which contains the NADPH-binding site, has an α/β structure and resembles domains found in other pyridine nucleotide-dependent oxidoreductases. The C-terminal domain consists almost entirely of α-helices and is of a previously unknown topology. In addition to having this novel fold, sequence comparison of KARI from various species revealed that the enzyme exists in two versions. There is a long version typified by the spinach enzyme, and a shorter form found in fungi and many bacteria that is missing a sequence of ~140 residues corresponding to the N-terminal half of the C-terminal domain (Dumas et al. 2001). Determination of the structure of a short form of KARI, from Pseudomonas aeruginosa (Ahn et al. 2003), revealed that it is very similar to the spinach enzyme, but with one crucial difference. The C-terminal domain of the spinach enzyme superimposes on a pair of C-terminal domains of P. aeruginosa KARI. Thus, the long form (defined by Ahn et al. [2003] as Class II) contains a duplication of the C-terminal domain found in the short (Class I) form of KARI. The active site of the enzyme is contained entirely within a single subunit of the spinach enzyme, but is formed from both members of a dimer in P. aeruginosa KARI.
Escherichia coli and several other Gram-negative bacteria contain a long form of KARI. However, the inserted extra 140 residues bears little sequence similarity to the insert of spinach KARI. For example, a BLAST search using the E. coli insertion fails to locate spinach, or any other plant, KARI. Similarly, a BLAST search using the spinach insertion does not locate E. coli KARI. Examining the sequence of the C-terminal 260 residues of E. coli KARI fails to reveal much evidence of an internal duplication. Finally, phylogenetic analysis shows that the fungal short form (e.g., in Saccharomyces cerevisiae, Neurospora crassa, and Pichia stipitis) clusters closer to the plant form (spinach, pea, and Arabidopsis thaliana) than does the bacterial long form. These observations raise the possibility that the long form of KARI might not be a single class and that the plant form typified by the spinach enzyme might be different in structure from the long form found in E. coli and related bacteria.
Earlier we reported the crystallization of E. coli KARI (McCourt et al. 2004). Although the enzyme proved to be exceptionally easy to crystallize, few of the many crystal forms observed would diffract to atomic resolution. Data from the best of them, which diffracts to 2.6 Å, have now been phased, and here we report the structure.
Results
Crystallization and structure determination
Previously, we have reported that KARI crystallizes readily under a wide range of conditions (McCourt et al. 2004). Most of the crystal types showed poor resolution and only two diffracted to stronger than 3 Å resolution. The better of these was used in the present study.
Structure determination by molecular replacement was challenging for several reasons. Firstly, the search-model proteins of spinach or P. aeruginosa KARI are of only low-sequence identity, ~19% and 24%, respectively, when compared with E. coli KARI. Secondly, there was no evidence to suggest that the association of the subunits into the quaternary structure resembled either the spinach (Biou et al. 1997; Thomazeau et al. 2000) or the P. aeruginosa (Ahn et al. 2003) models. Thirdly, the relative orientation of the N- and C-terminal domains was reported to be different in the spinach and P. aeruginosa enzymes (Ahn et al. 2003). Finally, it was not clear from solvent content calculations how many molecules were in the asymmetric unit.
Ultimately, the structure was solved by molecular replacement in AMoRe (Navaza 1994). Initially, a solution could not be obtained with either the spinach enzyme (Biou et al. 1997; accession code 1QMG) or the bacterial enzyme (Ahn et al. 2003; accession code 1NP3). Therefore, we built a model for residues 31–344 of E. coli KARI with the program ProModII and the Swiss model server (Peitsch 1996; Schwede et al. 2003). From this, a search model was made by superposition of the modeled protein and the common core of the two experimental X-ray structures, 1QMG and 1NP3. It was only this model that allowed correct placement of the monomers in the crystal. In total, four monomers could be placed, which resulted in a progressive reduction of Rcryst and an increase in the correlation coefficient (Table 1). The Matthews (1968) coefficient indicated that there might be six monomers in the unit cell. Attempts to place a fifth molecule did not further improve these statistics, leaving a high-solvent content (67%), but a reasonable crystal packing (Fig. 1A ▶).
Table 1.
Data collection and refinement statistics for E. coli KARI
| Crystal parameters | |
| Unit cell length (Å) | a = b = 226.87 c = 118.86 |
| Space group | P63 |
| Mosaicity (°) | 0.35 |
| Crystal dimensions (mm) | 0.6 × 0.2 × 0.2 |
| Diffraction dataa | |
| Temperature (K) | 100 |
| Resolution range (Å) | 32.5–2.60 |
| Observations (I〉0σ(I)) | 922,387 (62,533) |
| Unique reflections (I〉0σ(I)) | 106,858 (10,653) |
| Completeness (%) | 99.8 (98.5) |
| Rsymb | 0.074 (0.300) |
| 〈I〉/〈σ(I)〉 | 20.9 (5.1) |
| Monomers per asymmetric unit | 4 |
| Solvent content (%) | 66.9 |
| Matthews coefficient (Å3/Da) | 3.7 |
| Molecular replacementc | |
| Molecule 1 | 31.7 (53.9) |
| Molecule 2 | 36.7 (52.3) |
| Molecule 3 | 41.2 (50.4) |
| Molecule 4 | 46.4 (48.8) |
| Refinement | |
| Resolution limits (Å) | 32.5–2.60 |
| Rfactor | 0.2230 |
| Rfree | 0.2664 |
| rmsdd bond lengths (Å) | 0.007 |
| rmsd bond angles (°) | 1.22 |
| Ramachandran plot (%) | |
| Most favored | 88.5 |
| Additionally allowed | 11.0 |
| Generously allowed | 0.5 |
| Disallowed | 0.1 |
a Values in parentheses are for the outer resolution shell: 2.69–2.60 Å.
bRsym = ∑|I-〈I〉|/∑ 〈I〉, where I is the intensity of an individual measurement of each reflection and 〈I〉 is the mean intensity of that reflection.
c Values shown for successive molecules are correlation coefficient (Rcryst), both expressed as a percent.
d (rmsd) Root mean square deviation.
Figure 1.
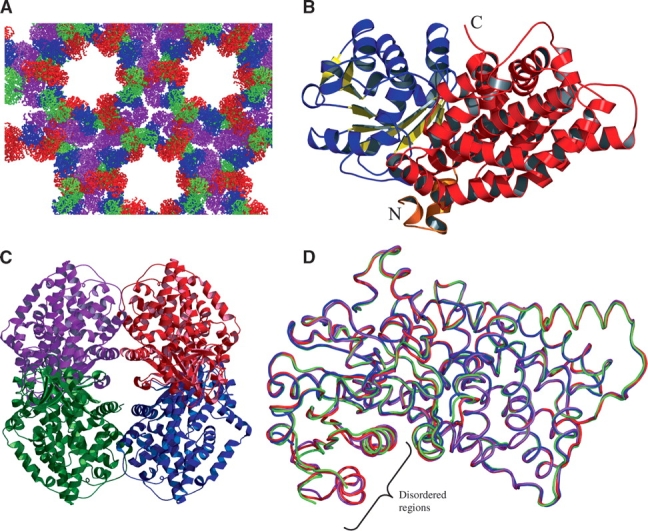
Crystal structure of E. coli KARI. (A) Packing of monomers in the crystal matrix. The four monomers in the asymmetric unit are color-coded A (magenta), B (blue), C (red), and D (green). (B) Monomer A, consisting of two domains: the α/β P-domain (residues 20–208) is shown in blue (α-helices) and yellow (β-strands), and the all α K-domain made up of residues 3–19 (orange) and 209–489 (red). (C) The biologically active subunit arrangement of the E. coli KARI tetramer, color-coded as in A. (D) Ribbon representation showing superimposition of monomers, color-coded as in A. Monomer B is disordered in the region of residues 37–91, whereas residues 44–49 and 75–85 are disordered in monomer D.
Overall structure
The final refined structure contains four monomers (Fig. 1C ▶), nine sulphate ions, and 416 water molecules in the asymmetric unit. Although NADPH was included in the crystallization buffer, no electron density consistent with the presence of this ligand was observed. The final Rfactor and Rfree values are 0.223 and 0.266, and the model has excellent stereochemistry (Table 1). A total of 88.5% residues occupy the most favored regions in the Ramachandran plot, and 11.0% are in the additionally allowed regions; only one residue (T245, monomer D) in the tetramer is in the disallowed region of the map. For two of four monomers in the asymmetric unit (monomers A and C), main-chain density can be traced from N3 to V489. Monomer D spans the same range, but has two gaps from G44 to G49 and from K75 to N85. Monomer B extends from N3 to A488 and is disordered in the region from G37 to T91. The overall structure of each monomer is similar (Fig. 1D ▶), with the most prominent variation at the A73–R76 surface loop. Given the great similarity with the root mean square deviation (rmsd) for Cα atoms ranging from 0.45 to 0.71 Å in pairwise comparisons, monomer A is taken as representative of all of the subunits.
The overall structure and the secondary elements are illustrated in Figure 1B ▶, which shows that there are two distinct domains. One domain spans C20–S208, and consists of a central nine-stranded β-sheet connected by loops and α-helices. Apart from P. aeruginosa KARI, the closest structural match to this domain detected by the EMBL-EBI Secondary Structure Matching server (Krissinel and Henrick 2004) is to the N-terminal region of an NADP+-dependent oxidoreductase (Warkentin et al. 2001) that acts upon the 5′-deazaflavin, F420, as shown in Figure 2A ▶. The position of NADP+ in this latter enzyme places its 2′-phosphate in close proximity to R76 of E. coli KARI. Mutagenesis of this residue (Rane and Calvo 1997) has been shown to change the substrate specificity of E. coli KARI so that it favors NADH over NADPH. Therefore, we suggest that NADPH is bound in a similar position and orientation in these two enzymes. We will refer to this domain as the “P-domain,” indicating that it contains the pyridine nucleotide-binding site.
Figure 2.

The active site of KARI. (A) Overlay of E. coli KARI (residues 38–209, blue; R76 in ball-and-stick) with Archaeoglobus fulgidus F420H2:NADP+ oxidoreductase (residues 1–44, 52–97, 113–180, red; NADP+ in stick). (B) Superimposition of the active-site residues of free E. coli KARI (red sticks, with labels) and spinach KARI (green sticks). Mn2+ (balls), and dihydroxymethylvalerate (yellow sticks) from the spinach KARI complex (1QMG) are also shown. (C) E. coli KARI showing the location of the eight conserved amino acids at the interface of the P-domain (blue) and the K-domain (red). (D,E) Comparison of the active sites regions of E. coli (D) and spinach (E) KARI, with NADPH from spinach KARI (1YVE) superimposed on both structures. In these electrostatic surface representations, the E. coli KARI active site is exposed in the resting state, while the active site in spinach KARI is completely buried.
The second domain (the “K-domain,” so named because it contributes most of the ketol-acid substrate-binding site and also because it contains a knot) is composed entirely of α-helices and connecting loops, with no β-strands. It is made up of residues S209–V489 plus the N-terminal segment of the protein (N3–K19). The structure of the K-domain shows strong evidence of duplication. While this is not readily evident when viewing the three-dimensional structure of the whole domain (Fig. 3A ▶), a structural alignment of G220–P343 with D356–G474 shows that these two segments have very similar folds (Fig. 3B ▶). The derived sequence alignment (Fig. 3C ▶), while not convincing of itself (21 identities in 124 positions), is persuasive when considered in combination with the structural overlay. Moreover, as illustrated in Figure 3D ▶, the positions of side chains at several conserved residues are very similar. These data leave little doubt that most of the K-domain of E. coli KARI has been derived by a duplication event involving 120–140 residues.
Figure 3.

Duplication within the K-domain of E. coli KARI. (A) Structure of the whole domain. (B) Structural overlay of two segments, G220–P343 with D356–G474. (C) Structure-based sequence alignment of G220–P343 with D356–G474. The regions highlighted in red and blue correspond to the red coil and the blue helix where the overlaid segments (B) are most different. The structures of the boxed sections in C are overlaid in D, illustrating the similar positions of conserved side chains.
Subunit arrangement
KARI from Salmonella typhimurium is a tetramer (Shematek et al. 1973; Hofler et al. 1975). The high degree of similarity to E. coli KARI (98.4% sequence identity) would suggest that E. coli KARI is also a tetramer. This was not observed initially in the molecular replacement solution, but reconstruction of the asymmetric unit revealed a plausible tetramer (Fig. 1C ▶). There are two types of interface within the tetramer: one that joins the A/D or B/C pairs (we refer to this as the bulge interface) and another that joins the A/C and B/D pairs (the flat interface). There are no contacts between the subunits across the diagonal (i.e., A/B or C/D). Both interfaces are formed entirely from residues comprising the K-domain (3–19 and 209–489).
There are three leucine residues (10, 14, and 17) along with 10 other residues (F5, N9, R11, Q13, Q301, D305, I308, D378, S379, and G380) that form a sharp protrusion (Fig. 4A ▶) that stabilizes the bulge interface (Table 2). Thus, the interacting surfaces bury a mixture of polar and non-polar residues as well as allowing the formation of dual salt bridges between R11 and D305. A detailed view of these interactions is presented in Figure 4B ▶. The total accessible surface area that is buried by this interaction is 1011 Å2 for the A/D pair and 1024 Å2 for the B/C pair, a value smaller than would be found typically in interacting surfaces for a protein of this size (Jones and Thornton 1996).
Figure 4.

The E. coli KARI dimer interaction surface. (A) Connolly surface of the A/D pair (A, dark gray; D, light gray) showing the interaction between these two subunits. (B) Detailed view of the interaction showing contacts (≤ 4 Å) (Table 2), shown in dark gray and light gray for monomers A and D, respectively. Leucine residues are shown as ball models, while other interacting residues are shown as stick models.
Table 2.
Contacts between the A/D and B/C monomer pairs
| Distance (Å)a | |||||
| Monomer A or B | Monomer D or C | A vs. D | B vs. C | ||
| F5 | Cz | L10 | Cδ | 3.9 | — |
| N9 | Nδ | D305 | Oδ | 3.5 | 3.7 |
| L10 | Cδ | L10 | Cδ | — | 3.5 |
| L10 | Cδ | Q13 | Oɛ | 3.8 | 3.2 |
| L10 | Cβ | I308 | Cγ2 | 3.7 | 3.9 |
| L10 | Cδ | S379 | O | 3.4 | 3.6 |
| R11 | Nɛ | Q301 | Cγ | — | 3.7 |
| R11 | NH1 | Q301 | Oɛ | 3.7 | — |
| R11 | NH2 | D305 | Oδ | 2.7 | 2.9 |
| R11 | Cγ | D378 | O | 3.9 | 3.7 |
| R11 | Cβ | S379 | Oγ | 4.0 | 3.9 |
| Q13 | Oɛ | L10 | Cδ | 3.4 | 3.0 |
| L14 | Cδ | L10 | Cδ | — | 3.9 |
| L14 | Cδ | L14 | Cδ | 3.6 | 3.8 |
| L14 | Cδ | D378 | O | 3.9 | — |
| L14 | Cδ | S379 | C | 3.9 | 3.7 |
| L14 | Cδ | G380 | N | 3.7 | 3.5 |
| L17 | Cδ | L10 | Cδ | 3.8 | — |
| Q301 | Cγ | R11 | Cz | 3.9 | — |
| Q301 | Nɛ | R11 | NH2 | — | 3.8 |
| D305 | Oδ | N9 | Nδ | 3.1 | 3.1 |
| D305 | Oδ | R11 | Nɛ | 2.8 | 2.8 |
| I308 | Cγ2 | L10 | Cδ | 3.8 | — |
| I308 | Cγ2 | L10 | Cβ | — | 3.7 |
| D378 | O | R11 | Cγ | 3.7 | 3.5 |
| D378 | O | L14 | Cδ | 3.9 | 3.9 |
| S379 | O | L10 | Cδ | 4.0 | 3.6 |
| S379 | Oγ | R11 | Cβ | — | 3.7 |
| S379 | Oγ | R11 | Cδ | 3.9 | — |
| S379 | C | L14 | Cδ | 3.9 | 3.8 |
| G380 | N | L14 | Cδ | 3.7 | 3.7 |
a Distances greater than 4 Å are not included. Where there are two or more contacts between the same pair of amino acids, only the closest contact is listed.
The contacting surfaces at the flat interface are more typical of those observed in subunit interactions of most proteins. There are three segments of polypeptide (263–279, 351–352, and 450–475) from each face that form bonds to make the flat interface. The A/C surface is stabilized by two ion pairs, nine hydrogen bonds, and 21 van der Waals’ contacts, while the B/D surface is stabilized by a similar set of interactions, but with anextra hydrogen bond and two more van der Waals’ interactions. The total surface area buried by these interfaces is 1769 Å2 for the A/C pair and 1778 Å2 for the B/D pair, values in keeping with those typically observed in proteins but in contrast to the 5500 Å2 buried by dimer formation in the P. aeruginosa KARI (Ahn et al. 2003), where the smaller C-terminal domains from different monomers interlock to form a unit that is analogous to a single K-domain in E. coli KARI. Spinach KARI also has a rather high subunit interface surface area of 4020 Å2.
Domain comparison with spinach and P. aeruginosa KARI
A comparison of the N-terminal section of E. coli KARI with that of spinach KARI (Fig. 5A ▶) shows that they have an rmsd of 1.40 Å for the 159 Cα atoms that can be superimposed, but there are some prominent structural differences, particularly in the region from E. coli KARI N3–Q36 (spinach KARI A83–K123) as illustrated in Figure 5B ▶. As mentioned earlier, the first half of this region is the N-terminal segment that forms part of the K-domain. In contrast, spinach KARI A83–K123 is part of its N-terminal pyridine nucleotide-binding domain. This difference can be easily appreciated by measuring the distance between Cα atoms between corresponding residues (Fig. 5C ▶). A similar comparison between the equivalent domains of E. coli and P. aeruginosa KARI (Fig. 5C ▶) shows that they are somewhat more similar, with an rmsd of 1.11 Å for 168 equivalent Cα atoms. The two bacterial KARI structures represent that of the free enzyme, while the spinach structure has the active site fully filled. While the spinach KARI A83–K123 region does not interact directly with any active-site ligands, it certainly undergoes structural alterations upon ligand binding (Halgand et al. 1999). Thus, we speculate that filling of the active site of E. coli KARI could induce a conformational change in this region toward the structure observed in the spinach enzyme or, since it forms part of the dimer interface, affect the quaternary structure of the E. coli enzyme.
Figure 5.

Comparison of KARI structures. (A) Stereo images of a superimposition of the N-terminal regions of E. coli (red) and spinach (green) KARI. (B) The substantial structural differences between the extreme N-terminal segments; E. coli KARI residues 3–31 (red) and spinach KARI residues 89–117 (green). (C) The distance between corresponding Cα atoms for the N-terminal regions of E. coli vs. spinach KARI (red) and E. coli vs. P. aeruginosa KARI (blue) is shown. (D) Illustration of a superimposition of the C-terminal regions of E. coli (red) and spinach (green) KARI, with common regions shown as a Connolly surface, and the labels identifying the C terminus and loops involved in the dimerization of each protein.
The C-terminal segment (S209–V489) of E. coli KARI is composed of 14 α-helices and overlays quite closely with spinach KARI around the active site, but there are some prominent differences at other parts of the polypeptide (Fig. 5D ▶). One notable difference between the two structures is a loop spanning F422–P432 in spinach KARI where there is no corresponding structure in the E. coli protein. This loop in spinach KARI has been shown to be responsible for formation of the homodimer (Wessel et al. 1998). Conversely, in E. coli KARI, the region F435–E463 does not have a structural equivalent in the spinach enzyme. This region forms the interface of the A/C dimer in the biologically active tetramer. It appears that this region in the K-domain of E. coli KARI plays a similar role to the F422–P432 loop region of spinach KARI, forming the biologically active unit. It should be noted that the E. coli KARI F435–E463 region contains the red loop (Fig. 3B ▶), where the two halves of the K-domain differ the most. Moreover, the blue helix (Fig. 3B ▶) contributes to the bulge interface. Thus, following the duplication event that formed the K-domain, its two halves have developed the greatest structural divergence in regions needed to form the two types of subunit interface.
In addition to these differences, P343–E353 in E. coli KARI is shortened and displaced sideways compared with the corresponding G445–G458 of the spinach protein, and the C-terminal tails (E. coli T482–V489 and spinach P575–A595) are different in length and conformation.
As described above, the rmsd values for the Cα atoms of the pyridine nucleotide-binding domains of E. coli and spinach KARI is 1.40 Å for 159 Cα atoms, while for the other domains this value is 1.58 Å for 213 Cα atoms. When all 372 of these atoms are superimposed, the rmsd is similar at 1.82 Å. Thus, the relative orientations of the two domains in E. coli and spinach KARI are almost identical. A similar comparison of the E. coli and P. aeruginosa enzymes yields rmsd values of 1.42 Å for 175 Cα atoms in the P-domain, 1.58 Å for 237 Cα atoms in the K-domain, and 2.13 Å for all 412 Cα atoms in the combined two domains. Thus, the relative orientation of the two domains is closer between free E. coli KARI and spinach KARI with a filled active site than it is between the two free enzyme structures.
Active site
The active site is at the interface between the P- and K-domains and is bordered by eight polar residues: H132, K155, E213, D217, E221, E389, E393, and S414 (Fig. 2C ▶). The first two residues are part of the P-domain, while the remainder are part of K-domain. All eight residues are well conserved across bacterial, fungal, and plant KARI sequences (Dumas et al. 2001), emphasizing their importance for activity. The vital role of each of these residues for catalysis has been shown by mutagenesis studies (Tyagi et al. 2005) where activity is lost when any of these amino acids is altered. This loss is >96% for all mutations, except when E213 is altered to aspartate, when 25% activity is retained; changing E213 to glutamine abolishes activity.
The position in space of the eight conserved residues were compared against the equivalent residues in spinach (Fig. 2B ▶) and P. aeruginosa (data not shown) KARI. Seven of these residues occupy similar positions in all three structures, while the position of the K155 side chain varies somewhat between the three structures. In the spinach KARI structures, this residue forms salt bridges with D217 and E213, while in the other two structures these two side chains are at the closest, 6 Å from the ɛ-amino group of K155.
When the six residues that belong to the K-domain are aligned in the three structures, the position of the histidine in the P-domain also aligns well, but as described previously, the position of K155 varies. The fixed position of the histidine again suggests that there is no general movement of the P-domain relative to the K-domain. The repositioning of the K155 side chain is linked to movement of the region between residues A153 and C156. Further evidence of this movement can be seen by the observation that the carbonyl oxygen of P154 forms a hydrogen bond with a water molecule that is bonded to the metal ion in the spinach KARI structure. This oxygen atom is too far away to form such a bond in either of the P. aeruginosa or E. coli structures.
To probe possible changes in the active site during catalysis, the coordinates for the metal ions and product from the 1QMG spinach structure and for NADPH (Fig. 2E ▶, taken from 1YVE because this ligand is partially degraded in 1QMG) were superimposed onto the protein coordinates of the E. coli enzyme. The position of NADPH (Fig. 2D ▶) is similar to that of NADP+ in the F420 NADP+-dependent oxidoreductase (Fig. 2A ▶) with the greatest displacement (~4.5 Å) in the disphosphate bridge connecting the two ribose moieties. A calculation of the molecular surface of E. coli KARI and an overlay of the ligands shows that the metal and substrate-binding sites are almost completely excluded from access to solvent (Fig. 2D ▶), but the binding site for NADPH is almost completely exposed. If NADPH were to be bound in this conformation, there would not be sufficient room to allow either the metal ions or substrate access to their binding sites. Thus, the conformation of the NADPH must be different when bound to the E. coli enzyme, or structural changes occur during the catalytic cycle. Changes must also occur to the protein after metal binding to allow the substrate access to the active site. The observations that the number of cysteine residues accessible to reaction with DTNB changes from five to two upon binding Mg2+ (Tyagi et al. 2005), and that crystals of free E. coli KARI dissolve when soaked in Mg2+ (R.G. Duggleby, unpubl.), supports this notion.
In general, the NADPH from spinach KARI shows a complementary conformation to the E. coli KARI surface. The only significant clash is the side chain of Q110, which pierces the adenine ring (Fig. 2D ▶ shows this ring buried in the surface). In spinach KARI, the equivalent residue is alanine, so there is no potential for a clash in this position. Q110 or the adenine ring therefore need to move, and possibly a hydrogen bond could form between the 6-amino group and the side chain of either Q110 or D113. In spinach KARI, the 2′-phosphate is held in place by hydrogen bonds between its phosphoryl oxygen atoms and the side chains of S167 and S165. The equivalent residues in E. coli KARI are S78 and R76. In the E. coli KARI structure, the S78 side chain would have to move by ~2 Å to form such a hydrogen bond; likewise, R76 would have to move by a similar amount to make the equivalent bond. Both movements look feasible, especially in light of our comparative analysis against the F420 NADP+-dependent oxidoreductase structure in this region. In spinach KARI, one of the 5′-phosphate oxygen atoms is held in place by a hydrogen bond to the amide nitrogen of T519 (D415 in E. coli KARI). The two structures superimpose well in this region, so little, if any, conformational change is required here upon NADPH binding. In spinach KARI, the amide nitrogens of S135 and Q136 participate in hydrogen bonding to the phosphoryl oxygen atoms of the nicotinamide phosphate. The equivalent atoms in A47 and Q48 of E. coli KARI would have to move by 1.7 and 3.6 Å, respectively, to form the same bonds. Furthermore, the nicotinamide group blocks access to the metal and substrate-binding sites in this conformation, so a rotation is required to enable catalysis. In summary, movements of ~2–4 Å in three regions would be required to accommodate the binding of NADPH into the free structure of E. coli KARI.
Discussion
In an earlier study we obtained crystals of E. coli KARI under 59 conditions (McCourt et al. 2004). Most of these crystals form within a few days and the commonly observed crystals belong to the space group P3121 with unit cell parameters of a = b = 148.2 Å and c = 211.2 Å. Crystals with related symmetry and space groups were also found, suggesting common crystal packing forces. While facile crystallization is normally desirable, in this work it has proved a hindrance because crystals form quickly but diffraction is weak or virtually absent, with no data set obtained to better than 2.5 Å resolution.
The crystals obtained in this study, prepared with NADPH added to the crystallization buffer, took many months to form. A possible reason for this is that NADPH was completely degraded over this period and that loss of NADPH is required for crystals to form in this buffer. Consistent with this proposal, no electron density was observed for NADPH when the structure was solved. This raises the question of why NADPH addition was necessary, and we did observe formation of some small crystals under the same conditions, but when NADPH was omitted (McCourt et al. 2004). We suggest that the presence of NADPH discourages growth of the “common” crystal form and that gradual NADPH degradation allows time for the alternative crystals, which diffract to a higher resolution, to appear.
When the first reported KARI crystal structure was published (Biou et al. 1997) it was apparent from amino acid sequence comparisons that this spinach enzyme structure might not be representative of all KARI structures. The plant enzyme contains a central stretch of ~140 residues that is missing from the protein in fungi and in most bacteria (Dumas et al. 2001). The shorter form is now (Ahn et al. 2003) termed Class I, while the long form is Class II. It was not until the structure of P. aeruginosa KARI was solved (Ahn et al. 2003) that it became clear that the central insert in the Class II enzyme is the structural equivalent of one C-terminal domain in a pair of a Class I KARI subunits. The C-terminal domain of the second Class I subunit corresponds to the C-terminal region of the Class II protein. Thus, the spinach monomer consists of an N-terminal domain with structural similarities to domains found in other pyridine nucleotide-dependent dehydrogenases, and a C-terminal domain derived by a duplication event. The evolutionary traces of this duplication are largely obliterated in the amino acid sequence, and it had not been recognized in the structure until an analysis by Taylor (2000) during a search for knots in proteins.
The KARI found in most bacteria is the Class I form typified by the P. aeruginosa enzyme (Ahn et al. 2002). However, some bacteria (such as E. coli) contain a type of KARI that resembles the plant Class II in length, but with a central insert that shows little homology to the plant insert. This raised the possibility of a third KARI class. Our determination of the structure of E. coli KARI clearly demonstrates that it has a characteristic Class II structure. Thus, despite the low-sequence homology between the central inserts of E. coli and spinach KARI, each is derived in the same way, from a duplication of the C-terminal one-third of the protein. This duplication is clearly evident when the two segments of E. coli KARI are overlaid (Fig. 3B ▶). The basic topology of these two segments is similar, and in some regions, even the positions of amino acid side chains are conserved (Fig. 3D ▶).
The similarity between spinach and E. coli KARI is even more striking when the geometry of the active site is examined. There is a constellation of eight polar amino acid side chains defining the active site (Fig. 2C ▶) with their identities and positions closely conserved between the spinach and E. coli proteins (Fig. 2B ▶). As might be expected, mutation of any one of these residues greatly decreases or obliterates catalytic activity (Tyagi et al. 2005).
In contrast to the unusual structural features of the K-domain, the P-domain folds into a structure that is reminiscent of that found in other pyridine nucleotide-dependent dehydrogenases. Although no nucleotide is bound in our structure, we have no doubt about where it would bind (Fig. 2A ▶). The 2′-phosphate that distinguishes NADPH from NADH would be within 4 Å of the guanidino group of R76, a residue that is known to control nucleotide specificity (Rane and Calvo 1997). This residue is at the end of a loop connecting two helices (Fig. 2A ▶) in a region that is disordered in monomers B and D (Fig. 1D ▶). We imagine that this region would become ordered in all four monomers when NADPH is bound.
The spinach KARI asymmetric unit is a tetramer, although the enzyme in solution is a dimer (Wessel et al. 1998), with each active site totally contained within a monomer (Biou et al. 1997). In contrast, the active site of P. aeruginosa KARI is formed from a pair of monomers and the enzyme is composed of six dimers arranged on the edges of a tetrahedron (Ahn et al. 2003). We expected that E. coli KARI would be a tetramer, based on its very high-sequence identity to the tetrameric S. typhimurium protein (Shematek et al. 1973; Hofler et al. 1975), although this implies a rather high solvent content in the crystal (McCourt et al. 2004).
As we began to solve the structure, we identified four monomers in the asymmetric unit. Attempts to locate further monomers failed and we conclude that there are four monomers only, with an open lattice in the crystal (Fig. 1A ▶) resulting in a solvent content of 67%. The arrangement of these monomers does not form a reasonable tetramer. However, by translating one of these dimers into an adjacent unit cell, a near symmetrical tetramer can be recovered (Fig. 1C ▶), and we propose that this is the biological unit.
Spinach KARI is a dimer in both structures thus far determined (although the asymmetric unit in the crystal consists of four monomers), while P. aeruginosa KARI is a dodecamer (but also has four subunits in the asymmetric unit). The tetrameric structure of E. coli KARI therefore represents a third variation in quaternary structure for these enzymes. Furthermore, the arrangements of the subunits that makeup these three are quite dissimilar. Analysis of the amino acid sequences and the residues that make up the interfaces shows the reasons for this. The segments that make up the flat interface in E. coli KARI are residues 263–279, 351–352, and 450–475. When compared with spinach KARI, only three of 45 residues are identical. These regions are where nearby insertions or deletions occur when the two sequences are compared. Thus, the conformation of the polypeptide in these regions is significantly altered. When the sequences of the K-domains of E. coli KARI and spinach KARI are compared, there is a single-residue deletion between 260 and 261, a three-residue deletion between 348 and 349,a two-residue insertion after 355, and an 11-residue insertion after 444. Thus, for all of these regions that form subunit interactions in E. coli KARI, there are major differences to spinach KARI that preclude such stabilizing interactions. In the K-domains of these two enzymes there is only one other insertion: three residues between 331 and 332 in E. coli KARI. This is part of a loop in the spinach enzyme structure that forms an embrace with the other subunit in dimer (Fig. 5C ▶). P. aeruginosa KARI is a shorter version of the enzymes, such that there are no equivalent residues to 351–352 and 450–475 and therefore no subunit interactions of the type that are observed in the E. coli enzyme.
The active site of E. coli KARI includes amino acid residues from both the P- and K-domains. None of the amino acids that form the active site are also involved in subunit interactions to form the tetramer. As described previously, the subunits are held in place by bulge-interface and flat-interface interactions. Such a configuration may serve two purposes. Firstly, to act as braces to keep the active site open and accessible to substrate and secondly, to facilitate breathing of the molecule during catalysis. It has been reported that in free spinach KARI, the N- and C-domains are mobile relative to each other and only upon the addition of NADPH do they become ordered in fixed orientations (Halgand et al. 1999). The C-terminal domains in the spinach KARI dimer make numerous contact points and are stable with respect to each other, but the N-terminal domains of spinach KARI are held only by the hinge linking the N-and C- terminal domains. This flexibility may be the reason that highly diffracting crystals of the free spinach enzyme have not been obtained (Dumas et al. 1994). On the other hand, the formation of the E. coli KARI tetramer creates additional links to stabilize the relative orientations of the P- and K-domains, thereby facilitating rigidity of the molecule.
Ahn et al. (2003) observe a difference of 7° in the relative orientation of the N- and C- terminal domains of P. aeruginosa KARI and spinach KARI when they are overlayed. They suggest that this difference is due to the fact that the P. aeruginosa KARI is crystallized as the free enzyme and spinach KARI is crystallized as a complex. If this were true, then the domains in E. coli KARI and P. aeruginosa KARI should superimpose to a high degree, as they are both crystallized as free enzyme. However, our observation is that the E. coli and spinach enzymes have the more similar domain orientations. Thus it is likely that the differences in domain orientation observed are due to the differing structural classes rather than conformational changes during the catalytic cycle. It may be shown ultimately that while KARI from different classes have similar amino acids in the active site, there are variations as the structures move from the resting state until they arrive at the first common transition state in the catalytic cycle.
The first step in the catalytic cycle is the independent binding of either Mg2+ or NADPH. The binding of substrate, catalysis, and release of products follows sequentially. The structure of E. coli KARI begins to show the molecular basis for this ordered mechanism. In the resting state, the active site is sufficiently structured and accessible to allow Mg2+ binding in the absence of NADPH. However, it is not immediately obvious how Mg2+ would bind in the presence of NADPH. There must be structural differences when NADPH is bound alone and when metal ions are also present. The binding of substrate follows after the two metal ions and NADPH have bound. A major conformational change occurs when Mg2+ binds, which opens access to the substrate-binding residues. In the two spinach KARI structures, where a product or a transition state analog is bound, the active site is completely closed. It would therefore seem likely that a similar closure occurs in the E. coli enzyme.
Conclusions
In this report, we have described the first crystal structure of a bacterial Class II KARI, from E. coli. The enzyme is a tetramer and each monomer is similar in structure to those of the tetrameric spinach enzyme. However, the tetramer interfaces are substantially different. Of particular interest is the structure of the larger domain comprising the C-terminal two-thirds of the protein. This domain shows strong evidence of a duplication, with the two halves having almost identical structures apart from the regions that form the two types of domain interface within the tetramer. This evidence for a duplication supports the earlier proposal by Ahn et al. (2003), who showed that the equivalent domain in the Class I KARI from P. aeruginosa is formed by two identical subunits.
The smaller domain is similar between the spinach, P. aeruginosa, and E. coli enzymes and is reminiscent of that found in other pyridine nucleotide-dependent dehydrogenases. This domain clearly contains the site where the NADPH substrate would bind. The active-site structure is highly conserved across all three enzymes, with only small positional differences in most of the residues. However, it is clear that some motion would be necessary to allow access by the substrates. Solving the structure of other E. coli KARI complexes that we have crystallized (McCourt et al. 2004) may help to define these movements.
Materials and methods
Protein expression, purification, and crystallization
Expression and purification of E. coli KARI was as described previously (Hill and Duggleby 1999), except that the version of the protein with a C-terminal hexahistidine tag was used in this study (McCourt et al. 2004). For long-term storage, the enzyme was kept at −70°C in 20 mM Na-HEPES buffer (pH 7.5). Prior to crystallization, the enzyme was thawed and diluted to 9 mg/mL by addition of an NADPH solution in the ratio of 10 mol NADPH per mol of enzyme subunits. For crystallization, the hanging drop method was used, where 3 μL of reservoir solution and 3 μL of protein/NADPH solution were combined in the drops and incubated at 17°C for 6 mo. The reservoir solution that yielded the crystals contained 1.6 M (NH4)2SO4 and 0.1 M Na-bicine (pH 9.0).
Data collection
Prior to data collection, crystals were transferred to a cryoprotectant solution that contained 2.1 M (NH4)2SO4 and 20% glycerol in 0.1 M Na-bicine (pH 9.0). Crystals were directly transferred in a loop to the cryostream that was set to 100 K. The data were collected with an FR-E X-ray generator operated at 45 kV and 45 mA, and an R-Axis IV++ imaging-plate area detector at the University of Queensland. The detector to crystal distance was 230 mm. A total of 300 oscillation images (width = 0.5°) were obtained with each image exposed for 180 sec. The images were scaled and merged using Crystalclear 1.3.5 (Rigaku/MSC) (Pflugrath 1999).
Structure determination and refinement
The structure was solved by molecular replacement using the program AMoRe (Navaza 1994). Data between 15 and 6 Å resolution were used in these calculations. The electron density map was interpreted using the program MAID (Levitt 2000), which allowed the automatic fitting of >90% of the backbone polypeptide and amino acid side chains. Manual model building was carried out using O (Jones et al. 1991). Refinement, including simulated annealing and bulk solvent correction, was undertaken using the CNS software suite (Brünger et al. 1988). Initially, the model was refined with strict noncrystallographic symmetry restraints, but in the final rounds these were removed. Stereochemistry data and R factors for the final model are presented in Table 1. The stereo-chemical quality of the protein model was analyzed with PRO-CHECK (Laskowski et al. 1993). Domain superimpositions were calculated using the program Lsq_improve in O (Jones et al. 1991). Monomer superimpositions were calculated in Insight2000.1 (http://www.accelrys.com) using the Cα atoms defined by Lsq_ improve. Figures were prepared with MOLSCRIPT (Kraulis 1991), Raster3D (Merritt and Bacon 1997), GraFit (http://www.erithacus.com), PyMOL (http://www.pymol.org), and WebLab (http://www.accelrys.com). The atomic coordinates and the structure factors have been deposited in the Protein Data Bank under the accession code 1YRL.
Acknowledgments
This work was supported by the Australian Research Council grant number DP0208682. We thank Karl Byriel for assistance with data collection and Ragothaman Yennamalli for setting up software.
Abbreviations
KARI, ketol-acid reductoisomerase
rmsd, root mean square deviation
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051791305.
References
- Ahn, H.J., Eom, S.J., Yoon, H.-J., Lee, B.L., Cho, H., and Suh, S.W. 2003. Crystal structure of class I acetohydroxy acid isomeroreductase from Pseudomonas aeruginosa. J. Mol. Biol. 328 505–515. [DOI] [PubMed] [Google Scholar]
- Biou, V., Dumas, R., Cohen-Addad, C., Douce, R., Job, D., and Pebay-Peyroula, E. 1997. The crystal structure of plant acetohydroxy acid isomeroreductase complexed with NADPH, two magnesium ions and a herbicidal transition state analog determined at 1.65 Å resolution. EMBO J. 16 3405–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1988. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54 905–921. [DOI] [PubMed] [Google Scholar]
- Dumas, R., Job, D., Douce, R., Pebay-Peyroula, E., and Cohen-Addad, C. 1994. Crystallization and preliminary crystallographic data for acetohydroxy acid isomeroreductase from Spinacia oleracea. J. Mol. Biol. 242 578–581. [DOI] [PubMed] [Google Scholar]
- Dumas, R., Biou, V., Halgand, F., Douce, R., and Duggleby, R.G. 2001. Enzymology, structure and dynamics of acetohydroxy acid isomero-reductase. Accounts Chem. Res. 34 399–408. [DOI] [PubMed] [Google Scholar]
- Halgand, F., Dumas, R., Biou, V., Andrieu, J.P., Thomazeau, K., Gagnon, J., Douce, R., and Forest, E. 1999. Characterization of the conformational changes of acetohydroxy acid isomeroreductase induced by the binding of Mg2+ ions, NADPH, and a competitive inhibitor. Biochemistry 38 6025–6034. [DOI] [PubMed] [Google Scholar]
- Hill, C.M. and Duggleby, R.G. 1999. Purified recombinant Escherichia coli ketol-acid reductoisomerase is unsuitable for use in a coupled assay of acetohydroxyacid synthase activity due to an unexpected side reaction. Protein Expr. Purif. 15 57–61. [DOI] [PubMed] [Google Scholar]
- Hofler, J.G., Decedue, C.J., Luginbuhl, G.H., Reynolds, J.A., and Burns, R.O. 1975. The subunit structure of α-acetohydroxyacid isomeroreductase from Salmonella typhimurium. J. Biol. Chem. 250 877–882. [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1994. Searching protein structure databases has come of age. Proteins 19 165–173. [DOI] [PubMed] [Google Scholar]
- Jones, S. and Thornton, J.M. 1996. Principles of protein–protein interactions. Proc. Natl. Acad. Sci. 93 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Krissinel, E. and Henrick, K. 2004. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60 2256–2268. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26 283–291. [Google Scholar]
- Levitt, D.G. 2000. A new software routine that automates the fitting of protein X-ray crystallographic electron-density. Acta Crystallogr. D Biol. Crystallogr. 57 1013–1019. [DOI] [PubMed] [Google Scholar]
- Lindqvist, Y., Schneider, G., Ermler, U., and Sundström, M. 1992. Three-dimensional structure of transketolase, a thiamine diphosphate dependent enzyme, at 2.5 Å resolution. EMBO J. 11 2373–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews, B.W. 1968. Solvent content of protein crystals. J. Mol. Biol. 33 491–497. [DOI] [PubMed] [Google Scholar]
- McCourt, J.A., Tyagi, R., Guddat, L.W., Biou, V., and Duggleby, R.G. 2004. Facile crystallization of Escherichia coli ketol-acid reductoisomerase. Acta Crystallogr. D Biol. Crystallogr. 60 1432–1434. [DOI] [PubMed] [Google Scholar]
- Merritt, E.A. and Bacon, D.J. 1997. Raster3D: Photo-realistic graphics. Methods Enzymol. 277 946–950. [DOI] [PubMed] [Google Scholar]
- Muller, Y.A. and Schulz, G.E. 1993. Structure of the thiamine- and flavin-dependent enzyme pyruvate oxidase. Science 259 965–967. [DOI] [PubMed] [Google Scholar]
- Peitsch, M.C. 1996. ProMod and Swiss-Model: Internet-based tools for automated comparative protein modeling. Biochem. Soc. Trans. 24 274–279. [DOI] [PubMed] [Google Scholar]
- Pflugrath, J.W. 1999. The finer things in X-ray diffraction data collection. Acta Crystallogr. D Biol. Crystallogr. 55 1718–1725. [DOI] [PubMed] [Google Scholar]
- Rane, M.J. and Calvo, K.C. 1997. Reversal of the nucleotide specificity of ketol acid reductoisomerase by site-directed mutagenesis identifies the NADPH binding site. Arch. Biochem. Biophys. 338 83–89. [DOI] [PubMed] [Google Scholar]
- Schwede, T., Kopp, J., Guex, N., and Peitsch, M.C. 2003. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 31 3381–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shematek, E.M., Diven, W.F., and Arfin, S.M. 1973. A kinetic study of α-acetohydroxy acid isomeroreductase from Salmonella typhimurium. Arch. Biochem. Biophys. 158 132–138. [DOI] [PubMed] [Google Scholar]
- Taylor, W.R. 2000. A deeply knotted protein structure and how it might fold. Nature 406 916–919. [DOI] [PubMed] [Google Scholar]
- Thomazeau, K., Dumas, R., Halgand, F., Forest, E., Douce, R., and Biou, V. 2000. Structure of spinach acetohydroxyacid isomeroreductase complexed with its reaction product dihydroxymethylvalerate, manganese and (phospho)-ADP-ribose. Acta Crystallogr. D Biol. Crystallogr. 56 389–397. [DOI] [PubMed] [Google Scholar]
- Tyagi, R., Lee, Y.-T., Guddat, L.W., and Duggleby, R.G. 2005. Probing the mechanism of the bifunctional enzyme ketol-acid reducto-isomerse by site-directed mutagenesis of the active site. FEBS J. 272 593–602. [DOI] [PubMed] [Google Scholar]
- Warkentin, E., Mamat, B., Sordel-Klippert, M., Wicke, M., Thauer, R.K., Iwata, M., Iwata, S., Ermler, U., and Shima, S. 2001. Structures of F420H2:NADP+ oxidoreductase with and without its substrates bound. EMBO J. 20 6561–6569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessel, P.M., Biou, V., Douce, R., and Dumas, R. 1998. A loop deletion in the plant acetohydroxy acid isomeroreductase homodimer generates an active monomer with reduced stability and altered magnesium affinity. Biochemistry 37 12753–12760. [DOI] [PubMed] [Google Scholar]