

Abstract
In a broad genomics analysis to find novel protein targets for antibiotic discovery, MurF was identified as an essential gene product for Streptococcus pneumonia that catalyzes a critical reaction in the biosynthesis of the peptidoglycan in the formation of the cell wall. Lacking close relatives in mammalian biology, MurF presents attractive characteristics as a potential drug target. Initial screening of the Abbott small-molecule compound collection identified several compounds for further validation as pharmaceutical leads. Here we report the integrated efforts of NMR and X-ray crystallography, which reveal the multidomain structure of a MurF–inhibitor complex in a compact conformation that differs dramatically from related structures. The lead molecule is bound in the substrate-binding region and induces domain closure, suggestive of the domain arrangement for the as yet unobserved transition state conformation for MurF enzymes. The results form a basis for directed optimization of the compound lead by structure-based design to explore the suitability of MurF as a pharmaceutical target.
Keywords: MurF, murein enzymes, peptidoglycan, multidomain structure, protein–ligand interaction, X-ray, NMR
The escalating rate of bacterial resistance to currently available antibiotics is a publicly recognized problem, and the need for novel therapeutic compounds is of increasing clinical importance. While the epidemiology of bacterial infections continually adapts to the environmental pressures applied by anti-bacterial agents, the tools to develop effective new compounds also continue to change, resulting in a paradigm shift for antibiotic discovery in the postgenomics era (Lerner and Beutel 2002). Knowledge of whole genomes offers a generalized approach to pharmaceutical efforts, screening for any bacterial protein that is necessary or essential for growth, and targeting those that are amenable to the influences of high affinity ligands. From this perspective, experiments were designed to screen for essential gene products in the genome of Streptococcus pneumoniae, an especially infectious member of the Gram-positive bacteria. While many proteins with unknown functions were identified, these efforts also highlighted the potential for targeting certain proteins with known functions, and one of the members in this category is MurF, a protein with substantial history in the scientific literature.
MurF belongs to a family of functionally related murein enzymes that participate in the biosynthesis of the bacterial cell wall, and other members include MurA, MurB, MurC, MurD, and MurE (Ikeda et al. 1990). The sequential nomenclature denotes the order of enzymatic action within the biosynthetic pathway of the peptidoglycan unit that comprises the cell wall and exhibits commonalities among bacterial strains (Bugg and Walsh 1992; van Heijenoort 2001). As this feature is both essential for bacteria and unique from human biology, the murein enzymes represent attractive targets for pharmaceutical investigation. Consistent with studies of other bacterial organisms, our screening efforts identified MurF as an essential gene product for the growth of S. pneumoniae. MurF utilizes ATP to catalyze the ligation of D-ala-D-ala dipeptide with the UDP-MurNAc-tripeptide to form the peptidoglycan UDP-MurNAc-pentapeptide monomer (Anderson et al. 1996). While MurA and MurB are quite distinct from MurF, there are structural similarities between MurF and the MurC, MurD, and MurE enzymes such that each act as ATP-dependent amino acid ligases in peptidoglycan biosynthesis and share similar enzymatic mechanisms relevant to understanding these proteins as pharmaceutical targets (El Zoeiby et al. 2003).
Our exploration of MurF as a potential pharmaceutical target began with screening the Abbott small molecule library for compounds that bind the S. pneumoniae protein using affinity selection coupled with mass spectrometry, and we report here the structural analysis of two compounds found to specifically inhibit the enzyme (Gu et al. 2004). NMR studies confirmed the specifity of binding to MurF and X-ray crystallography revealed the three-dimensional structure, yielding an observation that the protein–inhibitor complex adopts a dramatically different conformation than was found for an apo structure of MurF from Escherichia coli (Yan et al. 2000). These related structures form a comparison that is reminiscent of studies detailing large conformational changes in MurD, where the protein adopts a transition state structure through domain closure (Bertrand et al. 2000). In MurF, domain closure is apparently induced by the compound, which binds at an interface between the domains of the protein, and the structure provides an important basis for guiding the design of more potent inhibitory compounds. The integration of NMR and crystallographic efforts highlights the use of structural biology tools for the efficient exploration of pharmaceutical leads.
Results and Discussion
Lead validation by NMR-HSQC
Nuclear magnetic resonance experiments are a powerful means of screening for small molecule pharmaceutical leads in many drug discovery programs, and were especially informative in the present study (Hajduk and Burns 2002). Compounds were tested for their ability to bind MurF, monitoring shifts of HSQC protein spectra dependent upon the presence of the compound. Characteristic patterns of specific binding were observed with compounds 1 and 2, which contain similar chemical features (Fig. 1 ▶). Consistent with their similarity, perturbations in the protein spectra with the compounds were nearly identical. These spectral changes are exemplified in Figure 2A ▶ by differences highlighted in blue boxes in the presence and absence of compound 1, which indicate specific interaction with the protein (Fig. 2A ▶). Monitoring these chemical shifts during titration of compounds 1 and 2 yielded estimates for the binding constants of KD < 50 μM for both compounds. These values are consistent with measurements of inhibitory constants in an activity assay that yielded IC50 values of 1 μM and 8 μM for compounds 1 and 2, respectively (Gu et al. 2004). Because ATP is a cofactor in the ligase reaction, spectra were also recorded in the presence and absence of ATP, and again, differences were observed, strongly suggesting specific binding to the protein (Fig. 2B ▶). Interestingly, the changes in the protein spectra are different between compound 1 and ATP, indicating that compound 1 and ATP occupy two different binding sites.
Figure 1.

Chemical structures of compounds 1 (left) and 2 (right).
Figure 2.

1H/13C-HSQC spectra of MurF in the presence and the absence of compound 1 and ATP. (A) Spectra of 13CH3 (methyl)-labeled MurF are contoured in the presence (red) or the absence (black) of compound 1. (B) MurF spectra are contoured in the presence (red) or the absence (black) of ATP. The green boxes connected in A and B highlight peaks that are significantly perturbed upon addition of ATP but not compound 1, while the blue boxes connected in A and B highlight peaks that are significantly perturbed upon addition of compound 1 but not ATP, indicating two different binding sites.
Co-crystallization with compounds 1 and 2
To obtain information for structure-based design efforts, we screened conditions for crystallization of MurF. Although all attempts to crystallize preparations of the apo form of MurF failed, crystals were readily grown in co-crystallization setups with either of the compounds 1 or 2. Both compounds promoted crystallization under identical conditions that were optimized for X-ray diffraction studies, yielding high-resolution data that exhibited hexagonal symmetry for both complexes. Despite significant effort, no molecular replacement solution was obtained using the known apo structure of the MurF homolog from E. coli. A protein sample incorporating seleno-methionine was prepared and crystallized with each of the compounds under similar conditions. An initial electron density map was then experimentally determined by single wavelength anomalous X-ray diffraction on a co-crystal containing compound 1 that diffracted to 2.5 Å resolution, and an atomic model was readily built and refined against the data (Table 1). X-ray data for a seleno-methionine crystal containing compound 2 were also collected, and the structure was refined to 2.8 Å resolution.
Table 1.
X-ray phasing and refinement
| Compound 1 | Compound 2 | |
| X-ray diffraction data | ||
| Wave length (Å) | 0.9795 | 0.9796 |
| Resolution (Å) | 2.5 (2.59–2.50) | 2.8 (2.9–2.8) |
| Observations | 246,835 | 161,058 |
| Uniquea | 41,522 | 16,580 |
| Completeness (%) | 98 (97) | 99 (99) |
| I / σI | 13.3 (4.2) | 8.8 (3.4) |
| Rsym (%) | 5.0 (36) | 10.6 (50) |
| SAD Phasing (20–2.5 Å) | ||
| Se sites | 11 | |
| Z-Score (SOLVE) | 30.7 | |
| Figure of Merit (SOLVE) | 0.28 | |
| Figure of Merit (DM) | 0.73 | |
| Model refinement | ||
| Reflections (work/free) | 21,573/1166 | 15,665/851 |
| Completeness (work/free %) | 94.3/5.1 | 93.9/5.1 |
| Rfactor (work/free %) | 23.8/29.0 | 24.3/31.8 |
| Protein residues | 454 | 454 |
| Solvent molecules | 140 | 137 |
| Mean B factor(Å2) | 53 | 54 |
| RMSD ideal bond lengths (Å) | 0.008 | 0.007 |
| RMSD ideal bond angles (°) | 1.30 | 1.33 |
| RMSD ideal dihedral (°) | 22.8 | 22.8 |
| RMSD ideal improper (°) | 0.78 | 0.74 |
a Bijvoet pairs separated Rsym =∑|I-〈I〉|∑I, where I is the integrated intensity for a reflection. Figure of Merit = 〈∑P(α)eiα/∑P(α)〉, where P(α) is the phase probability at angle α. Rfactor = ∑|FP-FC|/∑FP,where FP and FC are the observed and calculated structure factor amplitudes, while Rfree. is calculated on 5% of the data excluded from refinement. Values in parentheses are for the highest resolution shell.
The crystals contain one protein molecule in the asymmetric unit consisting of 454 residues that comprise three domains (Fig. 3 ▶). The structures of the three domains individually are similar to those of the MurF homolog from E. coli (pdb code 1gg4), and indeed, the general descriptions of the fold for each of the three domains of the E. coli homolog suitably describe the corresponding structural elements for this newly determined structure (Yan et al. 2000). The N-terminal domain (residues 1–81) is unique to MurF protein homologs, and consists of a small α/β fold that contacts the larger central domain across a broad hydrophobic interface. The central domain (residues 82–302) and the C-terminal domain (309–454) adopt mononucleotide and dinucleotide (Rossmann) folds, respectively, and are connected by a short linker peptide that is poorly ordered in the crystal. Although the expression construct encodes an N-terminal His-tag fusion, this feature is not evident in the electron density map and assumed to be disordered within the crystal.
Figure 3.

Overall structure of MurF complex with compound 1 (stereo view). (A) The three structural domains are colored to highlight the N-terminal domain (blue), the central domain (green), and the C-terminal domain (red) with the linker peptide (yellow). (B) A corresponding view of the α-carbon trace is shown with representative amino acid numbering.
The similarity of the individual domains from the S. pneumonia and E. coli structures are evident in structural alignments, where overlap of the first two domains yields an RMSD value of 2.1 Å for 278 α-carbon atoms, while the C-terminal domains can be separately aligned with an RMSD value of 2.0 Å for 90 α-carbon atoms (Fig. 4 ▶). The amino acid sequences share 26% identity, and are aligned based on structural overlap, in which residues are aligned only if their α-carbons are within 3.5 Å of each other, and a gap is inserted where the α-carbon positions differ by more that 3.5 Å. Although the three domains display close structural similarities individually, the spatial arrangement of the domains differs substantially.
Figure 4.

Structural alignment of individual domains of MurF from S. pneumonia and E. coli. (A) The overlap of the N-terminal and central domains of MurF from S. pneumonia (yellow) and E. coli (gray) is shown with positions of identical residues highlighted in red. (B) Overlap of the C-terminal domains is shown separately. (C) The amino acid sequences are aligned according to the structural overlap, where residues are aligned only if their corresponding α-carbon atoms are within 3.5 Å; otherwise a gap has been inserted as a hyphen. Identical residues are noted with a period. Numbering corresponds to the S. pneumonia amino acid sequence.
Ligand structures exhibit domain closure
In contrast to the extended domain arrangement observed in the apo structure of the E. coli homolog, the three domains of MurF in this co-crystal structure occupy a compact assembly, and the electron density maps reveal the presence of compound between the domains (Fig. 5 ▶). The C-terminal domain is positioned to contact the N-terminal and middle domains, with compound located at the interface of the three domains and surrounded by protein interactions. This arrangement represents a large conformational change for the C-terminal domain relative to the corresponding position observed in the apo structure of the homolog(Fig. 6 ▶). In particular, with the first two domains of both structures aligned, Asn328 of the C-terminal domain is nearly 30 Å from the position of the corresponding conserved residue, Asn336, in the unliganded E. coli homolog, and this difference represents a relative change in both translational position and rotational orientation of the C-terminal domain. The linker peptide between the central and C-terminal domains tethers the domains together with an apparent hinge point for the domain positions at Gln300, located at the end of the central domain terminated by a helix. The extended arrangement of the apo structure is evidently in an “open” conformation, while the compact topology of the co-crystal suggests the protein has been captured in a “closed” state.
Figure 5.
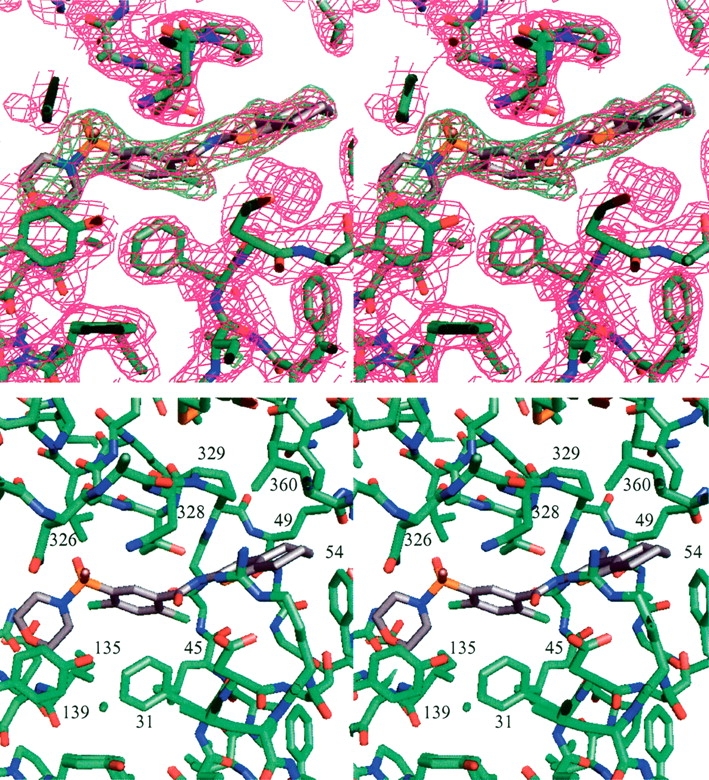
Details of the ligand binding site in the MurF complex with compound 1 (stereo view). (A) The 2Fo–Fc map is contoured at 1σ (magenta), and an Fo–Fc omit map calculated without ligand atoms is contoured at 3σ (green). (B) Additional details of the binding site are shown, and residues lining the ligand binding site are labeled.
Figure 6.

Overlap of MurF structures. The structure of MurF from E. coli (gray) is aligned with the N-terminal and central domains of the structure of MurF from S. pneumonia (colored by domains as in Fig. 3 ▶).
The compact conformation of the protein in the co-crystals is apparently dependent upon compound binding. In both cases the ligand is completely surrounded by protein contacts. The compound does not easily diffuse out of the crystallized protein, and cannot be displaced by other compounds in simple soaking experiments. The two compounds are strikingly similar, and perhaps not surprisingly, interact with the protein similarly, forming a significant portion of the contacts across the interface between the domains. The cyanothiophene is located centrally in the contact between the N- and C-terminal domains, with the nitrile suitably oriented to form a H-bond (3.1 Å) with the backbone amide of Arg49 (Fig. 5 ▶). The attached saturated ring (cyclohexyl for compound 1, and cyclopentyl for compound 2) extends the plane of the thiophene toward a patch of hydrophobic residues from the C terminus, contacting the side chains of residues Pro329, Leu360, and Leu367. Phe54 interacts with the compound from the opposing N-terminal side, and a small cavity below the compound contains solvent molecules that form bridging H-bonds between main-chain atoms for residues of the N-terminal domain. The amide-linked Cl-benzene and substituted sulfonamide are located at the interface between all three domains, laying on a hydrophobic shelf formed by Phe31 and Leu45 of the N-terminal domain and Tyr135 and Ile139 of the central domain, and most closely contacting residues Asn326, Asn328, and Thr330 of the C-terminal domain (Fig. 5 ▶). Because the protein did not crystallize in the apo form but readily crystallized in the presence of compound, the compounds arguably induce or stabilize the conformation through these interactions with the protein.
The MurF proteins belong to the larger structurally related family of murein synthetases that includes MurC, MurD, and MurE, sharing several invariant amino acid residues that are suggestive of a common enzymatic reaction mechanism throughout the family (Bouhss et al. 1999). Structural information is available from several studies on these enzymes, and general similarities are apparent. These proteins all contain a three-domain arrangement, albeit with significant differences in detail as might be expected for subfamily members with ~15% sequence identity. Elegant studies of the MurD protein provide descriptions of “open” and “closed” conformations that help explain the enzymatic reaction mechanism conserved throughout the family (Bertrand et al. 1999, 2000). Interestingly, the structures of MurD also exhibit large conformational differences between the positions of the C-terminal domain relative to the rest of the protein, wherein the closed form is thought to approximate the enzymatic transition state and the open form would represent a generic interdomain conformation without substrates or products. Although the closed conformations for MurD and MurF do not overlap exactly, the comparison is noteworthy (Fig. 7 ▶). Alignment of their central domains (RMSD of 1.9 Å for 139 α-carbons) yields a closer topological comparison for the C-terminal domains than with the structure of MurF from E. coli, but it is unclear how closely the ligand bound structure of S. pneumonia MurF approximates the domain arrangement of the transition state. The locations of the invariant residues suggest the MurF structure would need to undergo significant conformational changes at least locally to attain a transition state structure. In reporting the apo structure of MurF from E. coli, the investigators compare the structure with the closed form of MurD and suggest a large conformational change is required for catalysis (Yan et al. 2000). The present co-crystal structure supports this hypothesis by providing a novel example of MurF in a compact conformation, more closely approximating that observed for the closed state of MurD.
Figure 7.

Comparison of MurF with the closed-form of MurD. (A) An overlap of the structures of MurD (gray) and MurF (colored by domains as in Fig. 3 ▶) is shown based on alignment of the central domains. (B) Structural alignment of the C-terminal domains of MurD (gray) and MurF (red). (C,D) The locations of invariant residues conserved among MurC, MurD, MurE, and MurF enzymes are highlighted in magenta on the structures of MurF with compound 1 (C) and MurD with substrate (D).
Ligands occupy a substrate-binding site
To address the functional nature of the binding site observed for the compound in the MurF co-crystals, structural studies of other Murein enzymes again provide helpful comparisons. The four enzymes, MurC, MurD, MurE, and MurF catalyze sequential ATP-dependent ligations to the growing peptide chain of the developing peptidoglycan unit (van Heijenoort 2001). While the nonribosomal peptide ligation mechanism is evidently conserved among these enzymes, the increasingly larger substrate naturally requires variation for the unique portions of substrate recognition. MurF catalyzes the addition of D-Ala-D-Ala dipeptide to the C terminus of the UDP-N-acetylmuramoylalanine-D-glutamyl-lysine (UDPMurNAc-tripeptide), yielding the UDPMurNAc-pentapeptide unit to which lipids are attached to complete the peptidoglycan monomer (Anderson et al. 1996). By comparison, MurD acts two steps prior in the biosynthetic pathway by adding D-glutamate to UDPMurNAc, a significantly smaller substrate than the UDPMurNAc-tripeptide recognized by MurF (Bertrand et al. 1999). In each case, a peptide bond is formed with the growing peptidoglycan via activation of its carboxylate with an acyl-phosphate intermediate followed by nucleophilic attack by the incoming amino acid substrate (Falk et al. 1996).
In the report of the E. coli MurF structure, the investigators describe an X-ray experiment in which they soaked a crystal with two substrates, UDPMurNAc-tripeptide and D-Ala-D-Ala dipeptide, and observed electron density for the uridine–ribose moiety located on the surface between the N-terminal and central domains (Yan et al. 2000). Although limited information makes it difficult to compare with the S. pneumonia MurF in detail, the uridine–ribose binding site unambiguously overlaps with the corresponding ligand binding site observed for compounds 1 and 2. For MurD, interestingly, inclusion of ligands was important to capture the closed form in crystallization, requiring either ATP analogs and/or substrate (UDP-N-actyl-muramoyl-L-alanine), and their binding modes were readily established (Bertrand et al. 1999). While MurF differs in detail, the UDP-derivative binds MurD across the N-terminal domain and extends toward the ATP binding pocket in the middle domain, partly coinciding with the corresponding site for compounds 1 and 2 in MurF (Fig. 7C,D ▶). Comparisons can also be drawn from the structures of MurC and MurE complexes, differing again in detail, but offering homologous examples with topologically similar locations of substrate binding sites (Gordon et al. 2001; Mol et al. 2003). Unfortunately, similar efforts to soak crystals of S. pneumonia MurF did not reveal any evidence of substrates in the electron density maps of X-ray experiments. While much remains uncertain about the binding mode of substrates for MurF, the comparisons strongly suggest that compounds 1 and 2 occupy a portion of the substrate-binding region.
Topological comparisons also provide insight to additional features of the MurF structure in the vicinity of the active site. Structures of MurD identify an ATP binding site in the central domain, consistent with other proteins with mononucleotide folds containing a characteristic “Walker” sequence motif, and these features are conserved in the E. coli structure of MurF (Smith and Rayment 1996; Bertrand et al. 1999; Yan et al. 2000). The sequence of MurF from S. pneumonia also contains the characteristic motif (residues 104–112), but the conformation of this loop is unusual. Although the density in this region is relatively poor, the loop does not adopt a typical conformation for binding ATP but rather extends toward the C-terminal domain. X-ray data collected on crystals soaked with nucleotides did not yield evidence of binding, which is consistent with the observed atypical conformation that is apparently incompatible with ATP. Intriguingly, NMR data suggest compound 1 and ATP can bind simultaneously, suggesting that the crystal structure does not capture a conformation accessible in solution as observed by NMR. This difference is conceivably due to local conformational changes of the nucleotide-binding loop without influencing the interactions at the binding site for compound 1 or 2.
In conclusion, we have identified a novel class of small molecule compounds that bind MurF and determined the structural interactions of the protein–ligand complex. The compounds capture the protein in a topologically compact state that is reminiscent of the closed forms of transition state structures for related enzymes sharing similar catalytic mechanisms. While the observed structure is clearly not in a transition state conformation, the binding site for the compound overlaps with the expected binding site for substrate. The detailed interactions of the compound with the protein form the basis for further structure-based drug design. These studies highlight the coordinated efforts of NMR and X-ray crystallographic studies to validate pharmaceutical leads and yield valuable information for further directed exploration by medicinal chemistry.
Materials and methods
Protein expression and purification
A pET30 plasmid encoding a recombinant construct of an N-terminal fusion peptide of amino acid sequence MKHHHHHHDDDDK followed by the full-length sequence of MurF from S. pneumoniae was cloned by standard techniques and transformed into E. coli BL21(DE3) for expression. Normal growths were cultured in Terrific Broth (Sigma) with kanamycin (50 mg/L). Cultures for 13C-NMR studies were supplemented with [3-13C]-α-ketobutyrate and [3,3′-13C]-α-ketoisovalerate, whereas cultures for X-ray crystallography studies were supplemented with Se-methionine in minimal media. Cells were grown to mid exponential-phase at 37°C, at which point 1 mM IPTG was added and the temperature was shifted to 30°C. Cells were harvested 4.5 h post-induction and frozen at −85°C. A French pressure cell was used to lyse the cells in 50 mM Tris, 10% glycerol, and 1 mM dithiothreitol (buffer A, pH 8.0). The soluble portion was applied to a Q-sepharose anion exchange column and eluted using a 100- to 250-mM NaCl gradient in buffer A (pH 7.5). Ammonium sulfate was added to the protein pool for a final concentration of 2 M, and the pool was applied to an Me-HIC (Bio-Rad) chromatography column in 50 mM Tris (pH 7.5), 2 M ammonium sulfate, and 1 mM DTT. Protein was eluted with a gradient into buffer A (pH 7.7), and concentrated for a final step of gel filtration on Sephacryl S200 Hi-prep in 50 mM Tris (pH 7.5), 150 mM NaCl, 1 mM DTT.
NMR screening
NMR samples were composed of 13C-methyl labeled MurF in an H2O/D2O (9/1) solution containing 20 mM Tris, 5 mM DTT, 5 mM MgCl2 (pH 8.5) (Hajduk et al. 2000). Ligand binding was detected by acquiring 1H/13C-HSQC spectra utilizing a WATERGATE sequence for solvent suppression on 500 μL of 0.04 mM protein in the presence and the absence of added compound (Piotto et al. 1992). A Bruker sample changer was used on a Bruker DRX500 spectrometer equipped with a CryoProbe (Hajduk et al. 1999). Binding was determined by the observation of changes in the HSQC spectrum. Dissociation constants were obtained for selected compounds by monitoring the chemical shift changes of the protein resonances as a function of ligand concentration. Data were fit using a single binding site model, and a least-squares grid search was performed by varying the values of KD and the chemical shift of the fully saturated protein.
Crystallization and structure determination
Purified protein at 10–15 mg/mL in 50 mM Tris (pH 7.5), 150 mM NaCl, and 1 mM DTT was incubated with compound and crystallized at 4°C by the hanging drop method, using a reservoir containing 2.5 M ammonium sulfate, 10 mM magnesium acetate, and 50 mM MES (pH 5.6). Crystals were transferred to fresh reservoir solution with 25% (w/v) glycerol and rapidly frozen in liquid nitrogen. X-ray data were collected at the Advanced Photon Source of Argonne National Laboratory on the IMCA beamline 17-ID with an ADSC quantum 210 detector. Anomalous diffraction data were collected on a co-crystal of compound 1 and MurF containing seleno-methionine using a wavelength of 0.9795 Å, which was verified as the peak of fluorescence across the selenium absorption edge. Data were integrated and scaled using HKL2000 (Otwinowski and Minor 1997), and the diffraction exhibited hexagonal symmetry of the P6122 space group with cell parameters of a = b = 116.27 Å and c = 161.39 Å. For a monomer of 50 kDa, the Vm coefficient of 3.1 Å 3/Da suggests the asymmetric unit contains one protein molecule with ~60% solvent. Intensity differences of all Bijvoet pairs were used as input to the program SOLVE (Terwilliger and Berendzen 1999), which successfully located the positions of 11 outof the 12 selenium atoms expected for the recombinant protein. Subsequent density modification using DM (CCP4 1994) yielded an interpretable electron density map. Parallel calculations with P6522 clearly distinguished P6122 as the correct polar space group assignment. A protein model was built and refined using the programs O (Jones et al. 1991), QUANTA and CNX (Accelrys), targeting the measured structure factor magnitudes and HL coefficients containing the experimentally determined phase information. Figures were prepared using InsightII (Accelrys) and PyMOL (DeLano Scientific). The atomic coordinates and structure factors have been deposited in the Protein Data Bank with accession codes 2AM1 (compound 1) and 2AM2 (compound 2).
Acknowledgments
For crystal structure analysis, X-ray data were collected at beamline 17-ID in the facilities of the Industrial Macromolecular Crystallography Association Collaborative Access Team (IMCA-CAT) at the Advanced Photon Source. These facilities are supported by the companies of the Industrial Macromolecular Crystallography Association.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051604805.
References
- Anderson, M.S., Eveland, S.S., Onishi, H.R., and Pompliano, D.L. 1996. Kinetic mechanism of the Escherichia coli UDPMurNAc-tripeptide D-alanyl-D-alanine-adding enzyme: Use of a glutathione S-transferase fusion. Biochemistry 35 16264–16269. [DOI] [PubMed] [Google Scholar]
- Bertrand, J.A., Auger, G., Martin, L., Fanchon, E., Blanot, D., Le Beller, D., van Heijenoort, J., and Dideberg, O. 1999. Determination of the MurD mechanism through crystallographic analysis of enzyme complexes. J. Mol. Biol. 289 579–590. [DOI] [PubMed] [Google Scholar]
- Bertrand, J.A., Fanchon, E., Martin, L., Chantalat, L., Auger, G., Blanot, D., van Heijenoort, J., and Dideberg, O. 2000. “Open” structures of MurD: Domain movements and structural similarities with folylpoly-glutamate synthetase. J. Mol. Biol. 301 1257–1266. [DOI] [PubMed] [Google Scholar]
- Bouhss, A., Dementin, S., Parquet, C., Mengin-Lecreulx, D., Bertrand, J.A., Le Beller, D., Dideberg, O., van Heijenoort, J., and Blanot, D. 1999. Role of the ortholog and paralog amino acid invariants in the active site of the UDP-MurNAc-L-alanine:D-glutamate ligase (MurD). Biochemistry 38 12240–12247. [DOI] [PubMed] [Google Scholar]
- Bugg, T.D. and Walsh, C.T. 1992. Intracellular steps of bacterial cell wall peptidoglycan biosynthesis: Enzymology, antibiotics, and antibiotic resistance. Nat. Prod. Rep. 9 199–215. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (CCP4). 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50 760–763. [DOI] [PubMed] [Google Scholar]
- El Zoeiby, A., Sanschagrin, F., and Levesque, R.C. 2003. Structure and function of the Mur enzymes: Development of novel inhibitors. Mol. Microbiol. 47 1–12. [DOI] [PubMed] [Google Scholar]
- Falk, P.J., Ervin, K.M., Volk, K.S., and Ho, H.T. 1996. Biochemical evidence for the formation of a covalent acyl-phosphate linkage between UDP-N-acetylmuramate and ATP in the Escherichia coli UDP-N-acetylmuramate:L-alanine ligase-catalyzed reaction. Biochemistry 35 1417–1422. [DOI] [PubMed] [Google Scholar]
- Gordon, E., Flouret, B., Chantalat, L., van Heijenoort, J., Mengin-Lecreulx, D., and Dideberg, O. 2001. Crystal structure of UDP-N-acetylmuramoyl-L-alanyl-D-glutamate: meso-diaminopimelate ligase from Escherichia coli. J. Biol. Chem. 276 10999–11006. [DOI] [PubMed] [Google Scholar]
- Gu, Y.G., Florjancic, A.S., Clark, R.F., Zhang, T., Cooper, C.S., Anderson, D.D., Lerner, C.G., McCall, J.O., Cai, Y., Black-Schaefer, C.L., et al. 2004. Structure–activity relationships of novel potent MurF inhibitors. Bioorg. Med. Chem. Lett. 14 267–270. [DOI] [PubMed] [Google Scholar]
- Hajduk, P.J. and Burns, D.J. 2002. Integration of NMR and high-throughput screening. Comb. Chem. High Throughput Screen 5 613–621. [DOI] [PubMed] [Google Scholar]
- Hajduk, P.J., Gerfin, T., Boehlen, J.M., Haberli, M., Marek, D., and Fesik, S.W. 1999. High-throughput nuclear magnetic resonance-based screening. J. Med. Chem. 42 2315–2317. [DOI] [PubMed] [Google Scholar]
- Hajduk, P.J., Augeri, D.J., Mack, J., Mendoza, R., Yang, J., Betz, S.F., and Fesik, S.W. 2000. NMR-based screening of proteins containing 13C-labeled methyl groups. J. Am. Chem. Soc. 122 7898–7904. [Google Scholar]
- Ikeda, M., Wachi, M., Jung, H., Ishino, F., and Matsuhashi, M. 1990. Homology among MurC, MurD, MurE and MurF proteins in Escherichia coli and that between E. coli MurG and a possible MurG protein in Bacillus subtilis. J. Genet. Appl. Microbiol. 36 179–187. [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A Found. Crystallogr. 47 110–119. [DOI] [PubMed] [Google Scholar]
- Lerner, C.G. and Beutel, B.A. 2002. Antibacterial drug discovery in the post-genomics era. Curr. Drug Targets Infect. Disord. 2 109–119. [DOI] [PubMed] [Google Scholar]
- Mol, C.D., Brooun, A., Dougan, D.R., Hilgers, M.T., Tari, L.W., Wijnands, R.A., Knuth, M.W., McRee, D.E., and Swanson, R.V. 2003. Crystal structures of active fully assembled substrate- and product-bound complexes of UDP-N-acetylmuramic acid:L-alanine ligase (MurC) from Haemophilus influenzae. J. Bacteriol. 185 4152–4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Piotto, M., Saudek, V., and Sklenar, V. 1992. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 2 661–665. [DOI] [PubMed] [Google Scholar]
- Smith, C.A. and Rayment, I. 1996. Active site comparisons highlight structural similarities between myosin and other P-loop proteins. Biophys. J. 70 1590–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger, T.C. and Berendzen, J. 1999. Automated MAD and MIR structure solution. Acta Crystallogr. D Biol. Crystallogr. 55 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heijenoort, J. 2001. Recent advances in the formation of the bacterial peptidoglycan monomer unit. Nat. Prod. Rep. 18 503–519. [DOI] [PubMed] [Google Scholar]
- Yan, Y., Munshi, S., Leiting, B., Anderson, M.S., Chrzas, J., and Chen, Z. 2000. Crystal structure of Escherichia coli UDPMurNAc-tripeptide d-alanyl-d-alanine-adding enzyme (MurF) at 2.3 Å resolution. J. Mol. Biol. 304 435–445. [DOI] [PubMed] [Google Scholar]