

Abstract
Semisynthetic cephalosporins are synthesized from 7-amino cephalosporanic acid, which is produced by chemical deacylation or by a two-step enzymatic process of the natural antibiotic cephalosporin C. The known acylases take glutaryl-7-amino cephalosporanic acid as a primary substrate, and their specificity and activity are too low for cephalosporin C. Starting from a known glutaryl-7-amino cephalosporanic acid acylase as the protein scaffold, an acylase gene optimized for expression in Escherichia coli and for molecular biology manipulations was designed. Subsequently we used error-prone PCR mutagenesis, a molecular modeling approach combined with site-saturation mutagenesis, and site-directed mutagenesis to produce enzymes with a cephalosporin C/glutaryl-7-amino cephalosporanic acid catalytic efficiency that was increased up to 100-fold, and with a significant and higher maximal activity on cephalosporin C as compared to glutaryl-7-amino cephalosporanic acid (e.g., 3.8 vs. 2.7 U/mg protein, respectively, for the A215Y-H296S-H309S mutant). Our data in a bioreactor indicate an ~90% conversion of cephalosporin C to 7-amino-cephalosporanic acid in a single deacylation step. The evolved acylase variants we produced are enzymes with a new substrate specificity, not found in nature, and represent a hallmark for industrial production of 7-amino cephalosporanic acid.
Keywords: cephalosporin C, 7-amino cephalosporanic acid, protein engineering, directed evolution, site-saturation mutagenesis, enzymes, active sites, structure/function studies, protein sequencing, modification, mass spectrometry, protein structure prediction, kinetics
Semisynthetic cephalosporins are the most widely used antibiotics and are primarily synthesized from 7-amino cephalosporanic acid (7-ACA), which is usually obtained by chemical deacylation of the natural antibiotic cephalosporin C (CephC). The chemical route includes, however, several expensive steps and requires treatment of toxic wastes. A two-step enzymatic route can also be used that, in two separate reactors, uses D-amino acid oxidase and glutaryl-7-amino cephalosporanic acid (gl-7ACA) acylase activity (see Scheme 1, below): Although this route solves the environmental safety problems, it is also expensive and not entirely satisfactory for industrial production. Therefore, enzymatic conversion of CephC to 7-ACA is of great interest to cephalosporin antibiotics manufacturers (an annual worldwide market of ~400 million US dollars has been estimated) (Pilone and Pollegioni 2002). The greatest hindrance to enzymatic industrial production is that acylase takes glutaryl-7-ACA (gl-7ACA) as a primary substrate, and its specificity (and activity) is too low for CephC.
Glutaryl-7-ACA acylases are members of the N-terminal hydrolases (Ntn) class of hydrolytic enzymes. The gene structure of the open reading frame (ORF) of the members of this class consists of a signal peptide, followed by an α-subunit, a spacer sequence (which is not present in the acylase under investigation), and a β-subunit. The single, inactive, precursor polypeptide is post-translationally modified into a mature heterodimeric αβ enzyme by an autoproteolytic cleavage upon folding, generating a new N-terminal residue. The other members of this superfamily are penicillin G acylase, penicillin V acylase, class II glutamine amidotransferases, proteasome β-subunit, and glycosylasparaginase (Murzin et al. 1995).
According to their substrate specificity and sequence conservation, known glutaryl acylases have been divided into five classes: These enzymes have an activity to CephC that varies from 0% to 4% relative to gl-7ACA (Oh et al. 2003 and references therein). Members of class I (e.g., P130 from Pseudomonas sp. 130) and class II (e.g., N176 from Pseudomonas sp. N176) show the highest activity to CephC.
Various attempts have previously been undertaken to directly convert CephC into 7-ACA by a single-step enzymatic reactor: mutagenesis of penicillin G acylase to modify its substrate specificity (Oh et al. 2004); random mutagenesis followed by saturation mutagenesis of residue N266 of Pseudomonas SY-77 acylase (Otten et al. 2004); and site-saturation mutagenesis of residues identified by ligand-bound structures of Pseudomonas diminuta acylase (Oh et al. 2003). In fact, up to now only the three-dimensional structure of class I acylase from P. diminuta (synonymous Brevundimonas diminuta) has been resolved (Kim et al. 2000). Because of the availability of its structure in complex with gl-7ACA and glutarate, investigations have focused on the mutagenesis of this acylase, reaching an up to 7.9-fold increase in deacylation activity to CephC compared to the wild type (Oh et al. 2003). This represents a significant accomplishment in improving the deacylation activity on CephC but is far from being considered economically applicable for the industrial production of 7-ACA.
In this study, we used a synthetic gene (named VAC) designed on the basis of the primary sequence of glutaryl acylase from Pseudomonas N176 (Aramori et al. 1991) as the starting scaffold to develop an enzyme specifically more active on CephC and that had all the properties required for industrial bioconversion. This goal was achieved using a combination of random mutagenesis, molecular modeling, and site-saturation and site-directed mutagenesis approaches. The acylase enzymes that evolved represent the first known successful production of an effective CephC acylase, and their production shed light on some of the structural determinants responsible for substrate specificity in this class of enzymes.
Results
Construction of error-prone PCR (EP-PCR) libraries and selection of VAC mutants with altered substrate specificity
The conditions for the construction of the libraries were optimized using the synthetic VAC gene as template (Fig. 1 ▶). For each set of conditions tested, a library of ~10,000 clones was generated and up to 2000 independent clones obtained for each PCR condition were screened for acylase activity on CephC and gl-7ACA as substrates. Sequencing of at least 1000 bp of seven randomly picked clones from each bank revealed that condition 2 reached the optimal mutation frequency (ranging from 0.11% to 0.23%): The observed mutation frequencies resulted in ≤1-bp mutation per mutant for condition 1 and 1–3-bp mutations per mutant for condition 2. The mutant gene libraries were cloned in the pET24Δ plasmid and expressed in Escherichia coli BL21(DE3)pLysS cells.
Figure 1.

Nucleotide sequence of VAC cDNA and corresponding amino acid primary structure. The residues modified in the evolved VAC variants are highlighted in bold.
Among the mutants generated during the first round of mutagenesis, higher activity on CephC was detected in the crude lysate of four E. coli cultures expressing mutant clones M1–M4 (Table 1). The cDNAs coding for the isolated mutants contained one to three mutations and resulted in a single point substitution for mutants M1–M3 (F270M, A215Y, and D416Y) and two substitutions for mutant M4 (D416Y-H417Y) (Table 1). Starting from the M4 mutant, a second round of random mutagenesis was performed. By means of the activity assay, two mutant clones were selected: M5 and M6, both exhibiting a very low activity on gl-7ACA. The DNA sequence analysis showed the presence of three additional mutations in both clones, yielding two additional amino acid substitutions (Table 1). Finally, starting from the M2 mutant, a third round of random mutagenesis was performed. In this round, two clones (M7 and M8) could be isolated that showed a comparatively lower activity on gl-7ACA than on CephC. These mutants contained a total of four nucleotide mutations (Table 1).
Table 1.
DNA and amino acid substitutions in evolved acylase mutants (obtained by random mutagenesis and screening for enhanced activity on CephC)
| Clone | Base | Base substitution | Position | AA position | PCR conditions | AA substitution | |
| First round (from wild-type VAC) | |||||||
| M1 | 808 | T → A | 1 | } | 270 | 1 | F → M |
| 810 | T → G | 3 | |||||
| M2 | 643 | G → T | 1 | } | 215 | 1 | A → Y |
| 644 | C → A | 2 | |||||
| 645 | G → C | 3 | |||||
| M3 | 1246 | G → T | 1 | 416 | 1 | D → Y | |
| M4 | 1246 | G → T | 1 | 416 | 1 | D → Y | |
| 1249 | C → T | 1 | } | 417 | H → Y | ||
| 1251 | C → T | 3 | |||||
| Second round (from M4 variant) | |||||||
| M5 | 130 | A → G | 1 | 44 | 2 | I → V | |
| 145 | G → T | 1 | 49 | E → stop | |||
| 315 | C → T | 3 | 105 | Silent | |||
| 1246 | G → T | 1 | 416 | D → Y | |||
| 1249 | C → T | 1 | } | 417 | H → Y | ||
| 1251 | C → T | 3 | |||||
| M6 | 64 | T → C | 1 | 22 | 2 | S → P | |
| 147 | A → G | 3 | 49 | Silent | |||
| 1180 | A → C | 1 | 394 | T → P | |||
| 1246 | G → T | 1 | 416 | D → Y | |||
| 1249 | C → T | 1 | } | 417 | H → Y | ||
| 1251 | C → T | 3 | |||||
| Third round (from M2 variant) | |||||||
| M7 | 266 | A → C | 2 | 89 | 2 | E → A | |
| 643 | G → T | 1 | } | 215 | |||
| 644 | C → A | 2 | A → Y | ||||
| 645 | G → C | 3 | |||||
| M8 | 643 | G → T | 1 | } | 215 | 2 | |
| 644 | C → A | 2 | A → Y | ||||
| 645 | G → C | 3 | |||||
| 809 | T → C | 2 | 270 | F → S | |||
The amino acid numbering refers to the sequence of the starting acylase (Aramori et al. 1991); see also Figure 1 ▶.
Selected VAC variants identified in the three rounds of random mutagenesis were expressed and purified as reported in the Materials and Methods section. However, whereas ~0.8 mg of purified protein/g of E. coli cell paste was recovered for the M1–M3 mutants, a fivefold lower yield was obtained for the M6 and M7 mutants, a 15-fold lower yield was obtained for the M5 and M8 mutants, and ~0.02 mg of purified protein/g cell paste was recovered following the HiTrap chelating chromatography for the M4 variant. Western blot analysis indicates that the low amount obtained for these purified VAC enzymes is due to a low expression level. A significant increase in the expression yield of the M8 VAC mutant was obtained when the cells were grown for an additional 21 h after adding isopropyl β-D-thiogalacto-pyranoside (IPTG; up to 1.1 mg of purified enzyme/g cell paste). On the other hand, alterations in the expression conditions—i.e., changing the moment of induction, the concentration of inducer (≤1 mM), the temperature of growth after induction (15°, 25°, and 30°C), and the time of cell collection after induction (≤21 h)—did not improve the enzyme recovery for the M4 (D416Y-H417Y) VAC variant. Unfortunately, the low expression of the M4 variant does not allow its scale-up application.
To investigate the changes in substrate specificity, we determined the activity of VAC variants on gl-7ACA and CephC as substrate (see Table 2). The M4 mutant (D416Y-H417Y) shows the most significant increase in activity on CephC (sixfold), followed by M2 and M8 mutants (A215Y and A215Y-F270S); an approximately twofold decrease of the Km parameter is observed for M1 (F270M), M2, and M3 (D416Y) mutants compared to the wild-type VAC. Concerning the substrate gl-7ACA, the residual activity was practically negligible for M5 (I44V-E49stop-D416Y-H417Y) and M6 (S22P-T394P-D416Y-H417Y) mutants (paralleled by a significant increase in Km), whereas the changes observed for the VAC mutants with a higher efficiency on CephC (M1, M2, and M9) (see Table 2) were more limited.
Table 2.
Kinetic properties of purified wild type and mutants of VAC on CephC and gl-7ACA as substrate
| CephC | gl-7ACA | |||||
| Vmax (U/mg) | Km (mM) | Vmax/Km | Vmax (U/mg) | Km (mM) | Vmax/Km | |
| Wild type | 0.7 | 11 | 0.06 | 24.2 | 1.6 | 15.1 |
| EP-PCR | ||||||
| M1 (F270M) | 0.7 | 6.5 | 0.11 | 24.6 | 2.7 | 9.1 |
| M2 (A215Y) | 1.8 | 6.9 | 0.26 | 16 | 1.7 | 9.4 |
| M3 (D416Y) | 0.7 | 7.2 | 0.09 | 4.9 | 1.7 | 2.9 |
| M4 (D416Y-H417Y) | 4.2 | 13.0 | 0.32 | 1.2 | 9.0 | 0.1 |
| M5 (I44V-E49stop-D416Y-H417Y) | 0.13 | 32.0 | <0.01 | 0.06 | 25.0 | <0.01 |
| M6 (S22P-T394P-D416Y-H417Y) | 0.02 | 24.5 | <0.01 | 0.11 | 37.2 | <0.01 |
| M7 (E89A-A215Y) | 0.10 | 18.9 | 0.01 | 0.66 | 1.1 | 0.6 |
| M8 (A215Y- F270S) | 1.56 | 10.1 | 0.15 | 23.8 | 2.0 | 12.1 |
| Site-saturation mutagenesis | ||||||
| A215E | 0.16 | 3.2 | 0.05 | 2.0 | 1.0 | 2.0 |
| A215F | 0.21 | 7.7 | 0.03 | 3.3 | 0.9 | 3.9 |
| A215L | 0.75 | 9.5 | 0.08 | 6.2 | 1.7 | 3.6 |
| A215V | 0.78 | 8.3 | 0.09 | 12.5 | 1.2 | 10.4 |
| Y271V | 0.12 | 2.7 | 0.04 | 13.4 | 1.9 | 7.1 |
| Y271F | 0.32 | 8.0 | 0.04 | 3.3 | 1.2 | 2.8 |
| H296N | 0.27 | 5.2 | 0.05 | 4.3 | 2.8 | 1.5 |
| H296S | 0.67 | 6.7 | 0.10 | 2.0 | 0.7 | 2.9 |
| H296T | 0.20 | 4.8 | 0.04 | 0.7 | 2.0 | 0.3 |
| H296F | 0.05 | 4.1 | 0.01 | 1.7 | 1.9 | 0.9 |
| H309Y | 0.24 | 6.3 | 0.04 | 3.0 | 1.0 | 3.1 |
| H309S | 2.20 | 24.4 | 0.09 | 7.7 | 4.6 | 1.7 |
| H417Y | 0.05 | 12.3 | <0.01 | 14.8 | 4.3 | 3.4 |
| A215Y-H296S | 0.66 | 10 | 0.07 | 2.2 | 4.0 | 0.6 |
| H296S-H417M | 0.04 | 1.7 | 0.03 | 0.5 | 5.5 | 0.1 |
| Combination of multiple mutations | ||||||
| A215Y-H309S | 3.9 | 17.2 | 0.23 | 41.7 | 0.9 | 44.4 |
| H296S-H309S | 3.0 | 12.2 | 0.24 | 4.8 | 6.9 | 0.7 |
| A215Y-H296S-H309S | 3.8 | 17.8 | 0.21 | 2.7 | 2.5 | 1.1 |
The Vmax and Km values were determined by measuring the initial rate of hydrolysis on a range of substrate concentrations and with a fixed amount of enzyme (see Materials and Methods).
Construction of the three-dimensional model of VAC and docking of cephalosporin C
Since the three-dimensional (3D) structure of VAC was not determined, a model of the protein was built by comparative modeling using the Swiss-model Web facility (Guex and Peitsch 1997; Schwede et al. 2003). Acylase from Pseudomonas diminuta (PDB code 1JVZ), a class I acylase, was chosen as template for both VAC subunits (Kim and Hol 2001). Because of the low identity in the primary structure (18.8%), the sequence alignment between VAC and 1JVZ was performed using the program T-coffee (Notredame et al. 2000) and proofread manually. The resulting alignment was then submitted to the Swiss-model facility; the two subunits were individually modeled. Using this procedure, we built a 3D-structure model (Fig. 2A,B ▶) including 87% of the amino acids of the α-subunit (208 vs. 239 residues, i.e., lacking the N- and C-terminal residues) and ~100% of those of the β-subunit (534 vs. 535 residues). The lower degree of success for the α-subunit is due to the presence of several insertions in the primary structure of VAC compared to the corresponding subunit of 1JVZ acylase (239 and 155 residues, respectively). Results of the modeling procedure are summarized in the legend to Figure 2 ▶.
Figure 2.
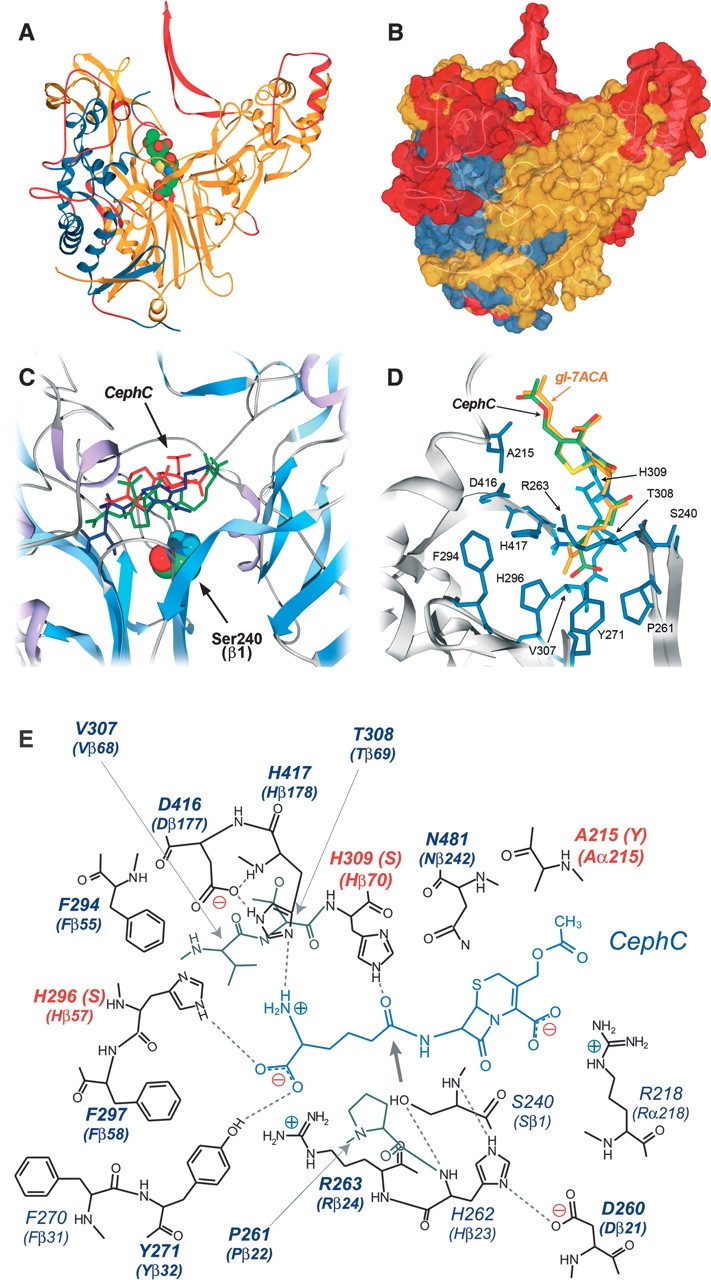
Three-dimensional model of VAC. (A) Ribbon representation and (B) surface representation of VAC model. (Blue) α-subunit; (yellow) β-subunit; (red) portion of the protein modeled with lower accuracy. The substrate (gl-7ACA) is colored by atom type, and the atoms are depicted by their van der Waals radii. Modeling statistics: 144 amino acids of the α-subunit (60.2%) and 465 amino acids of the β-subunit (86.9%) were modeled by homology; 64 and 69 amino acids were difficult to model for subunits α and β, respectively; 31 and 1 amino acids were not modeled for subunits α and β, respectively. (C) Results of CephC binding to VAC active site by the Autodock 3.0 program. The three substrate conformations possessing the lowest docked energy are depicted in red, blue, and green. (D) Model manually proofread for binding of CephC (green) and gl-7ACA (yellow) at the active site of the VAC model. (E) Schematic representation of the hydrogen-bond network in the CephC–VAC model complex. The positions subjected to saturation mutagenesis are shown in bold. The hydrogen bonds are marked as dotted lines; the thick arrow indicates the position of the site of CephC hydrolysis respect to the OH-group of S240. The residues modified in the best VAC variants are highlighted in red.
The binding mode of the substrate CephC in the VAC active site was simulated using Autodock3 software (Goodsell et al. 1996). Analysis of docking solutions showed that the substrate was positioned in the active site, but with the wrong orientation (i.e., with the amino-adipic side chain of CephC pointing toward the solvent) (see Fig. 2C ▶). Since biochemical studies demonstrate that VAC binds CephC in the configuration required for catalysis, we modeled both substrates (CephC and gl-7ACA) in the VAC active site (see Fig. 2D,E ▶). The resulting model shows that some residues are very close to the substrate molecules, in particular, H309 belonging to the large subunit that is at ~1.2 Å from the substrate: These steric clashes prevent Autodock from positioning the substrate in the active site with the correct orientation. Two observations suggest that the overall geometry of the active site can be taken into consideration. At first, the distance between the OH-group of S240 and the C15=O group of gl-7ACA (the site of hydrolysis) is 3.42 Å in the structure of P. diminuta acylase (Kim and Hol 2001) and 3.46 and 3.41 Å in our model with gl-7ACA and CephC, respectively. Second, the α-amino group of S240 is at hydrogen-bond distance from the (NE1) side chain of H262, and the nitrogen NE2 of this residue makes a hydrogen bond with the side chain of D260 (Fig. 2E ▶). The network of hydrogen bonds seen in the VAC model closely matches the network in the active site of penicillin G acylase structure, which is a strong indication that both enzymes adopt the same catalytic mechanism (Duggleby et al. 1995).
From the active-site model of VAC, the following positions were considered important for substrate binding and specificity (Fig. 2E ▶): 215, 261, 263, 271, 294, 296, 297, 307, 308, 309, 416, and 417.
Selection of VAC mutants obtained by site-saturation mutagenesis
Site-saturation mutagenesis was performed on the positions reported above using the QuikChange kit and starting from the wild-type VAC cDNA. For each position, ~200 clones were screened, a number that gives a probability of 96% that every single amino acid is introduced. As shown in Table 3, the saturation was also performed starting from selected single-point and double-point mutants obtained from the random and site-saturation mutagenesis approaches. The 15 variants most active on CephC were purified, and their kinetic parameters were compared to those of wild-type VAC. Interestingly, the increase in kinetic efficiency on CephC was due to a decrease in Km for mutants at position 215, as well as for variants at position 296, and to a threefold increase in Vmax for the H309S variant. In all cases, the decrease in kinetic efficiency on gl-7ACA was due to a decrease in Vmax (up to 36-fold for H296T), while the Km parameter was altered to a lesser extent (less than threefold); see Table 2.
Table 3.
Site-saturation mutagenesis of VAC variants
| Starting material | Amino acid position | No. of selected mutants | Selected mutants |
| Wild type | 215 | 250 | E, F, L, V |
| 261 | 230 | — | |
| 263 | 200 | — | |
| 271 | 242 | V, F | |
| 294 | 200 | ||
| 296 | 220 | N, S, T, F | |
| 307 | 220 | — | |
| 308 | 200 | — | |
| 309 | 220 | Y, S | |
| 416 | 290 | — | |
| 417 | 180 | Y | |
| H296S | 215 | 210 | Y |
| 271 | 200 | — | |
| 294 | 200 | — | |
| 297 | 200 | — | |
| 417 | 200 | M | |
| A215Y-H296S | 416 | 220 | — |
| D416Y-H417Y | 271 | 200 | — |
| H417Y | 263 | 215 | — |
The positions subjected to saturation mutagenesis were identified by modeling of CephC to the active site of the VAC model (see Fig. 2 ▶).
Introduction of multiple mutations by site-directed mutagenesis
The changes in substrate specificity introduced in VAC mutants obtained by EP-PCR and saturation mutagenesis were used as starting point for the production of enzyme variants containing multiple mutations. Up to now, the highest increase in kinetic efficiency on CephC was observed for the A215Y (M2), D416Y-H417Y (M4), and A215Y-F270S (M8) variants obtained by the EP-PCR procedure, and for the H296S, H309S, and A215V variants obtained by site-saturation mutagenesis (the highest activity with the H309S one) (see Table 2). With the exception of only the D416Y-H417Y mutant (whose expression level is too low for practical application), all these VAC variants show a Vmax value higher with gl-7ACA than with CephC (see Fig. 3 ▶; Table 2); therefore, they cannot be considered a “true” CephC acylase.
Figure 3.
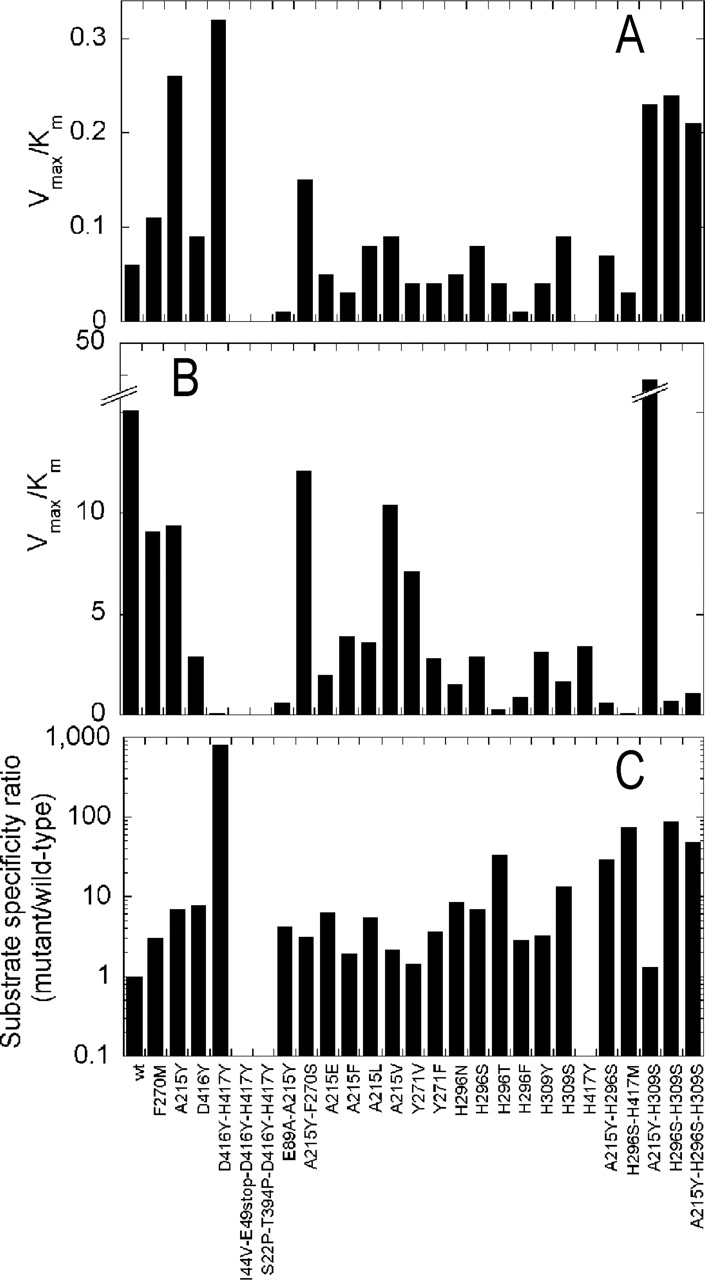
Catalytic efficiency (Vmax/Km ratios calculated from the data reported in Table 2) of wild type and mutants of VAC. The values have been determined on CephC (A) and gl-7ACA (B) as substrates, and the ratio of the specificity constants (C) on the two substrates [(Vmax/Km)CephC/(Vmax/Km)gl-7ACA] between the mutants and the wild-type VAC.
During the preliminary screening, no activity was detected in any of the multiple mutants obtained starting from the D416Y-H417Y mutant, probably as a result of the low expression yield of this mutant VAC (see above). The A215Y, H296S, and H309S substitutions were combined, and the double A215Y-H309S, A215Y-H296S, and H296S-H309S mutants, as well as the triple A215Y-H296S-H309S mutant, were prepared by site-directed mutagenesis; no significant changes in expression yield of these VAC variants were observed compared to the corresponding single-point mutants. Among the mutants produced, the highest activity on CephC was obtained for the A215Y-H309S variant, while the lowest Km was determined for the H296S-H309S one. It is noteworthy that the triple A215Y-H296S-H309S VAC variant shows a higher Vmax value with CephC than with gl-7ACA. All combined mutants exhibit an increase in kinetic efficiency on CephC compared to the wild-type VAC: This change is also paralleled by a significant decrease in the kinetic efficiency on gl-7ACA (up to 22-fold for the H296S-H309S mutant). The only exception is the A215Y-H309S mutant, which shows a significant increase in kinetic efficiency on gl-7ACA due to both a higher activity and substrate affinity (Table 2).
Properties of evolved VAC variants
All VAC mutant preparations showed a >90% purity (as indicated by SDS-PAGE analysis), were αβ-heterodimeric enzymes (83 ± 3 kDa according to gel-permeation chromatography at ~10 mg/mL protein samples), and were stable for several months when stored at −20°C. N-terminal sequencing of the two subunits isolated by SDS-PAGE electrophoresis indicates that the α-subunit lacks the N-terminal methionine (2TMA-ANTDRA…), while the sequence of the β-subunit is unchanged (240SNNWAVAPGR…).
Glutaryl acylases show a classical inhibition by “substrate,” in that their activity decreases at high substrate concentration: Such an inhibition can be observed for the wild-type VAC with both CephC and gl-7ACA as substrate (see Fig. 4A,B ▶). This represents a main limitation for bioconversion, which is usually performed in the 1%–5% (w/v) substrate concentration range (corresponding to ~25–120 mM) (Pilone and Pollegioni 2002). Interestingly, the VAC variants with increased activity on CephC are inhibited at higher substrate concentrations than wild-type VAC. In particular, the mutants obtained by a combination of EP-PCR and site-saturation mutagenesis do not show any substrate inhibition (see Fig. 4A,B ▶ for H296S-H309S). Analogously, the evolved VAC variants are less sensitive to inhibition by the product; for example, the Ki determined for the final product 7-ACA is higher for the A215Y-H309S mutant than for wild-type VAC (6.2 and 3 mM, respectively).
Figure 4.
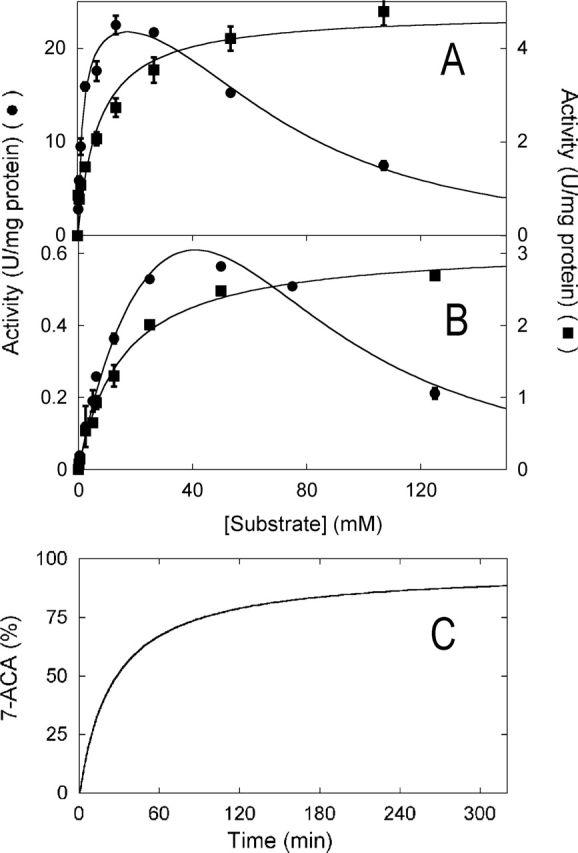
(A,B) Concentration dependence of the activity of wild-type and mutant VAC enzymes. Michaelis-Menten plot of the activity of the wild-type (•) and H296S-H309S (▪) variant of VAC on gl-7ACA (A) and CephC (B) as substrate. The lines represent the best fits of the experimental data obtained using a classical Michaelis-Menten equation for the H296S-H309S mutant and a substrate inhibition equation (when there is only one substrate) (Cleland 1983) for the wild-type VAC. Where the error bars are not shown, the error is smaller than the symbols used. (C) Time course of 7-ACA production by the H296S-H309S VAC variant. The experimental conditions are 100 mM potassium phosphate buffer (pH 8.5) (pH-stat with 2 N NaOH), 20 g/L (48 mM) CephC, and 250 VAC units/g CephC.
The pH dependence of the activity of evolved VAC variants on gl-7ACA in the pH range 5–9 resembles that determined for the wild-type enzyme: A 40% increase in activity is evident when shifting the pH from 8.0 to 9.0 (data not shown). On the other hand, the pH dependence of the activity significantly distinguishes the reaction of VAC on CephC from that on gl-7ACA: The activity measured at pH 9.0 on CephC is fourfold higher than at pH 8.0 for wild type, as well as for the H296S-H309S VAC variant. The stability of the wild type and variants of VAC after 30, 90, and 180 min of incubation at 25°C and in the pH range 5–10 is similar.
Experiments on the temperature dependence of the enzymatic activity show that the wild-type, A215Y, and A215Y-F270S forms of VAC exhibit an increase in activity up to 40°C, while the other VAC variants tested show a similar activity in the temperature range 20°–40°C. The effect of temperature on the protein stability, detected by measuring the residual activity after 30 min of incubation, was similar among the wild type and variants of VAC.
Cephalosporin C bioconversion
We investigated the ability of the best VAC variants obtained by the combined evolution approach to convert CephC to 7-ACA using a stirred reactor and the enzymes in the free form. The bioconversion time course was followed by the pH-stat assay and by quantification of the reaction products using HPLC chromatography. As demonstrated in Figure 4C ▶ for the H296S-H309S and at pH 8.5, an ~90% production of 7-ACA at times ≥180 min was observed under conditions resembling those used in the industrial application (at 20 g/L of CephC corresponding to ~48 mM, and 250 VAC units/g of CephC).
Discussion
In this paper, we describe the evolution of a glutaryl acylase to produce a catalyst for the productive hydrolysis of CephC to 7-ACA in a single enzymatic step. First, we identified a glutaryl acylase from Pseudomonas N176 (a class II acylase) as the starting protein scaffold, because its activity on CephC represents ~4% of that measured on gl-7ACA (Oh et al. 2003). In order to optimize the codon usage for E. coli expression, to facilitate the subsequent DNA manipulation, and to increase the activity on CephC, a synthetic gene (VAC) was designed whose nucleotide sequence shows a 77.5% identity with the original gene (Aramori et al. 1991). The VAC gene was then inserted in a shorter version of the pET24 expression plasmid (pET24Δ-VAC plasmid), which can be more readily manipulated and maintained in E. coli cells and which produces a chimeric protein containing a His-tag at the C terminus. A simple screening procedure was then set up based on a colorimetric assay and that can detect the activity of crude extracts from single colonies on gl-7ACA and CephC.
To introduce the genetic variability into the VAC cDNA, we used two different approaches and then combined the results. First, we performed three rounds of random mutagenesis by EP-PCR on the 1.4-kb fragment corresponding to the 5′-end of the VAC cDNA (encoding for the entire α-subunit and the 221 amino acids at the N-terminal part of the β-subunit) using two different amplification conditions (Table 1). Subsequently, we performed saturation mutagenesis on 12 positions to access all possible amino acid substitutions at the residues identified by a molecular modeling study and believed to offer opportunity for improvement in acylase activity on CephC. Finally, we combined the information from the two approaches producing VAC mutants containing multiple mutations and with a specificity constant (the ratio of kcat/Km for cephalosporin C with respect to glutaryl-7-ACA) increased up to 100-fold as compared to the wild type (Fig. 3C ▶).
Considering the mutants obtained by EP-PCR, the D416Y-H417Y mutant shows a threefold higher Vmax value, as well as kinetic efficiency ratio, on CephC than on gl-7ACA. Unfortunately, the conditions for an efficient expression in E. coli of this VAC variant were not identified. With this combinatorial method, the A215Y variant could be identified: The enzyme(s) bearing mutations at this position resulted in a better CephC/gl-7ACA activity ratio than the wild-type VAC. The presence of a tyrosine at this position may favor the acquisition of the correct orientation of CephC with respect to S240–OH (the nucleophile that carries out the enzymatic deacylation). That a bulkier side chain at position 215 is required for better interaction of VAC with CephC was confirmed by the lower Km values determined for all evolved A215 mutants (Table 2).
Site-saturation mutagenesis on positions identified by molecular modeling resulted in VAC variants with a higher kinetic efficiency on CephC because of the higher substrate affinity compared to the wild type (the only exception is the H309S mutant), indicating that such a procedure preferentially identified the positions affecting CephC binding. A significant (up to sixfold) increase in Vmax for CephC was obtained by combining the mutations identified by EP-PCR (A215Y) and by molecular modeling and site-saturation mutagenesis (H296S and H309S). The evolved VAC variants exhibit a >100-fold increase in the specificity constant (the Vmax/Km ratio between CephC and gl-7ACA) compared to the wild-type enzyme (see Table 2; Fig. 3C ▶). The A215Y-H296S-H309S triple mutant is a “true” CephC acylase, since it exhibits a higher Vmax on this substrate than on gl-7ACA. A total of three amino acid replacements introduced into wild-type VAC suffice to provide a 3.3-kJ/mol decrease of the free energy of activation changes of the reaction on CephC and a 6.3-kJ/mol increase of the same value on gl-7ACA (ΔΔG‡ values were calculated from the data of Table 2).
The substitutions introduced in the VAC variants selected for their improved ability to deacylate CephC appear to modify its properties acting in different ways:
Substitutions at positions 215 and 309 synergistically favor the binding of CephC in the best orientation for catalysis. In fact, while all substitutions at position 215 give a decrease in Km values, the H309S mutation increases the space surrounding the substrate, thus preventing the steric clashes predicted by the modeling analysis and result in an increase in the Vmax value (see Table 2).
The substitution at position 296 facilitates the binding of the adipyl side chain of CephC, decreasing the steric hindrance and interacting with the polar/charged D-amino acid group of this substrate (Fig. 2E ▶). Interestingly, and differently from that previously observed (Otten et al. 2004), while searching for VAC variants with higher activity for CephC, we identified a mutant (A215Y-H309S) with increased activity and affinity for gl-7ACA. This is not surprising, considering that these substitutions could break down some kinetic barriers (e.g., substrate/product diffusion) affecting the reaction of VAC with both substrates.
The ability of our VAC variants to perform the mono-step conversion of CephC to 7-ACA (see Scheme 1) was demonstrated by the bioconversion performed under conditions resembling those used industrially (Fig. 4C ▶). The high conversion yield is also due to the absence of substrate inhibition effect and by the lower inhibition effect exerted by the product 7-ACA.
In conclusion, the high activity on CephC and the high yield of CephC conversion appear to be a major advantage of the process we have successfully developed on a laboratory scale. The economics of this process renders this system competitive compared to chemical production as well as to the two-step enzymatic bioconversion system in the synthesis of 7-ACA. The VAC variants that evolved might represent a “revolution” in the industrial production of 7-ACA for the biosynthesis of semisynthetic cephalosporin antibiotics. Furthermore, the present results increase our knowledge of the structure–function relationships of this class of enzymes and generate additional information about the role of specific active site residues: Greater knowledge provides more options for further redesign.
Materials and methods
Design and cloning of the VAC gene
The VAC gene was designed according to the amino acid sequence of acylase from Pseudomonas N176 (Aramori et al. 1991). The following points were considered during the design:
The codon usage was optimized according to that of E. coli (Sambrook et al. 1989), which resulted in a significant change at the 5′-end of the new acylase gene with respect to the original one. In detail, this included elimination of the GGA codon for Gly, AGG and CGA for Arg, ATA for Ile, and CCC for Pro; and equilibration of the frequency of GAG and GAA codons for Glu, the TTT and TTC codons for Phe, the CAG and CAA codons for Glu, and the CAT and CAC codons for His (according to the frequency of use of E. coli).
The mutation M270F, which was reported to increase the activity on cephalosporin C (Ishii et al. 1995), was introduced.
The EcoRI and NdeI restriction sites at the 5′-end and XhoI at 3′-end of the gene were introduced.
Some restriction sites (e.g., BamHI, NdeI) were eliminated from the coding region, and a single BamHI restriction site at position 1370 was maintained.
The resulting VAC gene was synthesized by Thermo Hybaid and shows a 77.5% sequence identity with the Pseudomonas N176 gene. Enzymatic DNA modifications were carried out according to standard protocols (Sambrook et al. 1989). The synthetic gene, coding for the VAC gene (2341 bp) (see Fig. 1 ▶), was received in the pCR4TOPO vector and subcloned into a pET24b(+) vector (carrying the resistance for kanamycin) by digestion of both plasmids with NdeI and XhoI restriction enzymes. The 2.3-kb fragment corresponding to the VAC gene was ligated into the multiple cloning site of the pET24 vector between the NdeI and XhoI sites (yielding the pET24-VAC of 7.6 kb). With this subcloning strategy, six codons (for six additional histidines) can be added to the 3′-end of the VAC gene. A 2.6-kb-shorter version of the pET24 plasmid was obtained by a BglII/Tth111I double digestion of the pET24-VAC plasmid, followed by treatment with Klenow enzyme and ligation (pET24Δ-VAC plasmid).
Error-prone PCR
For the first round of error-prone PCR, the pET24Δ-VAC plasmid was used as template. Two oligonucleotides, 5′-CGA GATCTCGATCCCGCGAAA-3′ (Acy-Ext) and 5′-AAC CAACCGTTTCATGATGCTTCGGC-3′ (Acy-Up), were used as 5′ and 3′ primers. The amplified fragment of 1.6 kb corresponds to the 5′-end of the gene and contains NdeI and BamHI restriction sites. Following digestion with these two enzymes, the PCR products (1.4 kb, comprising the entire α-subunit and 211 amino acids at the N terminus of the β-subunit) were ligated with the 3.6-kb fragment of the pET24Δ-VAC vector digested with the same enzymes. The first round of error-prone PCR was performed using 20 ng of template DNA coding for the wild-type VAC in 100 μL of reaction mixture containing 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 10 mM 2-mercaptoethanol, 10% DMSO, 1.5 mM MgCl2, 0.4 μM each primer, 200 μM each dNTP, and 2.5 U of AmpliTaq. The second round of error-prone PCR was performed using 20 ng of template DNA coding for the D416Y-H417Y mutant (clone M4) in 25 μL of reaction mixture containing 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 2.5 mM MgCl2, 0.5 mM MnCl2, 0.1 μM each primer, 0.2 mM dGTP and dATP, 1 mM dCTP and dTTP, and 2.5 U of AmpliTaq. A third round of error-prone PCR was performed starting from the A215Y mutant (clone M2) (see Table 1) by using the same conditions as in the second round. In all cases, PCR amplification was carried out at 94°C for 30 sec, 52°C for 30 sec, and 72°C for 3 min, and 40 cycles in total for condition 1 and 20 cycles in total for condition 2. In all cases, the size and yield of the amplified DNA fragments were determined by gel electrophoresis.
Construction of a random mutant library
The PCR-generated fragments ligated to the NdeI/BamHI-digested pET24Δ-VAC vector were used to transform JM109 E. coli cells according to a modified version of the original procedure (Inoue et al. 1990). All colonies on the transformation plates were then collected for DNA extraction; 20 ng of the extracted DNA was used to transform competent BL21(DE3)pLysS E. coli expression cells. Single colonies from LB plates containing 30 μg/mL kanamycin and 34 μg/mL chloramphenicol were used to inoculate fresh LB medium and then incubated overnight at 37°C for the screening procedure.
Site-directed and site-saturation mutagenesis
Single-point mutations were generated by site-directed mutagenesis using the QuikChange Site-Directed Mutagenesis Kit (Stratagene) and the VAC cDNA subcloned into the pET24Δ plasmid. The introduction of the desired mutations was confirmed by automated DNA sequencing. Site-saturation mutagenesis at different amino acid positions was carried out using the same procedure as for site-directed mutagenesis and a set of degenerated synthetic oligonucleotides. The pET24Δ-VAC plasmid encoding for wild-type or mutant VAC proteins was used as template for mutagenesis at different positions (see Table 3). The PCR products were used to transform JM109 E. coli cells, transferred to BL21(DE3)-pLysS E. coli cells, and then used for the screening procedure as detailed above.
Screening for acylase activity
The library of mutants was analyzed by a screening procedure based on the colorimetric measurement of the 7-ACA produced starting from the substrates gl-7ACA or CephC (see below). A saturated E. coli culture (1 mL) was induced by adding 1 mM IPTG and then incubated at room temperature for 3 h. The culture was centrifuged at 11,000g for 1 min, and the cell pellet was resuspended by adding 400 μL of 100 mM potassium phosphate buffer (pH 8.0) and 1 μL of DNAse (1 mg/mL) and then subjected to vigorous vortexing. Cell lysis was performed by adding 50 μL of 1 N NaOH and was subsequently neutralized by adding 50 μL of 1 M KH2PO4. The crude extracts were centrifuged at 11,000g for 10 min, and then 125 μL of the supernatant was transferred to four wells (two for each substrate tested) of a 96-well Elisa plate. The activity was assayed on the crude extract by adding 50 μL of substrate solution (1.5% [w/v] gl-7ACA or 3% [w/v] CephC) and incubating the plates at 37°C for 10 min. The reaction was stopped by adding 100 μL of the stop solution (40% acetic acid) and then 50 μL of p-dimethylamino benzaldehyde (pDMAB, 1% [w/v] in methanol). After 10 min of incubation at 25°C, the absorbance at 415 nm was measured by a microtiter plate reader and compared with that of a culture expressing the VAC wild type and one not expressing VAC as control. The mutants that outperformed the control were selected and used for further analysis.
VAC expression and purification
For enzyme expression, the pET24Δ-VAC (wild-type and mutants) plasmids were transferred to the host BL21(DE3)-pLysS E. coli strain. Cells carrying the recombinant plasmids were grown at 37°C in LB Miller containing kanamycin (30 μg/mL final concentration) and chloramphenicol (34 μg/mL final concentration). Protein expression was induced at an OD600 = 0.8 (the only exceptions being the A215Y-H296S-H309S, D416Y-H417Y, D416Y, and H296S-H309S that were induced at saturation) by adding 0.6 mM IPTG. After induction, the cells were grown further at 25°C for 3 h (the only exceptions being the A215Y-H296S-H309S, D416Y-H417Y, and D416Y mutants that were collected after 5 h and the A215Y-H309S, A215Y-F270S, and H296S-H309S mutants that were collected 21 h after induction). Crude extracts were prepared by French Press lysis in 50 mM potassium phosphate buffer (pH 7.5), 0.7 μg/mL pepstatin, and 10 μg/mL DNase. The insoluble fraction of the lysate was removed by centrifugation at 39,000g for 1 h at 4°C; VAC is fully present in the soluble fraction.
Crude extracts (after the addition of 1 M NaCl final concentration) were loaded onto a HiTrap chelating affinity column (Amersham Biosciences) equilibrated with 50 mM sodium pyrophosphate buffer (pH 7.2), containing 1 M NaCl and 5% glycerol. The column was washed with this buffer until the absorbance value at 280 nm was that of the buffer. Then the bound protein was eluted with 20 mM sodium pyrophosphate buffer (pH 7.2), containing 500 mM imidazole and 10% glycerol. The fractions containing acylase activity were loaded on a Sephadex G25 column equilibrated with 20 mM potassium phosphate buffer (pH 8.0). In order to obtain a preparation with a higher purity, the enzyme was subsequently separated using a Superdex G200 HR10 column (25 mL) equilibrated with 20 mM potassium phosphate (pH 8.0), 250 mM KCl, and then the fractions containing the acylase activity were equilibrated with 20 mM ethanolamine (pH 8.7) and fractionated on a HiTrap DEAE column (1 mL; Amersham Biosciences) equilibrated with 20 mM ethanolamine (pH 8.7), and then eluted with a 0–1 M NaCl gradient.
VAC activity and kinetic measurements
VAC catalyzes the hydrolysis of gl-7ACA or CephC to 7-ACA. The primary amino group of 7-ACA reacts with pDMAB, giving a yellow Schiff’s base with a maximum of absorbance at 415 nm (Balasingham et al. 1972): One unit of acylase is defined as the amount of enzyme that converts 1 μmol of substrate per minute under the assay conditions. In detail: 0.5 mL of enzyme (in a phosphate buffer at pH 8.0) was incubated with 0.5 mL of substrate (pH 8.0) (1% [w/v] gl-7ACA or 2% [w/v] CephC) for 10 min at 37°C. The reaction was stopped with 3 mL of 20% acetic acid, and then 3 mL of this mixture was added at 0.5 mL of 0.5% (w/v) pDMAB (dissolved in methanol) and incubated for 10 min at 25°C, before measuring the absorbance at 415 nm.
The kinetic parameters were determined using a fixed amount of enzyme and different concentrations (0–150 mM) of substrate. The inhibition effect of 7-ACA and glutaric acid was determined by means of the same activity assay following the addition of varying concentrations of inhibitor.
The pH effect on enzymatic activity and stability was determined using a multicomponent buffer: 15 mM Tris, 15 mM sodium carbonate, 15 mM phosphoric acid, and 250 mM potassium chloride adjusted to the appropriate pH with HCl or KOH (Pollegioni et al. 1997). The activity was determined by incubating the enzyme with the substrate dissolved in the multicomponent buffer in the pH range 5–10 and according to the assay described above. To determine the effect of pH on the stability of VAC, the enzyme was incubated in the multicomponent buffer at different pH values, and aliquots were drawn at different times (up to 180 min) and used to measure the residual activity. The effect of temperature was investigated by measuring the enzymatic activity on protein samples incubated up to 30 min with the substrate at different temperatures (20°–50°C temperature range).
Enzyme characterization
Anti-VAC antibodies were produced in rabbits (Davids Biotecnologie) immunized with 0.7 mg of pure lyophilized VAC protein. Monospecific antibodies were produced from whole serum by ammonium sulfate precipitation, Accell chromatography, and an affinity chromatographic procedure (Pollegioni and Simonetta 1991). This latter step was performed using a HiTrap NHS-activated HP column (1 mL; Amersham Biosciences) containing 2 mg of immobilized VAC. Western blot analysis was performed for proteins separated by SDS-PAGE and then transferred to an immobilizing matrix (Immobilon-NC; Milliporeford) using the monospecific anti-VAC primary antibodies (30 μg/mL in 150 mM potassium phosphate at pH 7.2, 0.2 M NaCl) at 42°C for 1 h, followed by rabbit anti-IgG secondary antibody (Pollegioni and Simonetta 1991).
The molecular mass of native VAC was estimated by gel-permeation chromatography on a Superdex G200 HR10 column. The molecular mass of the two subunits of the enzyme was determined under denaturating conditions by SDS-PAGE. The sequence of the N-terminal end of the α- and β-subunits was determined using a Procise sequencer (Applied Biosystems) (Edman degradation) on protein samples separated by SDS-PAGE and transferred to a ProBlot membrane.
Molecular modeling studies
Sequence alignments were generated by T-coffee software (Notredame et al. 2000). The 3D model of VAC was built using the Swiss-model server (Guex and Peitsch 1997; Schwede et al. 2003). Automated ligand docking was performed by Autodock 3.0, a package based on a Montecarlo simulated annealing approach (Goodsell et al. 1996). The 3D structure of the substrate CephC was generated using the PRODRG2 server (Schuettelkopf and van Aalten 2004). Swiss PDBviewer 3.7 was used for the building of the 3D models of VAC mutants and for protein 3D structure visualization.
Bioconversion
The time course of bioconversion was determined by a pH-stat as well as by high performance liquid chromatography (HPLC) assay. The reaction mixture contained (in a final volume of 20 mL) 100 mM potassium phosphate buffer (pH 8.5) and 20 g/L of CephC at a 250 VAC units/g of CephC ratio. Reaction mixtures were incubated at 25°C, and aliquots were drawn at different times (up to 300 min). Samples were separated by HPLC on a C8-Aquapore RP-300 column 7μ (250 × 4.6 mm; Applied Biosystems) using 25 mM KH2PO4 (pH 3.0), 5% acetonitrile as elution buffer. The 7-ACA produced was detected following the absorbance at 254 nm.
Scheme 1.

Bioconversion of CephC into 7-ACA. The conversion can be obtained by the two-step process using D-amino acid oxidase and glutaryl-7-ACA acylase (right) or by the monostep using the newly evolved cephalosporin C acylase (left).
Acknowledgments
We thank Centro Grandi Attrezzature per la Ricerca Bio-medica dell’Università degli studi dell’Insubria, Varese, and Angelo Boselli for determining the amino acid sequences.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051671705.
Competing interests We declare competing financial interests (European patent 05000102.3). This work was supported by a grant from Anti-bioticos S.p.A.; R.V. and W.C. are employees of Antibioticos S.p.A.
References
- Aramori, I., Fukagawa, M., Tsumura, M., Iwami, M., Isogai, T., Ono, H., Ishitani, Y., Kojo, H., Kohsaka, M., Ueda, Y., et al. 1991. Cloning and nucleotide sequencing of new glutaryl 7 ACA and cephalosporin C acylase genes from Pseudomonas strains. J. Ferment. Bioeng. 72 232–243. [Google Scholar]
- Balasingham, K., Warburton, D., Dunnill, P., and Lilly, M.D. 1972. The isolation and kinetics of penicillin amidase from Escherichia coli. Biochim. Biophys. Acta 276 250–256. [DOI] [PubMed] [Google Scholar]
- Cleland, W.W. 1983. Substrate inhibition. In Contemporary enzyme kinetics and mechanism (ed. D.L. Purich), pp. 253–266. Academic Press, New York.
- Duggleby, H.J., Tolley, S.P., Hill, C.P., Dodson, G., and Moody, P.C.E. 1995. Penicillin acylase has a single-amino-acid catalytic centre. Nature 373 264–268. [DOI] [PubMed] [Google Scholar]
- Goodsell, D.S., Morris, G.M., and Olson, A.J. 1996. Automated docking of flexible ligands: Applications of AutoDock. J. Mol. Recognit. 9 1–5. [DOI] [PubMed] [Google Scholar]
- Guex, N. and Peitsch, M.C. 1997. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 18 2714–2723. [DOI] [PubMed] [Google Scholar]
- Inoue, H., Nojima, H., and Okayama, H. 1990. High efficiency transformation of Escherichia coli with plasmids. Gene 96 23–28. [DOI] [PubMed] [Google Scholar]
- Ishii, Y., Saito, Y., Fujimura, T., Sasaki, H., Noguchi, Y., Yamada, H., Niwa, M., and Shimomura, K. 1995. High-level production, chemical modification and site-directed mutagenesis of a cephalosporin C acylase from Pseudomonas strain N176. Eur. J. Biochem. 230 773–778. [PubMed] [Google Scholar]
- Kim, Y. and Hol, W.G. 2001. Structure of cephalosporin acylase in complex with glutaryl-7-aminocephalosporanic acid and glutarate: Insight into the basis of its substrate specificity. Chem. Biol. 8 1253–1264. [DOI] [PubMed] [Google Scholar]
- Kim, Y., Yoon, K., Khang, Y., Turley, S., and Hol, W.G. 2000. The 2.0 Å crystal structure of cephalosporin acylase. Struct. Fold. Des. 8 1059–1068. [DOI] [PubMed] [Google Scholar]
- Murzin, A.G., Brenner, S.E., Hubbard, T., and Chothia, C. 1995. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247 535–540. [DOI] [PubMed] [Google Scholar]
- Notredame, C., Higgins, D.G., and Heringa, J. 2000. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302 205–217. [DOI] [PubMed] [Google Scholar]
- Oh, B., Kim, M., Yoon, J., Chung, K., Shin, Y., Lee, D., and Kim, Y. 2003. Deacylation activity of cephalosporin acylase to cephalosporin C is improved by changing the side-chain conformations of active-site residues. Biochem. Biophys. Res. Commun. 310 19–27. [DOI] [PubMed] [Google Scholar]
- Oh, B., Kim, K., Park, J., Yoon, J., Han, D., and Kim, Y. 2004. Modifying the substrate specificity of penicillin G acylase to cephalosporin acylase by mutating active-site residues. Biochem. Biophys. Res. Commun. 319 486–492. [DOI] [PubMed] [Google Scholar]
- Otten, L.G., Sio, C.F., van der Sloot, A.M., Cool, R.H., and Quax, W.J. 2004. Mutational analysis of a key residue in the substrate specificity of a cephalosporin acylase. Chem. Biochem. 5 820–825. [DOI] [PubMed] [Google Scholar]
- Pilone, M.S. and Pollegioni, L. 2002. D-Amino acid oxidase as an industrial biocatalyst. Biocatal. Biotransformation 20 145–159. [Google Scholar]
- Pollegioni, L. and Simonetta, M.P. 1991. Immunochemical studies on Rhodotorula gracilis D-amino acid oxidase. Experientia 47 232–235. [DOI] [PubMed] [Google Scholar]
- Pollegioni, L., Blodig, W., and Ghisla, S. 1997. On the mechanism of D-amino acid oxidase. Structure/linear free energy correlations and deuterium kinetic isotope effects using substituted phenylglycines. J. Biol. Chem. 272 4924–4934. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., Fritsch, E.F., and Maniatis, T. 1989. Molecular cloning: A laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Schuettelkopf, A.W. and van Aalten, D.M.F. 2004. PRODRG—A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 60 1355–1363. [DOI] [PubMed] [Google Scholar]
- Schwede, T., Kopp, J., Guex, N., and Peitsch, M.C. 2003. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 31 3381–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]