

Abstract
The tryptophan repressor binding protein WrbA binds to the tryptophan repressor protein TrpR. Although the biological role of WrbA remains unclear, it has been proposed to function in enhancing the stability of TrpR–DNA complexes. Sequence database analysis has identified WrbA as a founding member of a flavodoxin-like family of proteins. Here we present crystal structures of WrbA from Deinococcus radiodurans and Pseudomonas aeruginosa and their complexes with flavin mononucleotide. The protomer structure is similar to that of previously determined long-chain flavodoxins; however, each contains a conserved inserted region unique to the WrbA family. Interestingly, each WrbA protein forms a homotetramer with 222 symmetry, unique among flavodoxin-like proteins, in which each protomer binds one flavin mononucleotide cofactor molecule.
Keywords: structural genomics, NYSGXRC, WrbA, TrpR binding protein
The WrbA protein copurifies and coimmunoprecipitates with the tryptophan repressor protein TrpR (Yang et al. 1993), and hence is often referred to as the TrpR binding protein. In numerous bacterial species, TrpR, when bound to tryptophan, binds several operators to prevent the transcription of genes involved in the biosynthesis of tryptophan (Rose and Yanofsky 1974). An early study reporting band-shift and endonuclease assays suggested that WrbA enhances the affinity and/or stability of DNA binding by TrpR (Yang et al. 1993). However, a subsequent study disputes this finding (Grandori et al. 1998). Thus, the effect of WrbA on TrpR-DNA binding is uncertain, and its biological function remains unclear. WrbA transcription is controlled by the stress response gene rpoS (Lacour and Landini 2004), indicating that WrbA is expressed not only in the stationary phase (Yang et al. 1993), but also under a variety of stress conditions.
Sequence database searches with WrbA revealed a family of flavodoxin-like proteins (Grandori and Carey 1994). Proposed structure homology based on this sequence analysis supports the hypothesis that WrbA may share the αβ twisted open-sheet fold characteristic of known flavodoxin structures (Grandori and Carey 1994). It has also been proposed based on these sequence analyses that members of the WrbA family contain a conserved insertion uncharacteristic of classical flavodoxins (Grandori and Carey 1994). Notably, prior studies have shown strong sequence similarity between proteins with quinone reductase activity and members of the WrbA family (Laskowski et al. 2002; Daher et al. 2005).
The presence of an N-terminal flavodoxin-like motif suggests the possibility of flavin mononucleotide (FMN) binding. Biochemical characterization of WrbA from Escherichia coli has shown that it binds one FMN molecule per monomer, although the binding constant was found to be weaker than that of many flavodoxins (Grandori et al. 1998). However, solution binding experiments indicated that WrbA did not bind FAD or riboflavin under the same conditions (Grandori et al. 1998). Ultracentrifugation experiments on E. coli WrbA indicate that it participates in a dimer–tetramer equilibrium (Grandori et al. 1998). In gel filtration experiments, FMN binding was found to have little or no effect on this multimeric equilibrium (Grandori et al. 1998). The WrbA from E. coli used for these characterization experiments shares 37% sequence identity with the putative WrbA from Deinococcus radiodurans and 40% sequence identity with the putative WrbA from Pseudomonas aeruginosa, for which the crystal structures are presented here. The sequences for the two structures presented here share only 29% sequence identity.
Here we report crystal structures of WrbA from D. radiodurans and P. aeruginosa, both in their apo forms and as complexes with FMN. These structures reveal a homotetramer that binds one FMN molecule per protomer. Each protomer adopts an αβ twisted open-sheet fold similar to known long-chain flavodoxin structures (Smith et al. 1983). There are no significant structural changes observed upon FMN binding. The region surrounding the α6 helix, located in the core of the WrbA tetramer, is responsible for the majority of tetramerization interactions. These studies also reveal the structure of a conserved insertion sequence unique to the WrbA family.
Results
Overall structure
We determined structures for WrbA proteins from D. radiodurans and P. aeruginosa, as apoproteins and in complex with FMN for both species. The structure of apo-WrbA from D. radiodurans was refined to a resolution of 2.0 Å; the resolution of the protein in complex with FMN is 3.1 Å. The structures from P. aeruginosa each contain over 20 disordered residues with resolutions of 2.6 Å and 2.8 Å for the apo and complex form, respectively. Detailed crystallographic data and PDB accession codes are reported in Table 1.
Table 1.
Data collection, solution, and refinement statistics
| Deinococcus radiodurans | Pseudomonas aeruginosa | |||
| PDB ID | WrbA 1YDG | WrbA w/FMN 1YRH | WrbA 1ZWK | WrbA w/FMN 1ZWL |
| Data collection statistics | ||||
| Collection site, Beamline | APS, ID-31/SGX-CAT | APS, ID-31/SGX-CAT | NSLS, X4A | NSLS, X29 |
| Detector type and model | CCD, MAR 165 | CCD, MAR 165 | ADSC Quantum 4 | ADSC Quantum 315 |
| Temperature (K) | 100° | 100° | 100° | 100° |
| No. of images | 90 | 70 | 180 | 100 |
| Oscillation angle (°) | 1 | 1 | 1 | 1 |
| Wavelength (Å) | 0.9793 | 0.9793 | 0.979 | 0.9791 |
| No. of unique reflections | 116,736 | 32,107 | 13,469 | 5616 |
| Redundancy | 3.6 | 3.8 | 6.8 | 6.2 |
| I/sigl | 13 | 5.6 | 23.4 | 14.8 |
| Resolution range (Å) | 20–2.0 (2.07–2.0) | 20–3.1 (3.21–3.1) | 20–2.6 (2.69–2.6) | 20–2.8 (2.9–2.8) |
| Completeness (%) | 97 (99.8) | 97.2 (97.0) | 99.9 (100) | 99.2 (93.3) |
| Rmerge (%) | 0.086 (0.290) | 0.170 (0.324) | 0.073 (0.35) | 0.11 (0.277) |
| Space group | P3221 | P3221 | P222 | P4222 |
| No. of molecules/asymmetric unit | 8 | 8 | 2 | 1 |
| Solvent content (%) | 52.6 | 52.9 | 58.9 | 55.86 |
| Structure refinement | ||||
| Resolution range (Å) | 20–2.0 (2.05–2.0) | 20–3.11 (3.19–3.11) | 20–2.6 (2.7–2.6) | 20–2.8 (2.87–2.8) |
| R | 0.189 (0.232) | 0.200 (0.230) | 0.229 (0.267) | 0.199 (0.233) |
| Rfree | 0.236 (0.256) | 0.233 (0.305) | 0.273 (0.385) | 0.236 (0.305) |
| % completeness for range | 96.9 | 97.0 (95.96) | 99.75 (97.16) | 98.78 (91.87) |
| No. of reflections in test set for Rfree | 5845 (436) | 1632 (124) | 732 (50) | 253 (15) |
| No. of reflections used in refinement | 116,506 (8192) | 32,055 (2135) | 13,467 (952) | 5602 |
| Overall average B factor (Å 2) | 22.25 | 32.47 | 57.1 | 57.5 |
| RMS deviations bond lengths (Å) | 0.01 | 0.012 | 0.011 | 0.01 |
| RMS deviations bond angles (°) | 1.182 | 1.159 | 1.271 | 1.334 |
| No. of protein atoms | 12,056 | 11,977 | 2443 | 1278 |
| No. of solvent atoms | 1293 | 161 | 51 | 35 |
| No. of other atoms | 50 | 248 | 10 | 31 |
| Refinement software | Refmac 5.2.0005 | Refmac 5.2.0005 | Refmac 5.2.0005 | Refmac 5.2.0005 |
| Ramachandran plot analysis | ||||
| Most favored regions (%) | 95.8 | 90.5 | 90.2 | 92.8 |
| Additionally allowed regions (%) | 4.2 | 9.2 | 8.7 | 6.5 |
| Generously allowed regions (%) | 0 | 0.3 | 1.1 | 0.7 |
| Disallowed regions (%) | 0 | 0 | 0 | 0 |
Data for the highest resolution shell is given in parentheses.
The structures show that WrbA forms a homotetramer and binds one FMN molecule per protomer (Fig. 1 ▶), in agreement with prior biochemical characterization studies of WrbA from E. coli (Grandori and Carey 1994). Each protomer adopts an αβ twisted open-sheet fold similar to long-chain flavodoxins. The majority of the tetramerization interactions are found in the helical region surrounding and including α6, although this helix is not known to function in the tetramerization of other flavodoxins. The structures also reveal an insertion following the β2-strand, which is conserved in members of the WrbA family but not in known flavodoxin structures (Fig. 2 ▶). This insertion region is comprised of α3 and α4 spanning from Glu43 to Thr70 in the structures of WrbA from D. radiodurans. This region lies close to the FMN binding site; however, it does not interact with the FMN molecule directly or contribute to the tetramerization of the molecule. This region is disordered in the structures from P. aeruginosa. The structure of WrbA from D. radiodurans also contains α7 spanning from Pro149 to Gly166. Long-chain flavodoxins contain an insertion corresponding to the α7 position, which splits the β5-strand. The function of this insertion in flavodoxins remains unclear; however, it is not believed to be structurally essential (López-Llano et al. 2004). This region is also disordered in the structures of WrbA from P. aeruginosa.
Figure 1.
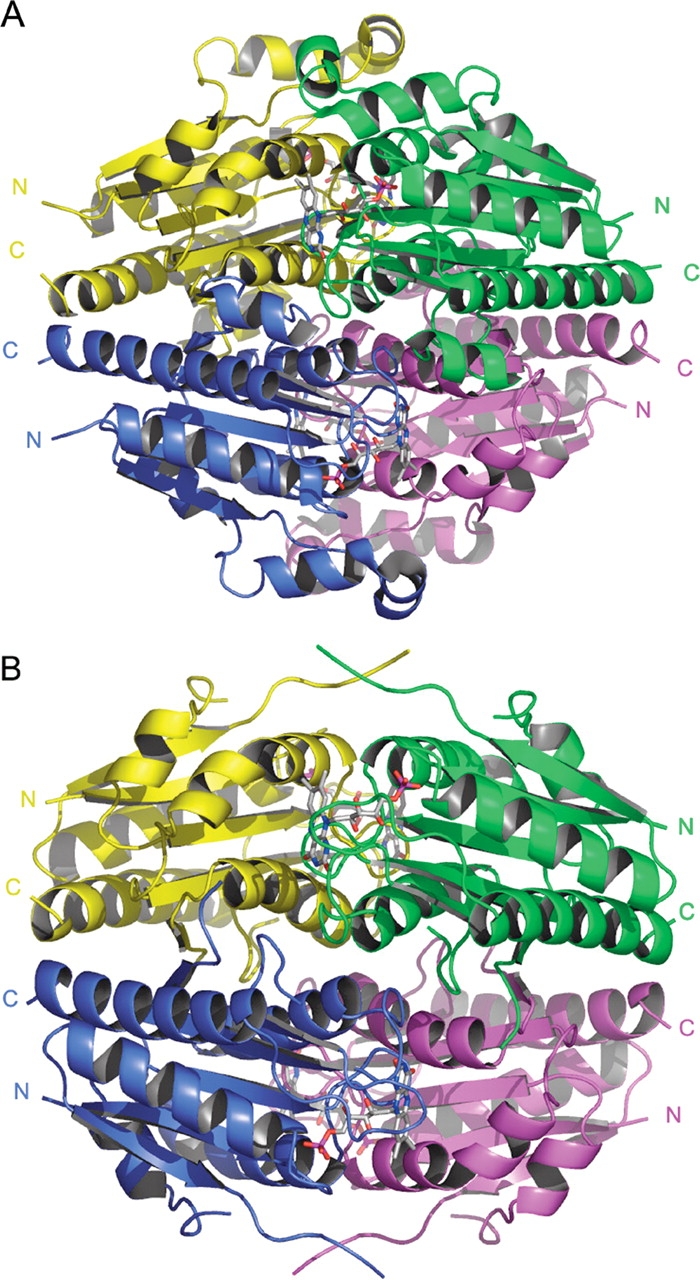
(A) Ribbon representation of the homotetramer WrbA from D. radiodurans in complex with FMN. (B) Ribbon representation of the homotetramer WrbA from P. aeruginosa in complex with FMN. Both tetramers have 222 symmetry and bind one molecule of FMN per protomer.
Figure 2.

(A) A protomer of WrbA from D. radiodurans in complex with FMN oriented similar to the blue subunit in Figure 1 ▶. The insertion of α2 and α3 following β2 is shown in red. Although this region lies close to the FMN binding site, it does not contribute direct contacts to binding of the FMN cofactor. The second insertion, common to long-chain flavodoxins, is α7, shown in red, and splits the β5 strand. (B) Alternate view of the protomer of WrbA from D. radiodurans. (C) Protomer of WrbA from P. aeruginosa in complex with FMN oriented similar to the blue subunit in Figure 1 ▶. Both regions corresponding to the insertions in WrbA from D. radiodurans are disordered in this structure. (D) Alternate view of the protomer of WrbA from P. aeruginosa.
A DALI search (Holm and Sander 1996) reveals a number of structures with significant similarity, many of which are flavoproteins. The three most similar structures found by this search were: 1E5D, a Rubredoxin-oxygen oxidoreductase (Frazão et al. 2000), the flavodoxin 5NUL (Ludwig et al. 1997), and the flavodoxin 1RCF (Burkhart et al. 1995). WrbA retains the basic flavodoxin αβ twisted open-sheet fold shared by these three flavoproteins. The RMS deviation of the Cα atoms of these structures from the unliganded WrbA structure from D. radiodurans structure are 1.8 Å, 2.0 Å, and 2.8 Å, respectively, across 148, 136, and 155 residue pairs. The unliganded structure from P. aeruginosa shows respective RMS deviations of 1.8 Å, 2.0 Å, and 2.6 Å across 144, 136, and 148 residue pairs. The unliganded structures of WrbA from D. radiodurans and P. aeruginosa show RMS deviation of 1.5 Å across 166 residue pairs. A sequence alignment between members of the WrbA family and members of the flavodoxin family is shown in Figure 3 ▶. The alignment was performed using all members of the WrbA family and all members of the flavodoxin family found in Swiss-Prot, but only a few examples of each are shown in the figure. Sequences of the three most structurally similar proteins—1E5D, 5NUL, and 1RCF—are also shown in the alignment.
Figure 3.

Multiple sequence alignment of the two protein structures presented here as well as all WrbA proteins and other flavodoxin proteins found in Swiss-Prot, using the ClustalX color scheme. All flavodoxins are shaded. The three most structurally related proteins according to a Dali search are also shown along with their PDB code. Red circles are used to indicate residues that interact with the FMN. Blue circles show residues that participate in hydrogen bond interactions which contribute to the tetramerization. Blue squares show residues that contribute to hydrophobic tetramerization interactions.
Tetramerization
The crystal structures reported here show that WrbA family proteins form tetramers with 222 symmetry, in agreement with analytical ultracentrifugation results indicating that E. coli WrbA participates in a dimer–tetramer equilibrium (Grandori et al. 1998). One of the observed dimer interfaces buries approximately 2423 Å2 surface area, 1212 Å2 per protomer (e.g., the yellow and blue protomer pair of Fig. 1 ▶), whereas the other interface (e.g., the magenta and blue pair of Fig. 1 ▶), buries only 1601 Å2, 800 Å2 per protomer, according to a surface area analysis using GRASP (Nicholls et al. 1991). Thus, it seems likely that the 2423 Å2 interface defines the more stable dimer, though this has not been experimentally verified. The bulk of the multimer interactions, across both dimer interfaces, form primarily in the regions of the α6 helix and the β5 sheet.
Although the insertion following β2 was correctly predicted by sequence alignment, the α6 helix region had also previously been proposed to be formed from an additional conserved insertion in the WrbA family (Grandori and Carey 1994). However, the structures reported here reveal that this region structurally aligns with the α4 helix of classical flavodoxin structures, located between β4 and β5 in most flavodoxins. WrbA is the first case in which this region is associated with tetramerization, which could account for the higher level of sequence conservation of WrbA proteins in this region. The α6 helix participates in interactions with each of the three other chains in the tetramer and is located at the core of the assembly (Fig. 4A ▶). Approximately 25%, 2486 Å2, of the accessible surface area of each protomer is buried upon tetramerization (Nicholls et al. 1991). Hydrophobic as well as polar and hydrogen-bonding interactions contribute to the tetramer interface (Fig. 4 ▶). Additionally, the first section of the β5 strand aligns as an anti-parallel β-sheet with its mate upon tetramerization, although this interaction is limited to two hydrogen bonds. The sequence alignment shows that a number of the residues involved in tetramerization—in particular His142, Gly144, Tyr152, Gly161, Gly162, Pro164, and Tyr165 (numbered according to WrbA from D. radiodurans)—are highly conserved among members of the WrbA family, but show little or no conservation among other flavodoxin-like proteins.
Figure 4.
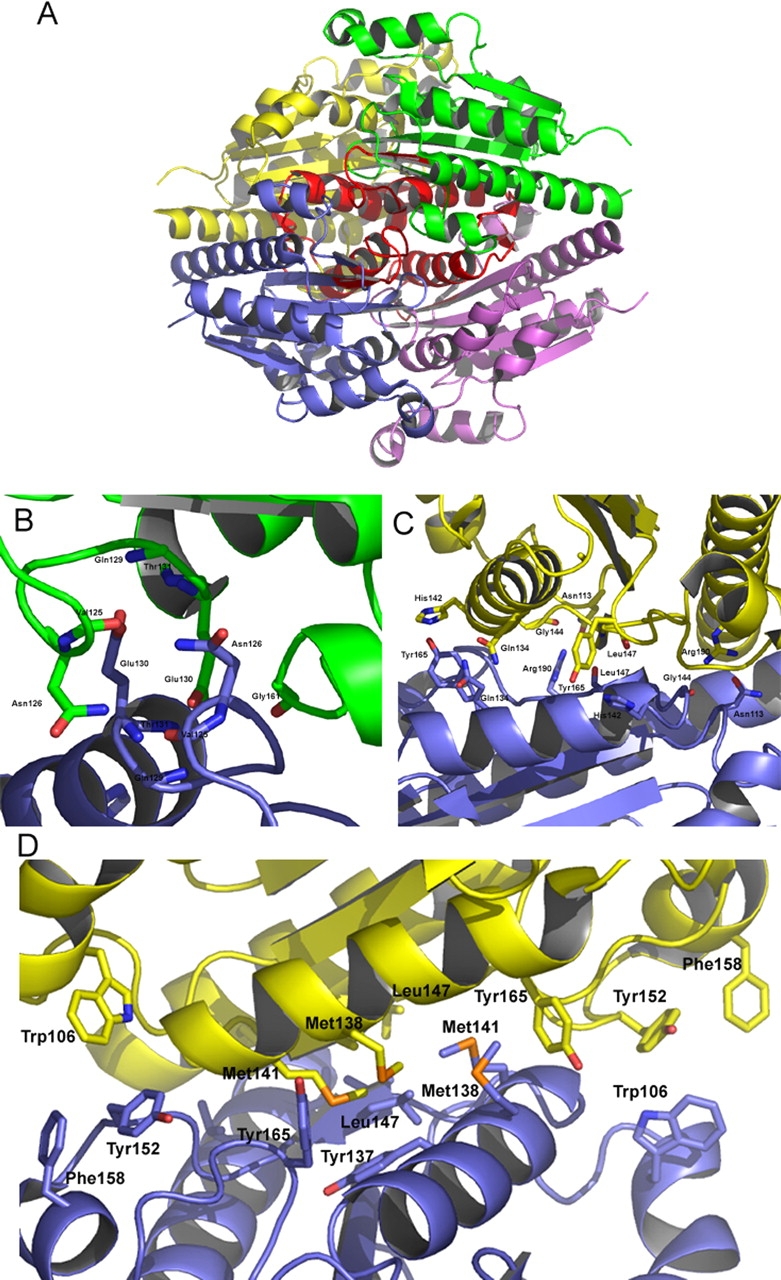
(A) A ribbon representation of WrbA from D. radiodurans highlighting the region surrounding and including α6. This region, shown in red, is located at the core of the tetramer and contributes to many of the tetramerization interactions. (B) Several residues involved in the hydrogen bond tetramerization interactions are shown. Notably, Glu130 is part of a highly conserved GGQE sequence, which interacts with the backbone amido groups of its mate’s GGQE. (C) Tyr165 interacts with His142, which contributes to properly orienting the histidine side chain to interact with FMN. Also of interest is Leu147, located on β5, which shows a two-bond anti-parallel β-sheet with its mate. (D) The hydrophobic residues contributing to the tetramerization are located along the α6 helix located at the center of the tetramer.
FMN binding site
Similar to other flavodoxins, the FMN binding site residues are located along the loop regions at the C-terminal apex of the central parallel β-sheet (Fig. 5A ▶) No significant structural changes are observed upon FMN binding. Residues corresponding to Ser13, Thr15, and Thr17 in the loop region of the D. radiodurans located between β1 and α1 are highly conserved residues among flavodoxins and display interactions with the phosphate moiety of FMN. Interestingly, WrbA from D. radiodurans is one of only a few members of the WrbA family in which these residues are conserved. WrbA from P. aeruginosa shows the sequence SRHGAT in this loop, while most members of the WrbA family show a strong conservation of the sequence SXYGH in this loop. Under the crystallization conditions used here for the D. radiodurans WrbA, this loop binds one sulfate molecule in the absence of FMN (Fig. 5B ▶). The side chain of Ser121 shows hydrogen bond interactions with the ribityl group of the FMN. The loop interactions with the phosphate moiety and ribityl group are similar to the interactions seen in the three similar flavoproteins—1E5D, 5NUL, and 1RCF—although the interactions with the isoalloxazine ring diverge considerably. Interactions with the isoalloxazine ring are mediated by residues Gln123, Asn124, and Gly127. The isoalloxazine ring is stacked over the whole length of the side chain from Arg87, and flanked on other sides by Phe88 and Trp106. Although the Trp106 is nearly 6 Åaway, and does not contact the FMN moiety in the WrbA structures, this residue is highly conserved and is oriented similarly to aromatic residues of quinone reductases (Foster et al. 1999). The analogous residues in quinone reductases form part of a hydrophobic substrate binding pocket adjacent to the FMN binding site. These three residues forming the FMN binding pocket— Arg87, Phe88, and Trp106—are highly conserved among members of the WrbA family. Interestingly, unlike other flavodoxins, His142 of a tetramermate chain also contributes to the binding of FMN. Further, His142 is oriented by interactions with the side chain from Tyr165 of a third chain. This feature implies that the binding of FMN is likely to be influenced by tetramerization.
Figure 5.
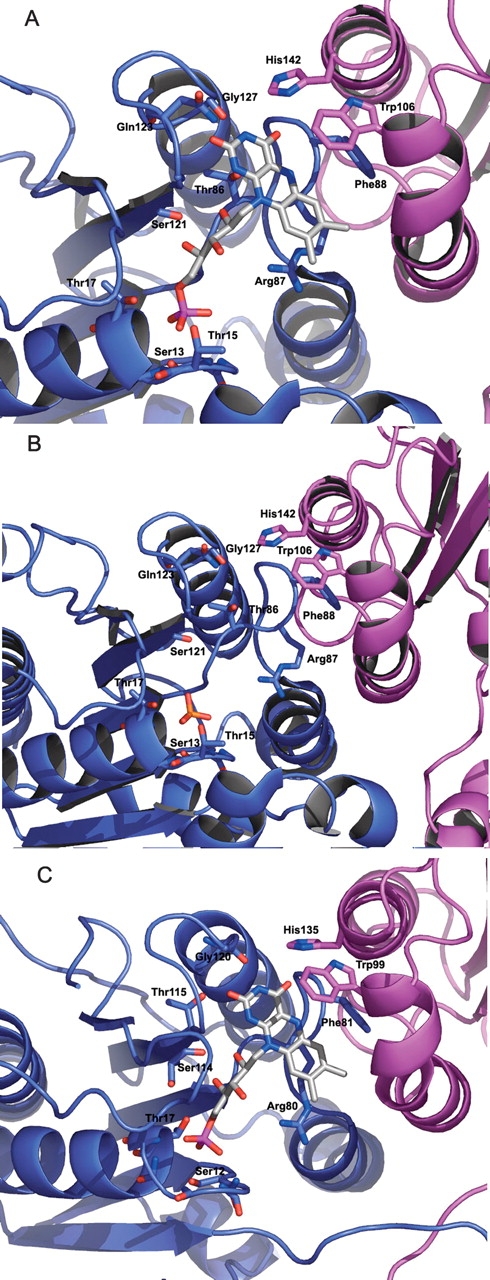
(A) FMN binding site of WrbA from D. radiodurans. The phosphate moiety shows interactions with a number of residues in the loop following β1. Note the side-chain interactions of Ser13 and Thr17, which are conserved in many flavodoxins. His142 from a tetramermate chain interacts with the isoalloxazine ring, thus also contributing to FMN binding. (B) The FMN binding site of WrbA from D. radiodurans crystallized in the presence of sulfate and the absence of FMN. The sulfate binds to the location where the phosphate moiety of FMN is found in the complex. (C) FMN binding site of WrbA from P. aeruginosa. The site is similar to that of D. radiodurans, despite some sequence divergence in the loop following β1. The isoalloxazine ring is similarly stacked around Arg80, Phe81, and Trp99. The FMN also interacts with His135 of a second tetramermate chain.
Discussion
Here we have presented crystal structures of the WrbA proteins from D. radiodurans and P. aeruginosa. These structures show that WrbA forms a 222 tetramer and binds one FMN molecule per protomer. We have confirmed predictions from sequence analysis that WrbA adopts a structure similar to that of long-chain flavodoxins. Further, we have shown that WrbA contains a conserved insertion following β2. The role of this insertion remains unclear, though it does not appear to be a necessary structural addition, and may play a functional role yet to be determined. These structures also show the FMN phosphate binding is similar to known structures; however, unlike other flavodoxins, residues from two chains contribute to interactions with the isoalloxazine ring indicating that tetramerization is likely to influence FMN binding. Tetramers are formed as dimers of dimers. The larger dimer interface, with 2423 Å2 buried surface area, involves no contacts with FMN. This is consistent with prior observations that the low-concentration dimeric gel filtration behavior of E. coli WrbA is unaffected by the presence of nucleotide (Grandori et al. 1998). The smaller dimer interface, burying 1601 Å2, involves intersubunit contacts mediated by FMN, and could thus conceivably be modulated by the presence of this cofactor. However, this is yet to be determined, since analytical ultracentrifugation studies to measure tetramer formation have not yet been compared in the presence and the absence of FMN (Grandori et al. 1998). The structures reported here do not provide clear insight into the identity of the WrbA binding site for TrpR. Nonetheless, these structural data help provide a framework for understanding biological functions for members of this protein family.
Materials and methods
The coding sequence of the wrbA gene from D. radiodurans was subcloned into a modified pET 26b vector, which encodes a C-terminal 6-His tag that was not removed for crystallization. In addition, due to cloning artifacts, the N terminus begins with the sequence MSLTA rather than the native MTA (with the threonine and alanine positions being correct in both cases). Selenomethionine (Se-Met) substituted protein was expressed in E. coli BL21 (DE3) cells using the defined media and autoinduction protocol developed by Studier (2005). The protein was purified by a two-step procedure involving nickel-affinity chromatography eluted with an imidazole gradient, followed by gel filtration on Superdex 75. The protein was concentrated to 27.7 mg/mL in 10 mM HEPES at pH 7.5, 150 mM NaCl, and 10% glycerol. Crystals were obtained by vapor diffusion in 1.2-μL hanging drops containing 0.6 μL of protein and 0.6 μL of well solution. The well solution consisted of 2.6 M ammonium sulfate, 20% glycerol, and 5 mM DTT at pH 4.5, and 20°C. Crystals were soaked with 10 mM FMN for 24 h to obtain structures for the holoprotein. The crystals were flash frozen at 100 K with the addition cryoprotectant consisting of 2.6 M ammonium sulfate and 50% glycerol. Although the oxidation state of FMN is not known with certainty, the crystallization conditions included DTT as a reducing agent. Crystals soaked with FMN for 24 h showed no reaction to a 5-mM increase in the concentration of DTT. Data were collected at the ID-31 beamline (SGX-CAT) at the Advanced Photon Source. The crystals belong to space group P3221 with cell dimensions of a=b=121.61 Å, c=207.93 Å for the apo form and a=b=122.33 Å, c=208.74 Å for the protein bound to FMN. The asymmetric unit consists of two tetramers, eight molecules total per asymmetric unit. Detailed crystallographic statistics can be found in Table 1.
The coding sequences of the wrbA gene from P. aeruginosa was subcloned into a modified pET 26b vector, with N-terminal MAHHHHHHSL tag that was not removed for crystallization. Selenomethionine substituted protein was prepared as described above. The protein was concentrated to 10.5 mg/mL in 10 mM HEPES at pH 7.5, 150 mM NaCl, and 10% glycerol. Crystals were obtained by vapor diffusion in 1.2 μL hanging drops containing 0.6 μL of protein and 0.6 μL of well solution. The well solution consisted of 16% PEG 3350, 0.12 M MgCl, 5 mM DTT, and Bis-Tris at pH 6.0 at 20°C. The crystals were flash frozen at 100 K in well solution supplemented with 30% glycerol. Crystals were soaked with 10 mM FMN for 24 h to obtain structures for the holoprotein. Data were collected at beamlines X29 and X4A at the NSLS. The crystal for the apo form belongs to space group P222 with cell dimensions of a=73.31 Å, b=73.33 Å, c=78.67 Å, and had two molecules per asymmetric unit. The FMN complex crystal belongs to space group P4222 with dimensions a=b=73.54 Å, c=78.17 Å, with one molecule per asymmetric unit. Detailed crystallographic statistics can be found in Table 1.
The data were processed and merged with Denzo (Otwinowski and Minor 1997). Selenium positions of the WrbA from D. radiodurans were located using Solve (Terwilliger and Berendzen 1999), and the initial model was traced with the program Resolve (Terwilliger 2000). O (Jones et al. 1991) was used to complete the model building. Refinement was performed with Refmac 5.2.0005 (Murshudov et al. 1997) using the CCP4i program suite (CCP 4 1994). Water molecules were added using Arp/wArp 6.1.1 (Perrakis et al. 1999). The molecular replacement solution for WrbA from P. aeruginosa was found by Phaser (Storoni et al. 2004) using the structure from D. radiodurans as a starting model. Molecular structure figures were made with Pymol (Delano Scientific), and the sequence alignment figure was made with Jalview (Clamp et al. 2004) and ClustalW (Thompson et al. 1994).
Acknowledgments
Use of the Advanced Photon Source was supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences (BES), under contract no. W-31-109-Eng-38. Use of the SGX Collaborative Access Team (SGX-CAT) beamline facilities at Sector 31 of the Advanced Photon Source was provided by Structural GenomiX, Inc., which constructed and operates the facility. Data for this study were measured at beamline X4 and X29 of the National Synchrotron Light Source. Financial support comes principally from the Offices of Biological and Environmental Research and BES of the DOE, and from the National Center for Research Resources of the NIH. This is a contribution of the New York Structural Genomics Consortium, NIH structural genomics pilot center, grant no. 1P50 GM62529.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051680805.
References
- Burkhart, B.M., Ramakrishnan, B., Yan, H., Reedstrom, R.J., Markley, J.L., Straus, N.A., and Sundaralingam, M. 1995. Structure of the trigonal form of recombinant oxidized flavodoxin from Anabaena 7120 at 1.40 Å resolution. Acta Crystallogr. D Biol. Crystallogr. 51 318–330. [DOI] [PubMed] [Google Scholar]
- Clamp, M., Cuff, J., Searle, S.M., and Barton, G.J. 2004. The Jalview Java Alignment Editor. Bioinformatics 12 426–427. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (CCP4). 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50 760–763. [DOI] [PubMed] [Google Scholar]
- Daher, B.S., Venancio, E.J., de Freitas, S.M., Bao, S.N., Vianney, P.V., Andrade, R.V., Dantas, A.S., Soares, C.M., Silva-Pereira, I., and Felipe, M.S. 2005. The highly expressed yeast gene pby20 from Paracoccidioides brasiliensis encodes a flavodoxin-like protein. Fungal Genet. Biol. 42 434–443. [DOI] [PubMed] [Google Scholar]
- Foster, C.E., Bianchet, M.A., Talalay, P., Zhao, Q., and Amzel, L.M. 1999. Crystal structure of human quinone reductase type 2, a metallo-flavoprotein. Biochemistry 38 9881–9886. [DOI] [PubMed] [Google Scholar]
- Frazão, C., Silva, G., Gomes, C.M., Matias, P., Coelho, R., Sieker, L., Macedo, S., Liu, M.-Y., Oliveira, S., Teixeira, M., et al. 2000. Structure of a dioxygen reduction enzyme from Desulfovibrio gigas. Nat. Struct. Biol. 7 1041–1045. [DOI] [PubMed] [Google Scholar]
- Grandori, R. and Carey, J. 1994. Six new candidate members of the α/β twisted open-sheet family detected by sequence similarity to flavodoxin. Protein Sci. 3: 2185–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandori, R., Khalifah, P., Boice, J.A., Fairman, R., Giovanielli, K., and Carey, J. 1998. Biochemical characterization of WrbA, founding member of a new family of multimeric flavodoxin-like proteins. J. Biol. Chem. 273 20960–20966. [DOI] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1996. Mapping the protein universe. Science 273 595–602. [DOI] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.-Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A. 47 110–119. [DOI] [PubMed] [Google Scholar]
- Lacour, S. and Landini, P. 1994. σS-dependent gene expression at the onset of stationary phase in Escherichia coli: Function of σS-dependent genes and identification of their promoter sequences. J. Bacteriol. 186 7186–7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski, M.J., Dreher, K.A., Gehring, M.A., Abel, S., Gensler, A.L., and Sussex, I.M. 2002. FQR1, a novel primary auxin-response gene, encodes a flavin mononucleotide-binding quinone reductase. Plant Physiol. 128 578–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Llano, J., Maldonado, S., Bueno, M., Lostao, A., Ángeles-Jiménez, M., Lillo, M.P., and Sancho, J. 2004. The long and short flavodoxins. J. Biol. Chem. 279 47177–47183. [DOI] [PubMed] [Google Scholar]
- Ludwig, M.L., Pattridge, K.A., Metzger, A.L., Dixon, M.M., Eren, M., Feng, Y., and Swenson, R.P. 1997. Control of oxidation-reduction potentials in flavodoxin from Clostridium beijerinckii: The role of conformation changes. Biochemistry 36 1259–1280. [DOI] [PubMed] [Google Scholar]
- Murshudov, G.N., Vagin, A.A., and Dodson, E.J. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53 240–255. [DOI] [PubMed] [Google Scholar]
- Nicholls, A., Sharp, K., and Honig, B. 1991. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11 281–296. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Perrakis, A., Morris, R., and Lamzin, V.S. 1999. Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 6 458–463. [DOI] [PubMed] [Google Scholar]
- Rose, J.K. and Yanofsky, C. 1974. Interaction of the operator of the tryptophan operon with repressor. Proc. Natl. Acad. Sci. 71 3134–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, W.W., Pattridge, K.A., Ludwig, M.L., Petsko, G.A., Tsernoglou, D., Tanaka, M., and Yasunobu, K.T. 1983. Structure of oxidized flavodoxin from Anacystis nidulans. J. Mol. Biol. 165 737–753. [DOI] [PubMed] [Google Scholar]
- Storoni, L.C., McCoy, A.J., and Read, R.J. 2004. Likelihood-enhanced fast rotation functions. Acta Crystallogr. D Biol. Crystallogr. 60 432–438. [DOI] [PubMed] [Google Scholar]
- Studier, F.W. 2005. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41 207–234. [DOI] [PubMed] [Google Scholar]
- Thompson, J., Higgins, D., and Gibson, T. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger, T.C. 2000. Maximum-likelihood density modification. Acta Crystallogr. D Biol. Crystallogr. 56 965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger, T.C. and Berendzen, J. 1999. Automated MAD and MIR structure solution. Acta Crystallogr. D Biol Crystallogr. 55 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, W., Ni, L., and Somerville, R.L. 1993. A stationary-phase protein of Escherichia coli that affects the mode of association between the Trp repressor protein and operator-bearing DNA. Proc. Natl. Acad. Sci. 90 5796–5800. [DOI] [PMC free article] [PubMed] [Google Scholar]