

Abstract
Structural genomics (SG) initiatives are expanding the universe of protein fold space by rapidly determining structures of proteins that were intentionally selected on the basis of low sequence similarity to proteins of known structure. Often these proteins have no associated biochemical or cellular functions. The SG success has resulted in an accelerated deposition of novel structures. In some cases the structural bioinformatics analysis applied to these novel structures has provided specific functional assignment. However, this approach has also uncovered limitations in the functional analysis of uncharacterized proteins using traditional sequence and backbone structure methodologies. A novel method, named pvSOAR (pocket and void Surface of Amino Acid Residues), of comparing the protein surfaces of geometrically defined pockets and voids was developed. pvSOAR was able to detect previously unrecognized and novel functional relationships between surface features of proteins. In this study, pvSOAR is applied to several structural genomics proteins. We examined the surfaces of YecM, BioH, and RpiB from Escherichia coli as well as the CBS domains from inosine-5′-monosphate dehydrogenase from Streptococcus pyogenes, conserved hypothetical protein Ta549 from Thermoplasm acidophilum, and CBS domain protein mt1622 from Methanobacterium thermoautotrophicum with the goal to infer information about their biochemical function.
Keywords: structural genomics, protein surface, surface pattern, protein function, pocket sequence, pocket shape, surface matching, functional genomics
Structural genomics initiatives seek to advance high-throughput methods of protein structure determination. By specifically targeting proteins without sequence similarity (the <30% identity rule), they are systematically filling in gaps in the universe of protein fold space. This strategy targets proteins with predicted function as well as proteins that are uncharacterized. For example, the Protein Structure Initiative (PSI) pilot projects have added to the protein structure body of knowledge at an unprecedented rate (77 [35 as preliminary data] deposited structures in the Protein Data Bank [PDB] [Berman et al. 2002] in 2001, 109 structures in 2002, 217 structures in 2003, and 404 in 2004). Sixty-six percent of these structures are unique. While innovations in protein structure determination become increasingly automated, the current structural bioinformatics approach resulted in only limited functional assignment of uncharacterized proteins (Sanishvili et al. 2003).
Instructural genomics, the majority of newly determined protein structures are, by design, unrelated to other known proteins. This challenges the limits of current methods of functional inference based on primary sequence and backbone structure. As a result, time-intensive, expert manual analysis, and experimental approaches are required to predict and verify the biochemical function of an un-characterized protein. This approach does not scale up as it struggles to keep pace with the accumulation of new structures. Development of automated approaches for functional inference will be highly valuable as structural genomics projects advance.
Protein biochemical function is typically associated with a specific 3D assemblage of residues involved directly or indirectly in binding and/or catalysis. Functional inference by sequence similarity must first identify a protein or protein family with a known function and relies on conservation of residues that have been shown to be crucial to protein function (Smith and Waterman 1981; Rost 2002; Tian and Skolnick 2003). More reliable is functional inference using structure when a structural homolog can be identified and functional sites directly compared (Orengo et al. 1999b; Todd et al. 1999). The biochemical function of the uncharacterized protein can then be inferred.
Similarity measures have been derived from a variety of algorithms based on both primary sequence analysis and three-dimensional structural analyses. Sequence analysis has proven to be valuable, and can provide functional inference for proteins sharing >70% sequence identity (Rost 2002; Tian and Skolnick 2003). Structural analysis has shared higher successes (Orengo et al. 1999b; Jaroszewski and Godzik 2000), but studies have also reported ambiguous results from similarities based on structural comparisons alone (Feng and Sippl 1996). It is now very well established that proteins sharing similar structures can perform different functions (Orengo et al. 1999a), and proteins with very different structures can perform identical function (Russell et al. 1998). Local spatial motifs, such as active site templates and metal binding sites derived from the three-dimensional patterns of proteins with known function or a set of highly conserved residues (Holm and Sander 1994; Wallace et al. 1997; Russell 1998; Di Gennaro et al. 2001), are very useful to identify potential functional sites in proteins. However, these methods are often restricted to a predetermined size and prior knowledge of functional site residues, which make it difficult to search for similar patterns across proteins in structural databases. The major problem in rigid template searches is associated with inherent flexibility of amino acid side chains as well as accommodation of functional site geometries to ligands and cofactors.
Recently a novel method, named pvSOAR (pocket and void Surfaces of Amino Acid Residues), of comparing the protein surfaces of geometrically defined pockets and voids was developed (Binkowski et al. 2003a, 2004). This method for protein surface analysis can capture the physicochemical texture and shape of a surface around functional residues. Pockets and void surfaces are analytically determined using the method of Computed Atlas of Surface Topography of Proteins (CASTp) (Binkowski et al. 2003b), which identifies and organizes all known protein surfaces from the PDB. pvSOAR systematically searches a query surface identified in the known protein structure against all protein surfaces based on a combination of the physicochemical texture, three dimensional side-chain arrangement, and orientation of residues for biochemical function, and reports statistically significant matches. The method is fast, robust, and is fully automated without the need of human intervention. By assessing the similarity between local surface sequence, local surface shape, and local surface orientation, pvSOAR was able to detect previously unknown relationships between aromatic aminotransferase and 17-β-hydroxy-steroid dehydrogenase and similarity between HIV-1 protease and heat-shock protein-90 (Binkowski et al. 2003a).
When pvSOAR is applied to a structural genomics protein structure, it can be an effective tool for functional analysis. It can be utilized to address broad questions about potential function or specific queries such as ligand preference or metal specificity. In this study, our starting point is biological knowledge gained from literature. Here, we present the results of surface pattern comparisons using several proteins from the Midwest Center for Structural Genomics (MCSG) pilot project. Specifically, we present a prediction of the active site location and propose metal substrate specificity of a hypothetical protein YecM from Escherichia coli. Next, we identify a putative CoA binding surface potentially important as a precursor to the known biotin synthesis pathway in BioH from E. coli. We then analyze two functional homologs, RpiB and RpiA from E. coli, that share no observable sequence or structural similarity. Finally, we propose two adenine nucleotide binding sites for the uncharacterized cystathionine β-synthase (CBS) domain.
Materials and methods
Functional inference and annotation using protein surfaces involve a number of key steps. An overview of the pvSOAR search methodology is shown in Figure 1 ▶. First, the three-dimensional coordinates of a protein structure are submitted to the CASTp Web server for pocket and void calculation and identification. Each pocket is assigned a unique identification number, roughly corresponding in order of increasing volume. A surface pocket is then used as a query template for searching against a library of identified surfaces. pvSOAR will return statistically significant hits based on several comparison metrics.
Figure 1.

An overview of the pvSOAR search methodology using YecM from E. coli. The structure of YecM (A) is submitted to the CASTp Web server (B) for pocket and void identification. A surface is identified, in this case CASTp pocket 18, and selected as the query template (C, green). The query is submitted to the pvSOAR Web server (D) and statistically significant surfaces matches are returned: neurolysin (PDB ID 1i1i, CASTp ID 85) from R. norvegicus (E, red), thermolysin (PDB ID 1lnd, CASTp ID 47) from B. thermoproteolyticus (F, orange), and from lethal toxin factor (PDB ID 1j7n, CASTp ID 198) from B. anthracis (G, yellow). The conserved residues between the query and library surfaces are superimposed (H). Figures were generated using PyMol (DeLano 2002) and CASTpyMol plugin.
In the absence of any functional annotation, as can be the case in structural genomics proteins, every surface of the query structure is searched against the entire pvSOAR surface library. The run time complexity of the pvSOAR algorithm and the increasing size of the PDB make this exhaustive search strategy undesirable. While structural genomics targets are all hypothetical proteins, it is becoming increasingly rare to solve structures that are completely unique. At the MCSG, only 7% of structures solved have new folds, meaning that most newly solved structures have some measurable similarity to other structures. Even when, as in many cases, the similarity is limited to other structural genomics targets with no known function, valuable information, such as conservation of structural features or residue composition, can be useful in selecting a query surface. Selection of the query surface pocket can also be aided by reviewing literature, when available, to identify residues of interest and mapping them to the surface containing it.
pvSOAR Surface Library
All pockets and void surfaces from proteins deposited in the PDB are exhaustively identified and organized by the CASTp server. The server is updated weekly to keep current with PDB releases. A pvSOAR surface library is composed of surfaces from the CASTp server. Currently, there are over 3,000,000 surfaces in the entire pvSOAR surface library, which represent all the proteins in the PDB. To reduce significant bias from similar surfaces across homologous protein families, a subset of the pvSOAR database is created from structures listed in the PDBSELECT (25%) (Hobohm and Sander 1994) dataset. This reduces the searchable database to 150,000 surfaces decreasing the search run time as well as decreasing the “noise” from homologous surfaces.
In some cases, functionally important residues can be identified, from PDB files (REMARK 800), and mapped to a particular surface. This allows for the creation of annotated surface libraries. This subset of annotated surfaces represents less than 1% of surfaces in the PDB, but in some cases can be used to create specialized surface libraries. The prevalence of bound nucleotides in the PDB has allowed a fairly comprehensive library to be constructed.
Comparison of protein surfaces
Protein surfaces are compared by local sequence composition, local shape, and local orientation between residues located on a geometrically defined pocket or void. pvSOAR is based on the methodology described in Binkowski et al. (2003a), and is used to identify similar surface regions in three-dimensional protein structures. A search is based on comparison of a query pattern of surface sequence and substructure against a database of surface patterns from known protein structures. pvSOAR only compares residues that are conserved between the query and the library surface. The level of conservation can be specified at run time to be rigid (identical residues) or flexible (Blosum62) (Henikoff and Henikoff 1992).
The statistical significance of similarity, in the form of P-values, between substructures as measured by the coordinate root mean square distance (cRMSD) are provided for discerning potentially biologically important results. The P-value of a local surfaces alignment is the probability of obtaining a cRMSD value by chance. The use of P-value allows for meaningful comparisons between alignments of differing number of residues, a task not possible by using the raw cRMSD value. In addition, a newly developed metric for substructure comparison, called orientation root mean square distance (oRMSD), is also provided, along with the corresponding statistical significance evaluations (Binkowski et al. 2003a). In an oRMSD measurement, spatial coordinates of residues from a pocket are first projected onto a unit sphere placed at the center of mass; the RMSD between the two sets of transformed residues is then measured. oRMSD provides a computationally feasible approach to identify similar surface patterns which undergo conformational changes.
Search heuristics
The exhaustive calculation of all pocket and voids surfaces in the PDB results in an informative but large search space. In some cases, a single surface search can identify a large number of statistically significant matches. We use the following criteria to select potentially biologically interesting hits. By default we utilize the smallest surface library that is relevant to the question. This involves creating a specialized surface library, when possible, or filtering surfaces by solvent accessible area or volumes (as identified by CASTp). For example, a library of known CoA binding surfaces was compiled for a study reported in a later section. Excluding large protein–protein interface pockets can also significantly reduce the search space and run time when searching for a catalytic triad. Our empirical results have indicated that <10−2 is typically the cutoff for biologically meaningful results. We therefore use this as a cutoff for automatically discerning true positive hits.
We also require greater than four residues be conserved between the two surfaces. Studies using three residue search motifs (e.g., catalytic triads) have been investigated and reported in Wallace et al. (1997) and Russell (1998). With the assumption that the greater surface environment surrounding functional residues are also important in defining biological function of molecules and in discriminating similar surfaces, we require four instead of three conserved residues.
Results
We describe examples of inferring biochemical function by detecting surfaces on the structures obtained from structural genomics proteins that are similar to known functional surfaces on other proteins. First, we present functional analysis from the results of a surface search with a proposed metal binding site on YecM from E. coli. Next, we identify a putative CoA binding surface potentially important as a precursor to the known biotin synthesis pathway in BioH from E. coli. In some cases, the biological functions of a protein from structural genomics are determined without knowledge of the mechanisms responsible for the activity. To this end, we show remote surface similarities between active sites on ribose 5-phospate isomerase (Rpi) structures. Finally, we identify a putative adenine nucleotide binding site for functionally uncharacterized CBS domain. We also propose novel adenine nucleotide binding sites and a potential mode for single domain binding.
Metal binding surface specificity of YecM from E. coli
YecM from E. coli (PDB ID 1k4n) is a conserved, un-characterized protein with sequence homologues found exclusively in bacteria (Fig. 1A ▶) (Zhang et al. 2003c). The protein was chosen as a structural genomics target because it exhibited no sequence similarity to any proteins of known structure.
Structure analysis by Zhang et al. (2003c) discovered that the structure of YecM shares remote structural similarity to an isomerase (PDB ID 1jc4, EC 5.1.99.1) and to several oxidorectases (PDB IDs 1mpy, 1cjx, 1han, EC 1.13.11.-). It also has more distant similarities to other proteins as well. A literature review of all structural homologs revealed a common bound divalent metal cation, leading to the prediction of YecM as a metal binding protein. Structural analysis of YecM revealed residues involved in proposed metal binding (His46, Glu101, His117, and Lys176). The preferred metal, based on geometric distances, was not able to be conclusively determined, but Co+2 or Zn+2 were suggested as good candidates.
The proposed coordinating residues of YecM are all located in a single well-formed surface pocket (CASTp ID 18, chain = A) (Fig. 1C ▶). An exhaustive comparison was performed between this pocket and a library of all known metal binding surfaces in the PDB to more accurately predict the metal cofactor. Figure 1 ▶ shows three metal bindings surfaces from neurolysin (PDB ID 1i1i, CASTp ID 85) from Rattus norvegicus (Fig. 1E ▶, red), thermolysin (PDB ID 1lnd, CASTp ID 47) from Bacillus thermoproteolyticus (Fig. 1F ▶, orange), and from lethal toxin factor (PDB ID 1j7n, CASTp ID 198) from Bacillus anthracis (Fig. 1G ▶, yellow) with strong similarity to the potential YecM metal binding surface (Fig. 1C ▶, green). All three surfaces have zinc bound through tetrahedral coordination from two histidine and two aspartic acid residues. pVSOAR surface comparisons had surface alignment P-values of 10−3 (cRMSD) and 10−4 (oRMSD), representing highly statistically significant matches. A superposition of the query surface residues and the conserved residues from the library surfaces is shown in Figure 1H ▶.
The pvSOAR search resulted in less significant (<10−1 and >10−3) matches from surfaces bound with other metals (i.e., Co, Mn, Fe, Mg). In one case (PDB IDs 1lna, 1lnb, 1lnc, 1lnd), the surfaces were all part of a study to determine the effect of different metals on the catalytic activity in thermoylsin (Holland et al. 1995). YecM had the strongest similarity to the native zinc binding surface (Fig. 1F ▶). A rank order listing of all significant hits has zinc binding surfaces in the top 30% of all matches. While the binding site of YecM could potentially bind different metals, the strong similarity to zinc metal binding surfaces indicate this as a strong candidate for the preferred metal.
Coenzyme A binding surface of BioH from E. coli
BioH from E. coli, a two-domain protein involved in biotin biosynthesis, was chosen as a structural genomics target because it had no sequence homologues in the PDB. Prior to the 1.7 Å structure (PDB ID 1m33) being determined, its function was unknown (Sanishvili et al. 2003). Secondary structural alignment using the DALI (Holm and Sander 1994) server showed backbone similarity to bromoperoxidase, aminopeptidase, epoxide hydrolases, haloalkane dehalogenase, and lyase. Structural bioinformatics analysis revealed a Ser82–His235–Asp207 catalytic triad similar to lipases (EC 3.1.1.3), with Ser82 identified as belonging to a sequence motif typical for acyltransferases and thioesterases (Sanishvili et al. 2003). Enzymatic assays showed significant esterase (carboxylesterase or thioesterase) and acetyltransferase activity. It also hydrolyzed p-nitrophenyl esters of fatty acids with a broad substrate preference for short-chain substrates. BioH was subsequently determined to be a new carboxylesterase in E. coli.
As a precursor to the known biotin synthesis pathway, BioH was proposed to condense coenzyme A (CoA) and pimelic acid into pimeloyl-CoA. BioH would function as a CoA donor to pimeloyl-acyl-carrier protein (pimeloyl-BioC), releasing pimeloyl-CoA. A complex of BioH–CoA has been identified through liquid chromatography-mass spectrometry (Tomczyk et al. 2002). To further elucidate the proposed role of BioH in biotin synthesis, the solvent accessible surfaces were searched against a library of CoA binding surfaces to identify the cofactor binding site.
A grouping of basic residues on the protein surface lie in a well-formed pocket (CASTp ID 120) with solvent-accessible surface area and volume of 179 Å 2 and 89 Å 3, respectively (Fig. 2A ▶). The best scoring hit from a search of this surface against known CoA binding surfaces in the PDB was from aminoglycoside 2′-N-acetyltransfer-ase [AAC(2′)-lc] from Mycobacterium tuberculosis (PDB ID 1m4g, E.C.2.3.1.-) (Fig. 2B ▶). AAC(2′)-lc catalyzes the CoA-dependent acetylation of the 2′-hydroxyl amino group of a broad spectrum of aminoglycosides (Vetting et al. 2002). There were 15 residues conserved between the two surfaces which superimpose for a cRMSD and oRMSD P-value of 7.6 × 10−3 and 1.55 × 10−6, respectively (Fig. 2C ▶). Included in the conserved surface residues are Arg138,142,155,159 and Lys162, which are conserved throughout many bacteria (Fig. 2A ▶) (Sanishvili et al. 2003). In Figure 2D ▶, a CoA ligand has been modeled into the surface of BioH, based on the surface superposition with AAC(2′)-lc.
Figure 2.
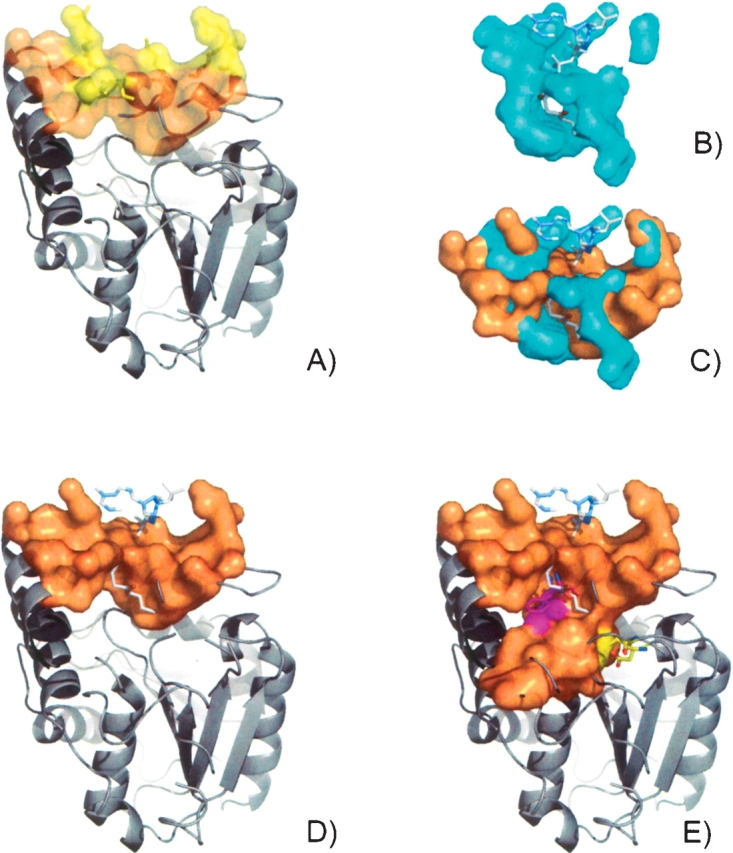
The surface pocket (CASTp ID 20, orange) on BioH from E. coli (PDB ID 1m33) contains a grouping of basic residues (yellow) (A). The CoA binding from aminoglycoside 2′-N-acetyltransferase (AAC(2′)-lc) from M. tuberculosis (PDB ID 1m4g, E.C.2.3.1.-) (B). CoA ligand has been modeled into the surface of BioH (D), based on the superposition with AAC(2′)-lc (C). Using a smaller solvent probe radius (1.2 Å) to define the pocket, the surface reveals an additional channel protruding into the domain interface which contains the buried catalytic triad (yellow) and Phe143 (magenta) (E).
To account partially for the possible rearrangement of residues in the active site such that additional residues become exposed, the BioH surface was recalculated using a smaller solvent probe radius (1.2 Å). The extended pocket reveals an additional channel protruding into the domain interface (Fig. 2E ▶). The solvent-accessible surface area and volume of the pocket are 387 Å 2 and 159 Å 3, respectively. The redefined surface also includes the buried catalytic triad residues (Fig. 2E ▶, yellow) and an invariant Phe143 (Fig. 2E ▶, magenta), which is thought to act as a binding facilitator for acyl substrates. When the ligand is modeled into the structure, the thiol group is positioned in close proximity to the catalytic triad (3.5 Å) and Phe143 (4 Å) (Fig. 1E ▶).
Detected surface similarities to known CoA binding proteins can help provide insight into the possible role of BioH in the biotin pathway. Based on similarity to AAC(2′)-lc, the newly identified CoA binding site could position the ligand directly into the hydrophobic crevice in the cap domain. It is possible that in the bound state, conformational changes could widen the crevice, which had shown preference for short-chain acyl substrates, allowing delivery of a pimeloyl unit to the catalytic site. The pimelic acid and CoA could then be condensed into pimeloyl-CoA.
Structural basis for the active site of RpiB
RpiB from E. coli (PDB ID 1nn4), has been shown to have ribose-5-phosphate isomerase activity (EC 5.3.1.6). However, uncertainty remained regarding the identity of the active site in the crystal structure (Zhang et al. 2003b). RpiA from E. coli (PDB ID 1o8b), a functional homolog of RpiB, exhibited neither sequence nor structural homology to RpiB. Both structures are comprised of α-helices and β-sheets (α /β), but Rpib belongs to the ribose/galactose isomerase RpiB/AlsB fold (SCOP c.121.1.1) while RpiA belongs to the NaqB/RpiA/CoA transferase-like fold (SCOP c.124.1.4) (Murzin et al. 1995). The active-site residues in RpiA were identified using mutagenesis and cocrystal structure with inhibitor (Zhang et al. 2003a). Interestingly, the grouping of residues forming the RpiA active site was not found on the RpiB. Moreover, the inhibitors of RpiA were ineffective against RpiB (Zhang et al. 2003b). A possible active site on RpiB was proposed based on multiple sequence alignments of the RpiB/LacAB family and structure analysis (Zhang et al. 2003a).
Recently, the structure of RpiB from M. tuberculosis (PDB ID 1USL) was determined (Roos et al. 2004). The tetrameric E. coli RpiB and M. tuberculosis RpiB share 30% sequence identity. The active site of RpiB from M. tuberculosis was proposed through a ligand docking study, which also provides insight on the active site of E. coli RpiB. While all three proteins perform the same function of interconverting ribose-5-phosphate and ribu-los-5-phosphate, their active sites lack significant similarities that were thought to exist. We utilize pvSOAR to conduct pairwise comparisons of the active sites to identify similar surface features which preserve their functionality despite any sequence or structural homology.
The surfaces containing the proposed catalytic residues are shown in Figure 3A–C ▶. The presumed active sites of two RpiBs are formed at the dimer interfaces, while the active site of RpiA is confined to residues within one subunit (although RpiA exist as a dimer). Between 16–20 amino acids form the active site pockets creating similar solvent-accessible surface areas (130, 137, and 165 Å 2) and volumes (67, 71, 79 Å 3) for E. coli RpiB (CASTp ID 67), RpiA (CASTp ID 49), and M. tuberculosis RpiB (CASTp ID 68), respectively. A pvSOAR comparison was carried out for each pair of surfaces. The conserved residues between M. tuberculosis and E. coli RpiB share almost identical surface geometry, with cRMSD and oRMSD P-values of 4.25 × 10−9 and 4.23 × 10−9, respectively. The phosphate binding residues are strongly conserved between the 12 conserved residue surfaces. Despite near identical orientation in the RpiB pocket surfaces, the putative catalytic base Cys75 of E. coli is a Glu75 in M. tuberculosis.
Figure 3.

The surfaces containing the proposed catalytic residues of RpiB from E. coli (PDB ID 1nn4, CASTp ID 67) (A) and M. tuberculosis (PDB ID 1usl, CASTp ID 67) (B) and RpiA from E. coli (PDB ID 1o8b, CASTp ID 49) (C). The distribution of amino acid residues in each surface (D).
Pocket surfaces of the two RpiB proteins showed more remote similarity to the RpiA. RpiB from E. coli shared six conserved residues that superimpose to cRMSD and oRMSD P-values of 1.78 × 10−2 and 2.18 × 10−3, respectively. RpiB from M. tuberculosis shared seven conserved residues that superimpose to cRMSD and oRMSD P-values of 1.01 × 10−1 and 1.26 × 10−3, respectively. Three residues are invariant among the surfaces of the three isomerases: an asparatic acid and two glycines (Fig. 3A,D,F ▶, highlighted in red). This aspartic acid was found to be the most important for catalysis (Zhang et al. 2003a). While glycine is not traditionally thought of as a functionally important residue, it may be important for these binding surfaces, allocating space for the arrangement of side chains from larger functional residues.
The most notable difference between RpiA and RpiB surfaces is the discrepancy in basic residue composition (Fig. 3D ▶). Both RpiB proteins contain three His and three Arg, while RpiA contains neither His nor Arg. It does, however, contain three Lys, which are located in the similar positions to His/Arg in RpiB. Allowing Lys/Arg/His substitutions in the pvSOAR calculations, the pockets were searched against each other (Lys/Arg/His all have positive substitution values in both PAM and BLOSUM matrices). The surface alignments of RpiA to RpiB had a 100-fold improvement in statistical significance, with cRMSD and oRMSD P-values of 4.17 × 10−4 and 1.55 × 10−4 for E. coli and 5.05 × 10−3 and 1.64 × 10−4 for M. tuberculosis.
While the residue composition of these pockets varies, the size and functional features of the surfaces are conserved. Surface patches of positively-charged residues for binding are conserved across all structures. The orientation of acidic and basic residues important for catalysis is also conserved. The similarities and differences between these two functional sites are striking, given that they perform the same biological function. Although experimental insight that these proteins all have the same substrate would suggest that they also have similar binding surfaces, RpiA and RpiB have no detectable sequence and structural similarity. Only surface analysis such as pvSOAR search is able to detect the functional similarity among the binding surfaces of these proteins. This indicates that pvSOAR can be useful in detecting convergent evolution as in this case of ribose-5-phosphate isomerase activity.
Adenine nucleotide binding in CBS domain proteins
CBS domains are small motifs of unknown function that are ubiquitous in all known species. The 60-residue domain is usually found in tandem pairs, which associate via hydrophobic interactions between homologous β-sheets and is often part of a larger protein (Bateman 1997). Point mutations in this region have been linked to several hereditary diseases in humans, including homocystinuria, retinitis pigmentosa, congential myotonia, idiopathic generalized epilepsy, hypercalciuric nephrolithiasis, classic Bartter syndrome, and Wolff-Parkinson-White syndrome (Scott et al. 2004). Recent work by Scott et al. (2004) provided the first insight into the general function of CBS domains pairs as cellular energy status sensors. They showed that pairs of CBS from AMP-activated protein kinase, IMP dehydrogenase-2, and chloride channel CLC2 bind adenosyl moieties such as AMP, ATP, or S-adenosyl methionine (SAM). Many question remain unclear about the biochemical role of CBS domains, and their active site remains uncharacterized.
Structures containing CBS domains from CBS domain protein mt1622 from M. thermoautotrophicum (PDB ID 1pbj), inosine-5′-monosphate dehydrogenase (IMPDH) from Streptococcus pyogenes (PDB ID 1zfj, E.C.1.1.1.205) (Zhang et al. 1999), and conserved hypothetical protein Ta549 from Thermoplasm acidophilum (PDB ID 1pvm) have been solved at MCSG (Fig.4A–C ▶). The three domains share sequence similarity (20%) inadequate for functional inference, yet all share a common fold. Solvent accessible surfaces on the structures were identified and searched against a library of AMP and ATP binding surfaces in the PDB to identify the domain binding site.
Figure 4.
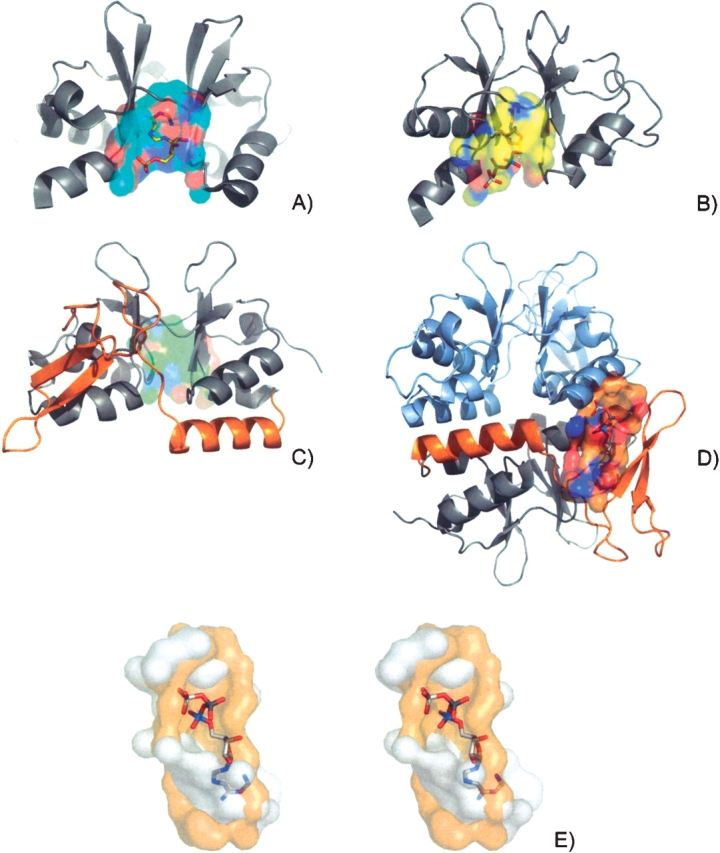
Structures containing CBS domains from CBS domain protein mt1622 from M. thermoautotrophicum (PDB ID 1pbj) (A), inosine-5′-monosphate dehydrogenase (IMPDH) from S. pyogenes (PDB ID 1zfj, E.C.1.1.1.205) (Zhang et al. 1999) (B), and conserved hypothetical protein Ta549 from T. acidophilum (PDB ID 1pvm) (C). Surfaces are colored by element type. The proposed nucleotide bindings surface of mt1622 (CASTp ID 9) has AMP modeled into it based on the superposition to a flavoprotein (PDB ID 1efp) (A). The proposed nucleotide bindings surface of IMPDH (CASTp ID 31) has ATP modeled into it based on the superposition to cyclin-dependent kinase 2 (PDB ID 1b38) (B). Ta549 contains an additional C terminus CBS domain (C, orange) opposite the tandem domain interface surface (C, green). The domain insert creates a novel surface that shares similarity to an ATP binding surface from saicarsynthase (PDB ID 1obd). An ATP molecule has been modeled into the surface of Ta549 (orange) based on the superposition of the conserved residues (D). A stereo representation of the surfaces from Ta549 (orange) and saicarsynthase (white) with the modeled ATP (E).
Tandem domain interface binding surface
The prevalence of CBS binding domains being found in tandem pairs has led to the belief that the binding surface is located in between these tandem pairs at the domain interface. Well-defined surfaces are indeed formed at the interface with solvent-accessible surface areas (150 and 210 Å 2) and volumes (150 and 141 Å 3) for mt1622 (CASTp ID 31) and IMPDH (CASTp ID 9), respectively (Fig. 4A,B ▶). The IMPDH CBS domain has a slightly larger interface pocket due mainly to a loop region (residues 93–97) that connects back to the catalytic domain. The surfaces share remote similarity to each other with only seven conserved residues aligning with cRMSD and oRMSD P-values of 3.03 × 10−2 and 6.04 × 10−4, respectively.
Both surfaces had many strong hits to diverse AMP and ATP binding surfaces from a wide variety of enzymes. Well-scoring hits were of the order of 10 × 10−3, with no observable correlation between nucleotide type and P-value. One of the best hits to mt1622 was an AMP binding surface from an electron transfer flavoprotein from Paracoccus denitrificans (PDB ID 1efp). The two surfaces share 10 conserved residues that superimpose to cRMSD and oRMSD P-values of 9.07 × 10−3 and 1.11 × 10−5, respectively. The AMP ligand has been modeled into the pocket of mt166 based on the surface superposition positioning the adenine deep inside the cleft with the phosphate group extending through the mouth (Fig. 4A ▶). One the best hits to IMPDH was from an ATP binding surface found in cyclin-dependent kinase 2 from Homo sapiens (PDB ID 1b38, E.C.2.7.1.37). The two surfaces share 16 conserved residues that superimpose to cRMSD and oRMSD P-values of 8.02 × 10−3 and 1.26 × 10−4, respectively. The ATP molecule has been modeled in to the surface of IMPDH and the ligand positions itself in a similar orientation to AMP (Fig. 4B ▶).
In agreement with Scott et al. (2004), it appears that both AMP and ATP could bind in the tandem CBS domain pockets, but without specific binding studies for these proteins, it is difficult to predict exactly with adenine nucleotide what is the natural ligand. However, when manually inspecting the nucleotide position in the superimposed surfaces, the geometry of the CBS domain from mt1622 appeared more favorable to AMP and IMPDH to ATP, but this could be artificially due to the larger surface.
Single-domain interface binding surfaces
Searching the surfaces of conserved hypothetical protein Ta549 CBS from T. acidophilum yielded unexpected results. The proposed tandem domain interface binding surface (CASTp ID 17, Chain A; Fig. 4C ▶, green) yielded equally significant matches to nucleotide binding surfaces as seen in IMPDH and mt1622, but the most significant hit was to an alternative surface formed by a C terminus domain insert (CASTp ID 30; Fig. 4D ▶, orange). The structure of Ta549 contains a 53-residue domain insert comprising an additional CBS domain unit not seen in the other structures. The overall structure contains six CBS domains, four of which are part of the expected tandem pair arrangement. The other domains exist as singletons with one on each chain, forming a trimerization domain.
A novel surface is formed from a sheet from the single domain and an adjacent helix from the CBS domain pair on the other chain. The surface is composed of 20 residues with a solvent-accessible surface area of 126 Å 2 and a volume of 59 Å 3 smaller than the tandem domain surface. The surface shares little similarity to the proposed IMPDH (15 conserved residues, cRMSD and oRMSD P-values of 1.18 × 10−2 and 3.10 × 10−4) and mt1622 (eight conserved residues, cRMSD and oRMSD P-values of 1.02 × 10−1 and 1.94 × 10−3) nucleotide binding surfaces. Results from the surface search revealed the most significant hit seen of all CBS domains searches. The ATP binding surface from saicar-synthase from Saccharomyces cerevisiae (PDB ID 1obd, E.C.6.3.2.6) shared 12 residues that superimpose to cRMSD and oRMSD P-values of 7.1 × 10−5 and 4.78 × 10−7, respectively. The ATP molecule has been modeled into the surface of Ta549 based on the surface superposition (Fig. 4D ▶).
An unresolved issue from Scott et al. (2004) was the discovery of two binding sites for both AMP and ATP in the CBS domain of the γ 2 subunit of AMPK. The discovery of the putative single domain binding surface can provide a hypothesis for multiple binding sites in a CBS binding domain. A single CBS domain may contain the physicochemical texture to allow nucleotide binding, but it needs to be stabilized. In most cases this is done by the second CBS domain, but it could also be stabilized by nearby secondary structure elements, either from a third CBS domain, as seen here, or from another domain, such as the catalytic domain of IMPDH (this alternative binding site was not observed). A stabilized single CBS domain could then potentially retain its function role. The presence of a second binding site could provide added complexity to the regulatory function of CBS domains in some organisms. This could also explain the lack of observed specificity in the tandem interface binding surface, as the preferred ligand may be dependent on allosteric effects of the single domain binding surface in some circumstances.
Discussion
Identifying similarities between protein surfaces can provide important insight into the functions of unknown proteins. In many cases, similarity in protein surface patterns can identify novel relationships in similar binding or catalysis that would go undetected when using traditional sequence or backbone structure comparisons.
In this study, we examined the surfaces of structural genomics proteins, YecM, RpiB, and BioH from E. coli and CBS domain proteins from CBS domain protein mt1622 from M. thermoautotrophicum, IMPDH from S. pyogenes, and conserved hypothetical protein Ta549 from T. acidophilum, with the goal to infer information about their biological activities. For YecM, we examined ion specificity for a proposed metal binding pocket. The comparison of RpiB to RpiA helped to understand how two seemingly different binding pockets could perform the same function. In BioH, the identification of a putative CoA binding surface provides insight on its function in the biotin synthesis pathway. Finally, the study of CBS domain proteins identified two different nucleotide binding surfaces. This study highlights the importance of geometrically defined protein surfaces in biologically activity and how their identification and comparison can facilitate interpretation of structures solved in structural genomics initiatives.
Acknowledgments
We thank Jeffrey Tseng for technical help. This work is supported by grants from the National Science Foundation (CAREER DBI0133856), National Institutes of Health (GM68958), Office of Naval Research (N000140310329), and Whitaker Foundation (TF-04-0023) to J.L., by a grant from the National Institutes of Health to A.J. (GM62414), and by the U.S. Department of Energy, Office of Biological and Environmental Research, under contract W-31-109-Eng-38.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051759005.
References
- Bateman, A. 1997. The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem. Sci. 22 12–13. [DOI] [PubMed] [Google Scholar]
- Berman, H.M., Battistuz, T., Bhat, T.N., Bluhm, W.F., Bourne, P.E., Burkhardt, K., Feng, Z., Gilliland, G.L., Iype, L., Jain, S., et al. 2002. The Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr. 58 899–907. [DOI] [PubMed] [Google Scholar]
- Binkowski, T.A., Adamian, L., and Liang, J. 2003a. Inferring functional relationships of proteins from local sequence and spatial surface patterns. J. Mol. Biol. 332 505–526. [DOI] [PubMed] [Google Scholar]
- Binkowski, T.A., Naghibzadeh, S., and Liang, J. 2003b. CASTp: Computed atlas of surface topography of proteins. Nucleic Acids Res. 31 3352–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binkowski, T.A., Freeman, P., and Liang, J. 2004. pvSOAR: Detecting similar surface patterns of pocket and void surfaces of amino acid residues on proteins. Nucleic Acids Res. 32 W555–W558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano, W.L. 2002. The PyMOL molecular graphics system. DeLano Scientific, San Carlos, CA. http://www.pymol.org.
- Di Gennaro, J.A., Siew, N., Hoffman, B.T., Zhang, L., Skolnick, J., Neilson, L.I., and Fetrow, J.S. 2001. Enhanced functional annotation of protein sequences via the use of structural descriptors. J. Struct. Biol. 134 232–245. [DOI] [PubMed] [Google Scholar]
- Feng, Z.K. and Sippl, M.J. 1996. Optimum superimposition of protein structures: Ambiguities and implications. Fold. Des. 1 123–132. [DOI] [PubMed] [Google Scholar]
- Henikoff, S. and Henikoff, J.G. 1992. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. 89 10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobohm, U. and Sander, C. 1994. Enlarged representative set of protein structures. Protein Sci. 3 522–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland, D.R., Hausrath, A.C., Juers, D., and Matthews, B.W. 1995. Structural analysis of zinc substitutions in the active site of thermolysin. Protein Sci. 4 1955–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1994. Searching protein structure databases has come of age. Proteins 19 165–173. [DOI] [PubMed] [Google Scholar]
- Jaroszewski, L. and Godzik, A. 2000. Search for a new description of protein topology and local structure. Proc. Int. Conf. Intell. Syst. Mol. Biol. 8 211–217. [PubMed] [Google Scholar]
- Murzin, A.G., Brenner, S.E., Hubbard, T., and Chothia, C. 1995. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247 536–540. [DOI] [PubMed] [Google Scholar]
- Orengo, C.A., Pearl, F.M., Bray, J.E., Todd, A.E., Martin, A.C., Lo Conte, L., and Thornton, J.M. 1999a. The CATH database provides insights into protein structure/function relationships. Nucleic Acids Res. 27 275–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orengo, C.A., Todd, A.E., and Thornton, J.M. 1999b. From protein structure to function. Curr. Opin. Struct. Biol. 9 374–382. [DOI] [PubMed] [Google Scholar]
- Roos, A.K., Andersson, C.E., Bergfors, T., Jacobsson, M., Karlen, A., Unge, T., Jones, T.A., and Mowbray, S.L. 2004. Mycobacterium tuberculosis ribose-5-phosphate isomerase has a known fold, but a novel active site. J. Mol. Biol. 335 799–809. [DOI] [PubMed] [Google Scholar]
- Rost, B. 2002. Enzyme function less conserved than anticipated. J. Mol. Biol. 318 595–608. [DOI] [PubMed] [Google Scholar]
- Russell, R.B. 1998. Detection of protein three-dimensional side-chain patterns: New examples of convergent evolution. J. Mol. Biol. 279 1211–1227. [DOI] [PubMed] [Google Scholar]
- Russell, R.B., Sasieni, P.D., and Sternberg, M.J. 1998. Supersites within superfolds. Binding site similarity in the absence of homology. J. Mol. Biol. 282 903–918. [DOI] [PubMed] [Google Scholar]
- Sanishvili, R., Yakunin, A.F., Laskowski, R.A., Skarina, T., Evdokimova, E., Doherty-Kirby, A., Lajoie, G.A., Thornton, J.M., Arrowsmith, C.H., Savchenko, A., et al. 2003. Integrating structure, bioinformatics, and enzymology to discover function: BioH, a new carboxylesterase from Escherichia coli. J. Biol. Chem. 278 26039–26045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott, J.W., Hawley, S.A., Green, K.A., Anis, M., Stewart, G., Scullion, G.A., Norman, D.G., and Hardie, D.G. 2004. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J. Clin. Invest. 113 274–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, T.F. and Waterman, M.S. 1981. Identification of common molecular subsequences. J. Mol. Biol. 147 195–197. [DOI] [PubMed] [Google Scholar]
- Tian, W. and Skolnick, J. 2003. How well is enzyme function conserved as a function of pairwise sequence identity? J. Mol. Biol. 333 863–882. [DOI] [PubMed] [Google Scholar]
- Todd, A.E., Orengo, C.A., and Thornton, J.M. 1999. Evolution of protein function, froma structural perspective. Curr. Opin. Chem. Biol. 3 548–556. [DOI] [PubMed] [Google Scholar]
- Tomczyk, N.H., Nettleship, J.E., Baxter, R.L., Crichton, H.J., Webster, S.P., and Campopiano, D.J. 2002. Purification and characterisation of the BIOH protein from the biotin biosynthetic pathway. FEBS Lett. 513 299–304. [DOI] [PubMed] [Google Scholar]
- Vetting, M.W., Hegde, S.S., Javid-Majd, F., Blanchard, J.S., and Roderick, S.L. 2002. Aminoglycoside 2′-N-acetyltransferase from Mycobacterium tuberculosis in complex with coenzyme A and aminoglycoside substrates. Nat. Struct. Biol. 9 653–658. [DOI] [PubMed] [Google Scholar]
- Wallace, A.C., Borkakoti, N., and Thornton, J.M. 1997. TESS: A geometric hashing algorithm for deriving 3D coordinate templates for searching structural databases. Application to enzyme active sites. Protein Sci. 6 2308–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, R., Evans, G., Rotella, F.J., Westbrook, E.M., Beno, D., Huberman, E., Joachimiak, A., and Collart, F.R. 1999. Characteristics and crystal structure of bacterial inosine-5′-monophosphate dehydrogenase. Biochemistry 38 4691–4700. [DOI] [PubMed] [Google Scholar]
- Zhang, R., Andersson, C.E., Savchenko, A., Skarina, T., Evdokimova, E., Beasley, S., Arrowsmith, C.H., Edwards, A.M., Joachimiak, A., and Mowbray, S.L. 2003a. Structure of Escherichia coli ribose-5-phosphate isomerase: A ubiquitous enzyme of the pentose phosphate pathway and the Calvin cycle. Structure (Camb.) 11 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, R.G., Andersson, C.E., Skarina, T., Evdokimova, E., Edwards, A.M., Joachimiak, A., Savchenko, A., and Mowbray, S.L. 2003b. The 2.2 Å resolution structure of RpiB/AlsB from Escherichia coli illustrates a new approach to the ribose-5-phosphate isomerase reaction. J. Mol. Biol. 332 1083–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, R.G., Duke, N., Laskowski, R., Evdokimova, E., Skarina, T., Edwards, A., Joachimiak, A., and Savchenko, A. 2003c. Conserved protein YecM from Escherichia coli shows structural homology to metal-binding isomerases and oxygenases. Proteins 51 311–314. [DOI] [PMC free article] [PubMed] [Google Scholar]