

Abstract
The phosphorylcholine esterase from Streptococcus pneumoniae, Pce, catalyzes the hydrolysis of phosphorylcholine residues from teichoic and lipoteichoic acids attached to the bacterial envelope and comprises a globular N-terminal catalytic module containing a zinc binuclear center and an elongated C-terminal choline-binding module. The dependence of Pce activity on the metal/enzyme stoichiometry shows that the two equivalents of zinc are essential for the catalysis, and stabilize the catalytic module through a complex metal-ligand coordination network. The pH dependence of Pce activity toward the alternative substrate p-nitrophenylphosphorylcholine (NPPC) shows that kcat and kcat/Km depend on the protonation state of two protein residues that can be tentatively assigned to the ionization of the metal-bound water (hydrogen bonded to D89) and to H228. Maximum activity requires deprotonation of both groups, although the catalytic efficiency is optimum for the single deprotonated form. The drastic reduction of activity in the H90A mutant, which still binds two Zn2+ ions at neutral pH, indicates that Pce activity also depends on the geometry of the metallic cluster. The denaturation heat capacity profile of Pce exhibits two peaks with Tm values of 39.6°C (choline-binding module) and 60.8°C (catalytic module). The H90A mutation reduces the high-temperature peak by about 10°C. Pce is inhibited in the presence of 1 mM zinc, but this inhibition depends on pH, buffer, and substrate species. A reaction mechanism is proposed on the basis of kinetic data, the structural model of the Pce:NPPC complex, and the currently accepted mechanism for other Zn-metallophosphoesterases.
Keywords: zinc-metallo hydrolases, choline-binding proteins, DSC, catalytic activity, pneumococcal Pce, binuclear zinc-center
Streptococcus pneumoniae is an important human pathogen that has an absolute nutritional requirement for choline (Tomasz 1967). Replacement of this amino alcohol in a synthetic medium by structural analogs, such as ethanolamine (EA cells), leads to severe alterations including cell shape, size, and physiology (Horne and Tomasz 1993). The presence of phosphorylcholine (PC) residues either attached to the cell-wall teichoic acid or bound to the membrane lipoteichoic acid has been shown to be an essential requirement for the optimal enzymatic activity of all but one of the pneumococcal murein hydrolases encoded by the bacterium or its phages (López et al. 2004). PC residues also appear to be involved with physiological functions of S. pneumoniae since they serve as anchors for surface-localized choline-binding proteins (CBPs) (Swiatlo et al. 2004) and as receptors for Dp-1 bacteriophage (López et al. 1982). In addition, they are also recognized by components of the host response, such as the human C-reactive protein (CRP) (Pepys and Hirschfield 2003) and the receptor of the platelet-activating factor (PAF) (Cundell et al. 1995). Moreover, it has been suggested that pneumococci with a lower choline content may escape the innate clearance mechanism in the blood-stream (Yother et al. 1988; Weiser and Kapoor 1999).
The pneumococcal phosphorylcholine esterase (Pce or CbpE, 602 amino acids in the mature protein) is a member of the CBP family that hydrolyzes about 30% of the total PC residues attached to the N-acetylgalactosamine moiety of teichoic and lipoteichoic acids (De las Rivas et al. 2001; Vollmer and Tomasz 2001), remodeling the content and distribution of choline on the bacterial envelope. Thus, Pce activity will indirectly modulate the activity of other CBPs and the pathogen–host interactions. The catalytic module of Pce (312 residues) is at the N terminus and is followed by a choline-binding module comprised of 10 sequence repeats of about 20 amino acids and by a long C-terminal tail of 85 residues without known similarity to sequences in the protein databases (De las Rivas et al. 2001; Vollmer and Tomasz 2001). Interestingly, the N-terminal catalytic region of Pce shows a weak similarity with enzymes of the metallo-β- lactamase fold family (Pfam entry PF00753), which also includes thiolesterases, competence proteins essential for natural transformation in bacteria, phospho-diesterases, and an oxygen oxydoreductase (ROO) from Desulfovibrio gigas (Daiyasu et al. 2001; Vogel et al. 2002). The enzymes from this family usually bind two zinc ions (Fe2+ in ROO from D. gigas) (Frazao et al. 2000) per molecule as cofactor and, depending on the enzyme, full activity can require the occupancy of only one or both metal-binding sites (Rasia et al. 2003, and references herein). Very recently, the structural determination of Pce by X-ray crystallography (Hermoso et al. 2005) has shown that Pce displays a novel modular structure, with a globular N-terminal catalytic module containing a binuclear Zn2+ catalytic center and an elongated choline-binding module. The catalytic module folds into an αβ/βα sandwich (Fig. 1A ▶), following the metallo-β-lactamase-like fold, which can be divided into two near-equivalent regions consisting of an anti-parallel β-sheet (five and four β-strands, respectively) followed by three αβ-motifs. The zinc ions (named Zn1 and Zn2) are bridged by the Oδ1 of D203. Zn1 is also coordinated by H85, H87, and N183, while Zn2 interacts with D89, H90, and H229 (Fig. 1B ▶). Pce also contains two Ca2+ ions at the interface of the catalytic and the choline-binding modules that appear to play a structural role in their arrangement. The presence of a PC molecule in the Pce crystals has also afforded the identification of the active site (Hermoso et al. 2005).
Figure 1.
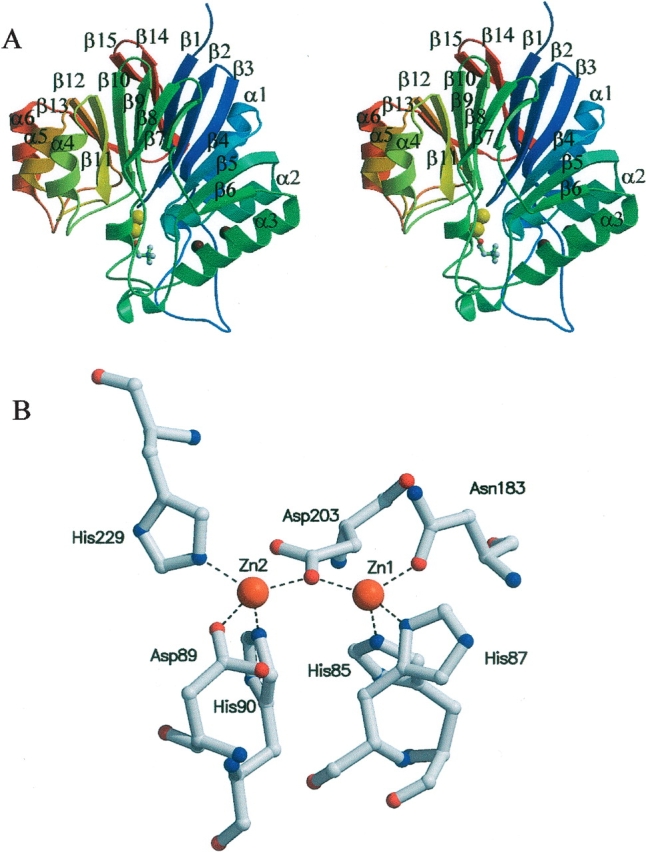
Structure of the Pce catalytic module. (A) Stereo view ribbon diagram of the catalytic module taken from Pce structure (PDB code 2BIB). The sequence is colored from blue at residue 1 to red at residue 301, and the secondary structure elements are labeled. The PC molecule at the active site is drawn in a ball-and-stick representation, while zinc ions are represented as yellow spheres. (B) Bimetallic cluster in the Pce active site. Broken lines indicate bonds between Zn2+ ions (orange) and Pce residues.
Considering that the zinc ions are directly involved in PC binding (Hermoso et al. 2005) and are probably also key to catalysis, here we have explored the influence of zinc binding on Pce activity. The enzyme catalysis has been characterized as a function of pH and the residues essential for Pce activity are proposed. In addition, the effect of zinc coordination on Pce structure and activity has been also analyzed.
Results
Dependence of Pce activity on metal content
The crystallographic structure of Pce has shown two zinc ions bound at the catalytic center (Hermoso et al. 2005). Since Pce crystals were grown in the presence of Zn2+ (Lagartera et al. 2005), the metal occupancy of the active site in protein samples isolated usingmetal-free buffers and extensively dialyzed against 20mM HEPES (pH 7.0) (buffer A) has been characterized. Under these conditions, 1.8 ± 0.1 equivalents of Zn2+ and 2.1 ± 0.1 equivalents of Ca2+ are bound per molecule of Pce. Similar metal-binding stoichiometries were found when 3 μMZnCl2 was added to the buffers used in the isolation procedure and Pce was further equilibrated in buffer A in the absence or in the presence of Zn2+ (Table 1). After extensive dialysis against 3 mM EDTA in buffer A to remove the bound metals, the Zn2+/Pce stoichiometry decreases to 0.66, while 1.7 equivalents of Ca2+ remain bound to the enzyme. As shown in Table 1, removal of 67% of the bound zinc is concomitant with a 60% reduction of Pce activity. These results strongly suggest that full activity requires the binding of both zinc ions. In contrast, supplementation of the reaction buffer with 3mMEDTA did not significantly reduce the holoenzyme activity, even after 6 h of incubation in these conditions. Remarkably, the apoenzyme (equilibrated in buffer A to eliminate EDTA) can be reconstituted in 3 or 10 μM Zn2+ containing buffers and recovers 90% of initial activity, suggesting that zinc depletion does not inactivate Pce irreversibly. Altogether, these results provide evidence that zinc ions sequestered by Pce during bacterial growth are bound so firmly to the enzyme that they remain attached during the isolation procedure, and its dissociation requires extensive dialysis against chelating agents. Although metal analysis yielded about 0.2 equivalents of iron per mole of Pce in some protein samples, the absence of the iron signal in the anomalous Fourier difference map obtained at the peak of the GdLIII absorption edge confirmed unequivocally the absence of iron in Pce structure (Hermoso et al. 2005).
Table 1.
Metal/enzyme ratio and activity at pH 7.0 of pneumococcal Pce purified in 3 μM ZnCl2 containing buffers
| Activity (%)a | Cation/Pce ratiob | ||||
| Dialysis buffer | HEPESc | HEPES 10 μM Zn2+ | HEPES 3 mM EDTA | Zn2+ | Ca2+ |
| HEPES | 100 | 100 | 100 | 2.0 | 2.1 |
| HEPES 3 μM Zn2+ | 100 | 100 | 100 | 2.0 | 2.1 |
| HEPES 10 μM Zn2+ | 100 | 100 | 100 | 2.2 | 2.2 |
| HEPES 3 mM EDTA | 40 | 90 | 40 | 0.66 | 1.7 |
a Whenever necessary, Pce samples were previously re-equilibrated in the buffer used for kinetic assays to avoid possible interferences.
b Cation/protein stoichiometries were measured by ICP-OE in the dialysis buffer (~10 μM Pce), and estimated errors are < 5%.
c Buffer A.
When Pce activity was assayed in the presence of higher Zn2+ concentrations, the initial rate of p-nitrophenylphosphorylcholine (NPPC) hydrolysis decreased as the metal concentration increased >100 μM (Fig. 2A ▶). The inhibition was found to be maximum around pH 7.0; however, the catalytic activity of Pce in ~1 mM Zn2+ was fully recovered when assayed in the presence of 3 mM EDTA in buffer A, indicating that this inhibitory effect is reversible (data not shown). The activity curves measured at several Zn2+ concentrations are consistent with a competitive inhibition mechanism (Fig. 2B ▶; Table 2). The simultaneous fitting of the activity curves measured at four Zn2+ concentrations in buffer A using equation 4 yielded values of Ki = 46 ± 2 μM, Km = 160 ± 40 μM and kcat = 9.9 ± 0.7 sec−1, in agreement with data derived from the Lineweaver-Burk plot (Fig. 2B ▶). The inhibitory effect of zinc at high concentration was buffer-dependent since no activity decrease was observed even at 1 mM Zn2+ upon substitution of buffers like HEPES or PIPES by Bis-Tris-propane (Fig. 2A ▶). In contrast, the kinetic constants for NPPC hydrolysis measured under noninhibitory conditions (3 μM Zn2+) are in excellent agreement (Fig. 3 ▶), indicating that Pce activity is not inhibited or stimulated by the buffers used in this study.
Figure 2.
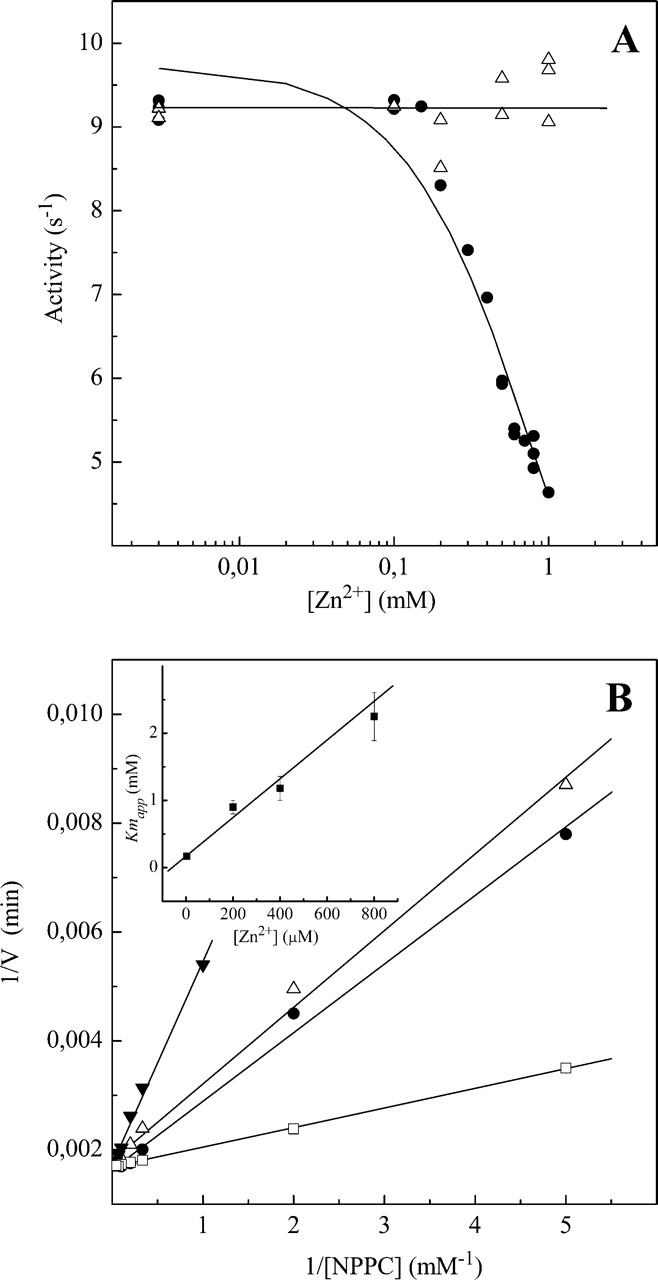
Inhibitory effect of Zn2+ on the NPPC hydrolysis by Pce at high metal concentration. (A) Dependence of Pce activity on Zn2+ concentration in HEPES (•) and Bis-Tris-propane (▵). Measurements were performed at 25°C using 3 mM NPPC and 0.1 μM Pce (pH 7.0) (I = 0.05). The continuous curve is the fitting of equation 4 at constant substrate concentration with Ki = 46 μM, Km = 140mM, and kcat = 9.9 sec−1. (B) Lineweaver-Burk plot of NPPC hydrolysis by Pce at increasing Zn2+ concentrations (□, 3 μM; •, 200 μM; ▵, 400 μM; ▾, 800 μM) in buffer A. The inset shows the zinc dependence of Kmapp. Experimental details as in A.
Table 2.
Kinetic parameters for hydrolysis of NPPC by pneumococcal Pce in buffer A at different Zn2+ concentrations
| [Zn2+] (μM) | Km (mM) | kcat (s−1) | kcat/Km (mM−1 s−1) |
| 3 | 0.17 ± 0.02 | 9.8 ± 0.2 | 57 ± 6 |
| 200 | 0.9 ± 0.1 | 10.5 ± 0.2 | 12 ± 1 |
| 400 | 1.2 ± 0.2 | 10.0 ± 0.3 | 8.3 ± 0.6 |
| 800 | 2.2 ± 0.4 | 9.9 ± 0.2 | 4.5 ± 0.2 |
Measurements were carried out at 25°C (I = 0.05).
Figure 3.

pH dependence of Pce kinetic constants for the hydrolysis of NPPC. Measurements were carried out at 25°C in the buffers indicated in the experimental section supplemented with 3 μMZn2+ (I = 0.05). The continuous curves are the fittings of equations derived for the two deprotonation-state model (Fig. 4 ▶) to the experimental data with the parameters shown in Table 4. (A) Logarithmic plot of kcat. (B) Logarithmic plot of kcat/Km. (C) Logarithmic plot of Km.
pH dependence of NPPC hydrolysis
The analysis of Pce metal content at pH values varying from 5.5 to 9.0 (Table 3) showed that the occupancy of the bimetallic active center remains constant over the whole pH interval at a free Zn2+ concentration as low as 3 μM. The kcat shows a sigmoidal increase over the pH range 7–9 (Fig. 3A ▶) and decays below pH 6.0, while the catalytic efficiency (kcat/Km) has a bell-shaped pH profile (Fig. 3B ▶) due to the large increase of Km above pH 7.4 (Fig. 3C ▶). The activity of protein samples assayed at low pH is fully recovered upon a 1:100 dilution with pH 8.0 buffers. Therefore, the pH dependence of Pce activity should be due to the reversible titration of protein residues, since NPPC has no ionizable groups in this interval of pH. The slope of the logarithmic plot of kcat/Km versus pH approaches values of 0.8 ± 0.1 and −0.50 ± 0.1 at the acidic and basic limbs (Fig. 3B ▶), indicating that Pce activity depends on the protonation state of two residues, one of them dissociating at acidic pH and the other at basic pH. The simplest reaction scheme (Fig. 4 ▶) that satisfies the observed pH dependence of NPPC hydrolysis requires three protonation states (EH2 ↔ EH ↔ E) over the pH interval from 5.5 to 10. The shape of the pH-rate profile indicates that the activity falls below pH 6.0. Thus, the first deprotonation would be essential for Pce activity, although the second one is required to reach the maximum activity (kcatSE = kh;kcatSEH = kl).
Table 3.
pH dependence of metal/Pce stoichiometry in the wildtype enzyme and the H90A mutant
| Protein | Mediuma | Zn2+b | Ca2+ |
| Wild type | Bis-Tris (pH 5.5) | 2.2 | 2.4 |
| Wild type | HEPES (pH 7.0) | 2.0 | 2.1 |
| Wild type | Bis-Tris-propane (pH 8.0) | 2.3 | N.D.c |
| Wild type | Bis-Tris-propane (pH 9.0) | 2.0 | 2.5 |
| H90A | Bis-Tris (pH 5.5) | 1.2 | 3.1 |
| H90A | HEPES (pH 7.0) | 2.4 | 1.9 |
| H90A | Bis-Tris-propane (pH 9.0) | 2.1 | 1.9 |
a Unless otherwise stated the [Zn2+]free in all buffers was 3 μM.
b Estimated errors in metal content are below 5%.
c N.D., not determined.
Figure 4.

Reaction scheme for the hydrolytic action of Pce on NPPC. The constants are defined in the text and P indicates product formation.
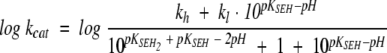 |
(1) |
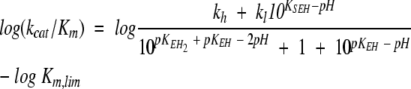 |
(2) |
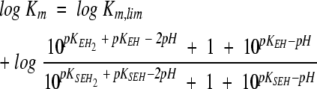 |
(3) |
Table 4 summarizes the catalytic parameters and the ionization constants derived from the theoretical analysis of the experimental data shown in Figure 3 ▶ in terms of equations 1–3, which were derived from the reaction scheme depicted in Figure 4 ▶ (Tipton and Dixon 1979). The values obtained for each ionization constant using the different fitting functions are in reasonable agreement, supporting the proposed model. The first deprotonation results in an active enzyme (kl = 7.8 sec−1) and enhances substrate binding (Fig. 3C ▶). In contrast, the second ionization further increases the Pce activity (kh = 26.5 sec−1) but markedly reduces the binding of NPPC (Fig. 3C ▶). The ionization constants of the groups relevant for activity, which were similar in the free enzyme (pKEH2 ≅ pKEH ≅ 7.2), are remarkably different in the NPPC:Pce complex (pKEH2S ≅ 5.3; pKEHS ≅ 8.5). Therefore, substrate binding would stabilize the single deprotonated form (EHS).
Table 4.
Kinetic parameters and ionization constants influencing the activity and of Pce catalyzed hydrolysis of NPPC at 25°C (I = 0.05)
| Parameter | Wild type | H90A |
| kl (s−1) | 7.8 ± 0.5a | 4.6 ± 0.2a |
| kh (s−1) | 26.5 ± 0.5a | 0.6 ± 0.1a |
| Km,lim (mM) | 2.2 ± 0.2b | |
| pKEH2 | 7.3 ± 0.3c 7.1 ± 0.6b |
|
| pKEH | 7.3 ± 0.3c 7.1 ± 0.4b |
|
| pKEH2S | 5.5 ± 0.3a 5 ± 1b |
5.7 ± 0.2a |
| pKEHS | 8.12 ± 0.05a 8.9 ± 0.1c 8.6 ± 0.1b |
8.03 ± 0.07a |
a From kcat fit.
b From Km fit.
c From catalytic efficiency fit: kl, khKm,lim were fixed at the values derived from the kcat and Km fits.
H90A Pce mutant
The double bridging of the two metal cations by a water molecule or an hydroxide molecule and an amino acid side chain (usually bearing a carboxylate group) is a characteristic feature of the metal coordination in zinc cocatalytic centers (Auld 2001a). Indeed, no bridging carboxylate group has been found in enzymes like metallo-β-lactamases, which are active in the mono-zinc form (Zn1), although they can accommodate a second metal ion (Zn2) with varying effects on the catalytic activity (Auld 2001a). Thus, the metal coordination pattern (Fig. 1B ▶) at the active site of Pce suggests that it behaves as a cocatalytic center. In order to explore the effect of perturbing the Zn2 coordination on Pce activity, the H90 has been mutated to alanine. This residue is the less-conserved donor of Zn2 within the family of proteins adopting the metallo-β-lactamase fold, but its position in the Pce structure is maintained through a complex concerted network of hydrogen bonds involving two highly conserved residues (D33 and T84; Pce numbering), as well as D203 (the metal bridging residue) and D89 (Zn2 donor). Table 5 summarizes the pH dependence of the kinetic parameters of the Pce H90A mutant for NPPC hydrolysis. Substitution of the H90 residue by alanine decreases kcat and kcat/Km to 12% and 1%, respectively, of those for the wild-type enzyme at neutral pH. The pH dependence of kcat parallels that of the wild-type Pce, although the rate constants are around one order of magnitude lower. The analysis of the kcat versus pH profile in terms of equation 1 yields pKa values of 5.7 (EH2S ↔ EHS) and 8.03 (EHS ↔ ES), similar to those obtained for the wild-type enzyme. In contrast, substrate binding depends weakly on Pce’s ionization state and the Km approaches the limiting value at basic pH for the wild-type enzyme, whereas the efficiency in NPPC hydrolysis is maximum around pH 8.0.
Table 5.
Kinetic parameters for the hydrolysis of NPPC by the S. pneumoniae H90A Pce mutant at different pH values
| pHa | Km (mM) | kcat (s−1) | kcat/Km (mM−1 s−1) |
| 6.0b | 1.1 ± 0.1 | 0.43 ± 0.02 | 0.38 ± 0.2 |
| 7.0c | 1.7 ± 0.4 | 1.13 ± 0.07 | 0.7 ± 0.1 |
| 7.6b | 2.6 ± 0.3 | 1.47 ± 0.04 | 0.56 ± 0.05 |
| 8.0d | 1.0 ± 0.1 | 2.84 ± 0.07 | 2.8 ± 0.2 |
| 8.25d | 0.75 ± 0.06 | 2.95 ± 0.06 | 3.9 ± 0.2 |
| 9.0b | 1.8 ± 0.3 | 4.5 ± 0.2 | 2.5 ± 0.3 |
| 10.0b | 3.9 ± 0.8 | 4.7 ± 0.4 | 1.2 ± 0.1 |
aMeasurements were made at 25°C in Bis-Tris-propane,b HEPES,c and HEPBSd at 3 μM Zn2+ (I = 0.05).
Since the H90A mutant still binds two equivalents of Zn2+ at neutral pH and above (Table 3), the drastic reduction of the mutant activity indicates that Pce activity depends not only on the metal stoichiometry, but also on the geometry of the bimetallic zinc cluster. However, the loss of one imidazole ligand upon mutation of H90 to alanine reduces Pce’s affinity for Zn2+ to an extent that almost abolishes its binding to the site Zn2 at acidic pH (Table 3). The protonation of either the metal donors or the residues helping to orientate them can account for this further reduction in Zn2+ affinity at lower pH. Similar effects have also been observed in other metallo-β-lactamases (Rasia and Vila 2002; Yamaguchi et al. 2005). In contrast to the behavior of the wildtype enzyme, the activity of the H90A mutant in HEPES does not decay upon addition of 2 mM Zn2+. These results suggest that substitution of H90 by alanine reduces the metal-mediated inhibition caused in HEPES or PIPES.
Zn2+ dependence of Pce hydrolytic activity on pneumococcal cell walls
The previous data on the Zn2+ dependence of Pce activity were obtained using NPPC as substrate, but it appeared interesting to confirm that such a dependence was also present when using its physiological substrate, the pneumococcal cell wall. Therefore, radioactively labeled pneumococcal cell walls have been also used as substrate for both the wild-type Pce and the H90A mutant (Table 6). In agreement with the results observed using NPPC as substrate, the activity of the H90 mutant on cell walls is 10% that of the wild-type enzyme, suggesting that H90 also plays a critical role for the hydrolysis of Pce natural substrate. Nevertheless, the effect of 1 mM Zn2+ on cell-wall hydrolysis by Pce was unexpected, since both the wild-type enzyme and the H90A mutant were drastically inhibited (~90%) in the presence of this cation. This result suggests that the interaction of Zn2+ and cell-wall components plays an additional role on cell-wall hydrolysis. The cell wall:Zn2+ complex might produce a double detrimental effect on enzyme activity either by blocking substrate binding to the catalytic center or by reducing the affinity of teichoic and lipoteichoic acids toward the choline-binding module. It has been established that the binding of teichoic acid to the choline-binding module of Pce is critical for its activity on cell walls, since the isolated catalytic module is practically inactive on this substrate (Hermoso et al. 2005). Therefore, we cannot discard that high concentrations of Zn2+ could also impair the anchoring of Pce onto the cell wall, thus promoting an additional reduction of Pce activity. Interestingly, the major pneumococcal autolysin is also drastically inhibited by this cation (Höltje and Tomasz 1976).
Table 6.
Enzymatic activity of wild-type Pce and H90A mutant on pneumococcal cell walls at different Zn2+ concentrations
| Protein | 3 μM Zn2+ | 0.5 mM Zn2+ | 1 mMZn2+ | 2 mM Zn2+ |
| Wild type | 796 | 68 | 11 | <1 |
| H90A | 66 | 7 | <1 | <1 |
Measurements were made at 25°C in buffer A. Activity is expressed in units per mg. One unit corresponds to 700 cpm of [H3-methyl] choline released in 10 min. Estimated errors are around 5%.
Stabilization of protein structure by Zn2+
In addition to the role played by zinc cations in the hydrolytic activity of Pce, we have explored their influence on the stability of the catalytic domain structure. To fulfill this aim, thermal denaturation studies of wildtype Pce and the H90A mutant were carried out by DSC. The heat-induced denaturation of Pce was irreversible and the heat-capacity profile of the wild-type enzyme equilibrated with 3 μM Zn2+ in buffer A exhibits two peaks with Tm values of 39.6 and 60.8°C and a shoulder around 56.3°C (Fig. 5 ▶, trace a). The enthalpy changes associated with the low and high temperature peaks are 69 kcal mol−1 and 210 kcal mol−1, respectively. Denaturation of the Pce catalytic module occurs >60°C (Fig. 5 ▶, inset) and it can be therefore correlated with the high-temperature peak, while the low-temperature one would arise from denaturation of the choline-binding module. The shoulder observed at the low-temperature side of the main thermal transition could result from denaturation of the N-terminal region of the choline-binding module, which is in close contact with the catalytic module and contributes to extending the catalytic-binding surface toward the third repeat of the cell-wall binding module (Hermoso et al. 2005). This interface is stabilized by the coordination of the two Ca2+ ligands, which are tightly held in Pce structure and remain bound after extensive dialysis against EDTA. Substitution of alanine for histidine reduces the high-temperature peak by about 10°C (Tm1 = 44.2°C and Tm2 = 50°C) in the Pce H90A mutant denaturation profile (Fig. 5 ▶, trace b). This effect is, however, significantly lower than the destabilization found upon Zn2+ dissociation from the metallic cluster, since the DSC transitions of the apoenzyme, which still retains the Ca2+ cations, are centered at 30.7 and 36.2°C (Fig. 5 ▶, trace c). The irreversibility of Pce denaturation precludes the estimation of the transition enthalpy change associated with each peak by deconvolution of the endotherms. Thermal denaturation results reveal that zinc ions are crucial for the stability of the catalytic module. They are also consistent with the presence of the zinc ions bound to the Pce H90A mutant at neutral pH (Table 3) and indicate that this mutation does not affect the native conformation of the enzyme, but does perturb the first and second coordination shells of the metal, decreasing the thermal stability of the catalytic module.
Figure 5.
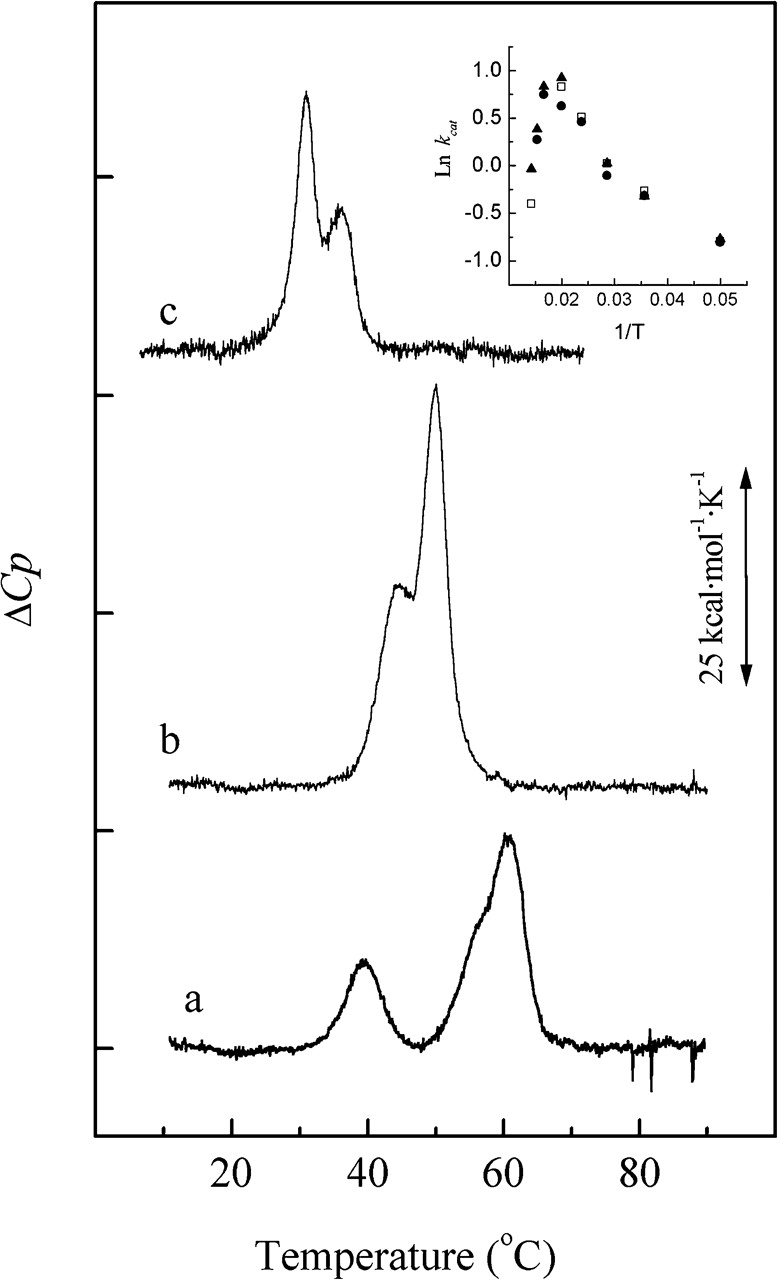
Thermal denaturation of wild-type Pce and the H90A mutant. Measurements were made in buffer A. Traces a and c correspond, respectively, to the holoenzyme (3 μM Zn2+) and the apoenzyme (2 mM EDTA) and trace b is the DSC curve of the mutant (3 μM Zn2+). The inset depicts the Pce activity against NPPC as a function of temperature in buffer A supplemented with 3 μM Zn2+.
Discussion
The biological role of choline during human pneumococcal infection is a subject of current interest. Choline may enhance pneumococcal attachment to the human cells by direct interaction with the receptor of PAF (Cundell et al. 1995) and also acts as a target for CBPs involved in cellular adhesion (Rosenow et al. 1997; Sánchez-Beato et al. 1998) and division (Crain et al. 1990; Hammerschmidt et al. 1997; Brooks-Walter et al. 1999; Tu et al. 1999) that may act as virulence factors. In this context, the pneumococcal Pce phosphorylcholine esterase, which modifies the distribution and the content of PC residues on the bacterial envelope and is able to hydrolyze PC residues present in human molecules such as PAF and other phospholipids (Hermoso et al. 2005), should play a critical role in pneumococcal virulence.
We have demonstrated here that Pce is a metallo-phosphodiesterase with a binuclear zinc cluster that exhibits maximum efficiency around neutral pH. Two Zn2+ and two Ca2+ cations are tightly bound per protein molecule, regardless of whether the buffers used in the Pce purification contained Zn2+ or not. Pce is a Zn2+-dependent enzyme, since depletion of the Zn2+-bound cation by extensive dialysis against EDTA yields an inactive enzyme that still retains the two Ca2+ cations. Indeed, the data reported here (see Table 1) are consistent with the notion that the saturation of the two Zn2+ sites is required for activity. Besides, the retention of calcium ions agrees with these ions being buried within the interface between the catalytic and the choline-binding modules of Pce and corroborates previous findings pointing to a structural role for the two Ca2+ (Hermoso et al. 2005). With the exception of iron traces in some Pce preparations, no significant amount of other divalent metals (see Materials and Methods) were found in the protein samples, and anomalous X-ray diffraction data unambiguously demonstrate the absence of iron in our Pce crystals (Hermoso et al. 2005). Nevertheless, we have tested the effect of reconstituting the Pce apoenzyme (5% residual activity upon 24 h dialysis against 20 mM EDTA) using Fe(II) and found that activity was about three times the value obtained for the zinc-reconstituted enzyme under the same conditions. Therefore, although crystallographic data and the metal-bound analysis suggest that Pce is a zinc metallo-hydrolase, we cannot discard the possibility that, depending on the physiological circumstances, the enzyme might contain binuclear zinc–iron or iron–iron centers as already reported for other metallo-hydrolases of the β-lactamase fold (Schilling et al. 2003). This would be in agreement with the data very recently reported (Garau et al. 2005) that showed the presence of two iron ions in a crystal of the isolated catalytic module of Pce, although the reported activity was several orders of magnitude lower than ours. Nevertheless, it is worth mentioning that the fast oxidation of ferrous into ferric ion, which almost completely inactivates the enzyme, complicates the interpretation of the results and raises some questions about the real impact of iron centers in the physiological form of the enzyme. In contrast with other transition metals whose activity can be conditioned by their oxidoreductive properties, zinc will provide stability in a biological medium whose redox potential may be in constant flux.
Pce activity is competitively inhibited by zinc ions at high concentration. This effect may arise from the acquisition of additional equivalents of zinc mediated by protein residues involved in Pce activity or located in the proximity of the active site, reminiscent of that observed for the inhibition of zinc dependent and independent enzymes by zinc ions (Auld 2001b). Coordination of the inhibitory metal in zinc metallo-enzymes might involve anions present in the buffering solution (Larsen and Auld 1991), and substitution of buffers bearing a sulfonate moiety like PIPES or HEPES by a cationic buffer such as Bis-Tris-propane makes the inhibition undetectable even at 1 mM Zn2+. Nevertheless, more complex mechanisms cannot be discarded, and the presence of buffering species (MES, cacodylate, or acetate) bound to the metal cluster of enzymes from the metallo-β-lactamase fold family has been also reported (Fitzgerald et al. 1998; Cameron et al. 1999).
The pH dependence of Pce activity suggests the presence of two deprotonation events relevant for the hydrolytic activity on NPPC. The pKa values of the free enzyme are in the range reported for histidine residues, but they could also represent a carboxylate, a Zn2+ bound water, or a combination of both (Auld et al. 1986; Bounaga et al. 1998). The currently accepted model for the reaction mechanism of zinc-metallo-phosphatases (Lipscomb and Sträter 1996; Wilcox 1996) assumes that the enzyme-catalyzed hydrolysis involves an associative mechanism with a nucleophilic attack on the phosphorus opposite to the leaving group (so-called in-line attack), resulting in a trigonal-bipyramidal phosphorus intermediate with the entering and leaving groups in the axial positions (Knowles 1980). A survey of the ligand-binding pattern in zinc-dependent enzymes has shown that the bridging of the two metal ions by water or a hydroxide molecule and by an amino acid side chain is a characteristic feature of zinc cocatalytic centers (Auld 2001a). The absence of a solvent molecule bridging the zinc atoms in the crystallographic structure of the Pce:PC complex (Hermoso et al. 2005) may be due to the occupancy of the catalytic site by the reaction product, since the binding of substrate-analogs to phospholipase C results in the dissociation of the bridging hydroxide from the metallic cluster (Hansen et al. 1992, 1993). However, the presence of a PC molecule in the crystallized form of Pce allowed us to model a NPPC molecule in the catalytic site (Fig. 6 ▶; see Materials and Methods). Interestingly, the p-nitrophenyl leaving group of NPPC can only be allocated at the position occupied by the PC phosphate oxygen interacting with H228. The phosphate moiety directly binds the metal ions, and this contributes to neutralize its negative charge, making the phosphorus atom more susceptible to nucleophilic attack. Meanwhile, the trimethylammonium cation is stabilized by D89 (electrostatic interaction) and W123 (cation-π interaction) (Fig. 6B ▶). The superposition of the metal clusters of the human glyoxalase II and the Pce:NPPC model depicted in Figure 6C ▶ shows that the hydroxide/water molecule bridging the zinc atoms in the human enzyme is positioned in the appropriate orientation for a nucleophilic attack on the NPPC ester bond, the hydroxide-P-O angle being of 161°, while the p-nitrophenol leaving group is pointing toward the solvent. The attacking nucleophile for the Pce-catalyzed hydrolysis reaction of NPPC may be, therefore, a water/hydroxide bound to the binuclear metallic center. Moreover, according to the structure of the Pce:NPPC model, the carboxylate moieties of D203 (Oδ1) and D89 (Oδ1 and Oδ2) would be within hydrogen-bonding distance of the metal-bound water (Fig. 6C ▶) and they could play a critical role in orienting the metal-bound water/hydroxide, holding it in a fixed position that reduces the entropic barrier for nucleophilic attack on the substrate (Christianson and Cox 1999) and modifies its pKa. The Km decrease accompanying the first deprotonation event suggests the implication of D89 in this process since it is also interacting electrostatically with the positively charged trimethylammonium group of the substrate (Fig. 6B ▶). Moreover, the homologous residue in glyoxalase II (Cameron et al. 1999; Zang et al. 2001) and metallo-β-lactamases (Rasia and Vila 2002; Yamaguchi et al. 2005) has been proposed to orient the nucleophilic water/hydroxide for attack. Hence, the pKa around 5.3 in the substrate-bound state of Pce might be regarded as the ionization of the metal-bound water hydrogen-bonded to D89. pKa values varying from four to six have been reported for the zinc-bound water in different cocatalytic centers (Auld 2001b), since deprotonation would be determined by direct and second-shell ligands. Thus, higher coordination numbers and negatively charged donors should increase the pKa value of the metal-bound water, whose acidity will be also modulated by binding of hydrogen-donating or accepting groups to the zinc ligands. The inspection of the Pce structure (Hermoso et al. 2005) shows that H228 is at hydrogen-bond distance (2.74 Å ) of the oxygen involved in the scissile bond (Fig. 6B ▶) and can contribute to substrate binding. Hence, the alkaline dependence of the Km may reflect the state of ionization of this residue and its interaction with the substrate. Moreover, upon the hydrolysis of the intermediate deprotonation of H228 can also accelerate PC dissociation from the active site, explaining the observed dependence of kcat on a second group with a pKa ~8.5. The breakdown of the pentacoordinated intermediate formed during the hydrolysis of teichoic and lipoteichoic acids by Pce requires the protonation of the departing N-acetyl-galactosamine moiety. There are no obvious solvent molecules or amino acid side chains to be proposed as a general acid catalyst from the structures of the Pce:PC complex or the Pce:NPPC complex model. Garau et al. (2005) have proposed a mechanism based in purple acid phosphate (Klabunde et al. 1996) where the attacking nucleophile would be a water molecule putatively bound to the iron ion located at the position of Zn1, whereas H228 would act as an acidic catalyst. Nevertheless, the water molecule and the leaving group do not seem to be located in the orientation (~180°) required for a SN2-type catalytic mechanism. In addition, the pH dependence of kcat with pH does not support the role proposed for H228 since the deprotonation of the imidazole ring should decrease the Pce activity, which is in contrast with the increase observed by us at basic pH.
Figure 6.

Three-dimensional model of the Pce:NPPC complex. (A) Structure of Pce in complex with NPPC (spheres). Pce molecular surface is colored according to the protein modules (N-terminal catalytic module, salmon; choline-binding module, blue). (B) The contact network at the active site. Broken lines indicate relevant bonds of NPPC (green) or Zn2+ ions with Pce residues. (C) Close-up view of the model of NPPC:Pce (gray) superimposed with human glyoxalase II (blue; PDB code 1QH3) in stick representation. Gray broken lines are as in B, while orange broken lines indicate putative relevant bonds of Pce residues and NPPC to the solvent molecule bound to the binuclear metallic cluster of glyoxalase II. H228 from Pce was omitted for clarity.
Interestingly, we have observed that the substitution of alanine for H90 not only removes one of the Zn2 donors, but it can also alter the concerted network of interactions between the metal ion cofactor and its first and second coordination spheres. The side chain ofH90 is at hydrogen-bond distance of D203 (the metal-bridging residue) and D89 that are also hydrogen bonded, respectively, toH85 and H229, which also act as ligands to Zn2+. In addition, the highly conserved residues D33 and T84 help to maintain the positioning of H90; the former is hydrogen bonded to H90 and T84, while the latter is also within hydrogen-bond distance of Zn1 donor H85. Indeed, a change in the active site geometry would account for the important loss of the catalyticmodule stability (Fig. 5 ▶) and the drastic decrease in activity (Table 5) shown by the H90A mutant that still binds two equivalents of zinc at neutral pH. Nevertheless, the decrease of the metal affinity derived from the loss of one imidazole donor for Zn2 is evident at acidic pH, where the Zn2+/enzyme stoichiometries for the mutant and the wild-type enzyme are 1.2 and 2.2, respectively. A similar effect has been recently described for the IMP-1 metallo-β-lactamase from Serratia marcenscens (Yamaguchi et al. 2005). The pH dependence of kcat shows that the relevant groups for the catalysis are still present in the H90A mutant. However, the maximum activity of the mutant is sixfold less than that of the wild-type enzyme, and the Km approaches the limiting value of the wildtype Pce. This is probably due to alterations in the positioning of the zinc ions and the D89 side chain, which are directly involved in the substrate recognition (Hermoso et al. 2005).
As shown above, the zinc ions play an important role in the stabilization of the Pce catalytic module, since they are coordinated to residues that are widely separated along the overall sequence of the catalytic module, though close together in the tertiary structure. As in the case of H90, the interaction between H229, D19 (salt-bridged to K225), and T252 highlights the importance of the contacts between the metal ions and their first and second spheres of coordination. Consequently, the dissociation of the cofactor drastically destabilizes the catalytic module structure, decreasing its denaturation transition temperature to 30.7°C.
Finally, the following reaction mechanism can be proposed on the basis of kinetic data, the structural model of the Pce:NPPC complex, and the currently accepted mechanism for other Zn-metallo-phosphoesterases. As the substrate is drawn to the active site by electrostatic interactions between the phosphate group and the zinc ions and helped by the interactions of the choline moiety with D89 and W123, one of the free phosphoryl oxygens binds to a zinc ion, displacing the bridging solvent molecule and making the phosphorus more susceptible to attack due to phosphate polarization. The solvent molecule, oriented by hydrogen bonding to D89 and, probably, D203, would lay approximately in-line with the scissile P-O bond and could attack the phosphorus atom. The interaction with the zinc ions would stabilize the intermediate and, after product formation, the electrostatic repulsion between PC and the p-nitrophenolate would facilitate the release of the latter from the active site, followed by PC dissociation. This mechanism would also account for the ability of Pce for degrading PC-containing compounds with large leaving groups such as pneumococcal teichoic acids or PAF (Hermoso et al. 2005) that might be crucial in pneumococcal pathogenesis.
Materials and methods
Site-directed mutagenesis and protein purification
Recombinant of the wild-type pce gene was made by cloning into plasmid pT7-7 the PCR fragment amplified with the oligonucleotides LytD-N2 (5′-CCGAATTCAAGGAGATTAAC ATATGCAAGAAAGTTCAGGAAATAAAATCC-3′, where the EcoRI and NdeI restriction sites are underlined) and LytD-C (5′-TTGGATCCCTACTACTGTTCTGATTCCGATTTG- 3′, where the BamHI restriction site is underlined), which are complementary, respectively, to the 5′-end of the signal peptide and the 3′-end of the stop codon of the pce gene. Primers PceHA5′ (5′-CCACAGTGATGCTATTGGAAATGTT- 3′) and PceHA3′ (5′-AACATTTCCAATAGCATCACTGT GG-3′) were the oligonucleotides used for the H90A mutant construction. The DNA sequence was confirmed with an automated Abi Prism 3700 DNA sequencer (Applied Biosystems). Wild-type Pce and the H90A mutant were produced from Escherichia coli BL21 (DE3) (pRGR12) (De las Rivas et al. 2001) or E. coli BL21 (DE3) (pAPM01), respectively. Recombinants were incubated at 37°C in Luria-Bertani medium containing ampicillin (0.1 mg/mL) up to an A600 of 1.0 and, at this time, isopropyl-thio-β-D-galactopyranoside (50 μM) was added. The incubation proceeded for 16 h at 25°C to minimize the presence of inclusion bodies. Both enzymes were purified by affinity-chromatography in DEAE-cellulose equilibrated in 20 mM sodium phosphate (pH 7.0) with or without 3 μM ZnCl2 following the procedure previously described (Lagartera et al. 2005). The purity of the isolated samples was routinely analyzed by SDS-PAGE and the proteins were stored at −20°C. Protein concentration was determined spectrophotometrically using a molar absorption coefficient of 194,020 M−1cm−1 at 280 nm (De las Rivas et al. 2001). Before being used, the enzymes were dialyzed for 24 h against the appropriate buffer (5 × 500 mL) at 4°C. The apoenzyme was obtained by dialysis against buffer A (20 mM HEPES at pH 7.0) containing EDTA following the same procedure, and the remaining activity was measured using NPPC (p-nitro- phenylphosphorylcholine) as substrate. When required, EDTA was removed by additional dialysis against buffer A with or without zinc. Typically, reconstitution of the EDTA-free apoenzyme was performed by incubating for 60 min in buffer A supplemented with 10 μM ZnCl2, but reconstitution using Fe2+ as metal cofactor was carried out with 10 μM Fe(NH4)2(SO4)2 in 20 mM Bis-Tris-propane (pH 6.0) adding 10 mM DTT to maintain the ferrous oxidation state.
Kinetic studies
Pce activity was determined using NPPC or radioactively labeled pneumococcal cell walls as substrate (De las Rivas et al. 2001; Vollmer and Tomasz 2001). The initial rates of NPPC hydrolysis were measured in a Shimadzu UV-2100 spectrophotometer using a molar absorption coefficient of 17,528 M−1cm−1 for the reaction product (p-nitrophenolate) at 410 nm (pH 11). Typically, 500 μL of NPPC (0.025–25 mM) were allowed to equilibrate at the selected temperature for 7 min. Then, the reaction was initiated by addition of 6 μL of a Pce stock solution (final concentration around 0.1 μM) and stopped by addition of 20 μL of 2.5 M NaOH after 4 min. Km and kcat values were determined by non-linear least-square fitting of initial rate values versus substrate concentration in terms of the Michaelis-Menten equation. Measurements at different pHs were performed in the following buffers: Bis-Tris (5.5–6.0), PIPES (6.5– 6.85), Bis-Tris propane (6–10), HEPES (6.5–8.0), Tris-HCl (7.6–8.8), and HEPBS (8.0–8.4). KCl was added to keep constant the ionic strength at 0.05. Unless otherwise stated, buffers contained 3 μM ZnCl2. In switching from one buffer to another, we have used pH overlap in order to exclude that any of them could be inhibiting the enzyme activity.
Zn2+ dependence of the Pce activity on NPPC
The Zn2+ dependence of Pce activity was initially characterized at pH 7.0 measuring the hydrolysis of NPPC by protein samples equilibrated against buffer A in the absence and in the presence of either 10 μM Zn2+ or 3 mM EDTA, using saturating concentrations of substrate. Inhibition studies of Pce activity at high Zn2+ concentration were carried out at several pHs in the buffers listed above. The kinetic parameters and the inhibition constant, Ki, by Zn2+ were determined by simultaneous nonlinear least-square analysis of activity versus substrate curves measured at several Zn2+ concentrations using the equation (equation 4) derived for competitive inhibition or Lineweaver-Burk plots.
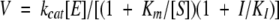 |
(4) |
Determination of the metal content of Pce
The metal content of Pce was determined by inductively coupled plasma spectrometry (ICP-OE) with a Perkin-Elmer Optima 2000DV spectrometer, and initially covered seven elements (Zn, Ca, Fe, Mg, Co, Cu, and Mn). All solutions were prepared with fresh milli-Q water and plasticware. Pce samples (≅ 10 μM) were equilibrated against the appropriate buffer, and the metal/protein stoichiometry was calculated from the difference in the metal concentration of the protein sample and the final dialysis solution using the calibration curves run in parallel. Values are the average of two to three independent measurements, each of which was carried out in triplicate.
Differential scanning calorimetry
DSC measurements were performed using a Microcal MCS instrument (Microcal, Inc.) under an extra constant pressure of 2 atm. Standard MCS and Microcal Origin software were used for data acquisition and analysis. The excess heat-capacity functions were obtained after subtraction of the buffer– buffer baseline. The second scan of previously heated samples showed that thermal denaturation of Pce was irreversible.
Docking of NPPC in Pce structure
A model of the Pce:NPPC complex was built using the structures of the Pce:PC and the NPPC:m3c65-antibody (heavy chain) complexes (PDB codes 2BIB and 1DL7, respectively). Firstly, the phosphorylcholine moiety of NPPC was fitted onto the PC bound at the active site of the Pce. Then, the p-nitrophenyl moiety was manually positioned with the program O (Jones et al. 1991) in order to avoid sterical clashes with the Pce molecule. To preserve the original conformation of the p-nitrophenyl moiety in the NPPC molecule, only torsions of the phosphate moiety were allowed. Interestingly, the p-nitrophenyl leaving group could only be positioned on the phosphate oxygen of PC interacting with H228 (Fig. 6B ▶). The model of the NPPC:Pce complex was subsequently energy minimized using the CNS program (Brunger et al. 1998).
Materials
Restriction enzymes and other DNA-modifying enzymes were from Amersham. All primers were synthesized on a Beckman model Oligo 1000M synthesizer. ICP-OE standard solutions (Alfa Aesar) were used for preparation of metal derivatives and ICP measurements. Unless otherwise stated, all chemicals were from Sigma and of the best quality available.
Acknowledgments
This work was supported by Dirección General de Investigación Científica y Técnica Grants BIO2002-02887, BIO2003- 09152, and BMC2003-00074. L.L. and A. G. were supported by fellowships from the Ministerio de Educación y Ciencia. We thank Dr. I. de la Mata for helpful discussions, Dr. A. Torrecillas and the S.U.I.C. staff from the Universidad de Murcia for ICP-OE measurements, and Dr. D. Laurents for linguistic revision of the manuscript.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051575005.
References
- Auld, D.S. 2001a. Zinc coordination sphere in biochemical zinc sites. Biometals 14 271–313. [DOI] [PubMed] [Google Scholar]
- ———. 2001b. Zinc sites in metalloenzymes and related proteins. In Handbook of metalloproteins (eds. I. Bertini et al.), pp. 881–959. M. Dekker Inc., New York.
- Auld, D.S., Larsen, K., and Vallee, B.L. 1986. Active site residues of Carboxypeptidase A. In Zinc enzymes (ed. I. Bertini), pp. 131–154. Birkhauser, Boston, MA.
- Bounaga, S., Laws, A.P., Galleni, M., and Page, M.I. 1998. The mechanism of catalysis and inhibition of the Bacillus cereus zinc-dependent β-lactamase. Biochem. J. 331 703–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks-Walter, A., Briles, D.E., and Hollingsead, S.K. 1999. The pspC gene of Streptococcus pneumoniae encodes a polymorphic protein, PspC, which elicits cross-reactive antibodies to PspA and provides immunity to pneumococcal bacteremia. Infect. Immun. 67 6533–6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger, A.T. Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54 905–921. [DOI] [PubMed] [Google Scholar]
- Cameron, A.D., Ridderström, M., Olin, B., and Mannervik, B. 1999. Crystal structure of human glyoxalase II and its complex with a glutathione thiolester substrate analogue. Structure 7 1067–1078. [DOI] [PubMed] [Google Scholar]
- Christianson, D.W. and Cox, J.D. 1999. Catalysis bymetal-activated hydroxide in zinc and manganese metalloenzyme. Annu. Rev. Biochem. 68 33–57. [DOI] [PubMed] [Google Scholar]
- Crain, M.J., Waltman, W.D., Turner, J.S., Yother, J., Talkington, D.F., McDaniel, L.S., Gray, B.M., and Briles, D.E. 1990. Pneumococcal surface protein A (PspA) is serologically highly variable and is expressed by all clinically important capsular serotypes of Streptococcus pneumoniae. Infect. Immun. 58 3293–3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cundell, D.R., Gerard, N.P., Gerard, C., Idanpaan-Heikkila, I., and Tuomanen, E.I. 1995. Streptococcus pneumoniae anchors to activated human cells by the receptor for platelet-activating factor. Nature 377 435–438. [DOI] [PubMed] [Google Scholar]
- Daiyasu, H., Osaka, K., Ishino, Y., and Toh, H. 2001. Expansion of the zinc metallo-hydrolase family of the β-lactamase fold. FEBS Lett. 503 1–6. [DOI] [PubMed] [Google Scholar]
- De las Rivas, B., García, J.L., López, R., and García, P. 2001. Molecular characterization of the pneumococcal teichoic acid phosphorylcholine esterase. Microb. Drug Res. 7 213–222. [DOI] [PubMed] [Google Scholar]
- Fitzgerald, P.M.D., Wu, J.K., and Toney, J.H. 1998. Unanticipated inhibition of the metallic-β-lactamase from Bacteriodes fragilis by 4-morpho-lineethanesulfonic acid (MES): A crystallographic study at 1.85 Å resolution. Biochemistry 37 6791–6800. [DOI] [PubMed] [Google Scholar]
- Frazao, C., Silva, G., Gomes, C.M., Matias, P., Coelho, R., Sieker, L., Macedo, S., Liu, M.Y., Oliveira, S., Teixeira, M., et al. 2000. Structure of a dioxygen reduction enzyme from Desulfovibrio gigas. Nat. Struct. Biol. 7 1041–1045. [DOI] [PubMed] [Google Scholar]
- Garau, G., Lemaire, D., Vernet, T., Dideberg, O., and Di Guilmi, A.M. 2005. Crystal structure of phosphorylcholine esterase domain of the virulence factor choline binding protein E from Streptococcus pneumoniae. J. Biol. Chem. 280 28591–28600. [DOI] [PubMed] [Google Scholar]
- Hammerschmidt, S., Talay, S.R., Brandtzaeg, P., and Chhatwal, G.S. 1997. SpsA, a novel pneumococcal surface protein with specific binding to secretory immunoglobulin A and secretory component. Mol. Microbiol. 25 1113–1124. [DOI] [PubMed] [Google Scholar]
- Hansen, S., Hansen, L.K., and Hough, E. 1992. Crystal structures of phosphate, iodide and iodate-inhibited phospholipase C from Bacillus cereus and structural investigations of the binding of reaction products and a substrate analogue. J. Mol. Biol. 225 543–549. [DOI] [PubMed] [Google Scholar]
- Hansen, S., Hough, E., Svensson, L.A., Wong, Y.-L., and Martin, S.F. 1993. Crystal structure of phospholipase C from Bacillus cereus complexed with a substrate analogue. J. Mol. Biol. 234 179–187. [DOI] [PubMed] [Google Scholar]
- Hermoso, J.A., Lagartera, L., González, A., Stelter, M., García, P., Martínez-Ripoll, M., García, J.L., and Menéndez, M. 2005. Insights into the mechanism of bacterial pathogenesis from the crystal structure of the teichoic-acid phosphorylcholine esterase Pce from Streptococcus pneumoniae. Nat. Struct. Mol. Biol. 12 533–538. [DOI] [PubMed] [Google Scholar]
- Höltje, J.V. and Tomasz, A. 1976. Purification of the pneumococcal N-acetylmuramyl- L-alanine amidase to biochemical homogeneity. J. Biol. Chem. 251 4199–4207. [PubMed] [Google Scholar]
- Horne, D.S. and Tomasz, A. 1993. Possible role of a choline-containing teichoic acid in the maintenance of normal cell shape and physiology in Streptococcus oralis. J. Bacteriol. 175 1717–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Klabunde, T., Sträter, N., Fröchlich, R., Witzel, H., and Krebs, B. 1996. Mechanism of Fe(III)-Zn(II) purple acid phosphatase based on crystal structures. J. Mol. Biol. 259 737–748. [DOI] [PubMed] [Google Scholar]
- Knowles, J.R. 1980. Enzyme-catalyzed phosphoryl transfer reactions. Annu. Rev. Biochem. 49 877–919. [DOI] [PubMed] [Google Scholar]
- Lagartera, L., González, A., Stelter, M., García, P., Kahn, R., Menéndez, M., and Hermoso, J.A. 2005. Crystallization and preliminary diffraction studies of the pneumococcal teichoic acid phosphorylcholine esterase, Pce. Acta Crystallograph. Sect. F Struct. Biol. Cryst. Commun. 61 221–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen, K. and Auld, D.S. 1991. Characterization of an inhibitory metal binding site in carboxypeptidase A. Biochemistry 30 2613–2618. [DOI] [PubMed] [Google Scholar]
- Lipscomb, W.N. and Sträter, N. 1996. Recent advances in zinc enzymology. Chem. Rev. 96 2375–2433. [DOI] [PubMed] [Google Scholar]
- López, R., García, E., García, P., Ronda, C., and Tomasz, A. 1982. Choline- containing bacteriophage receptors in Streptococcus pneumoniae. J. Bacteriol. 151 1581–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López, R., García, E., García, P., and García, J.L. 2004. Cell wall hydrolases. In The pneumococcus (eds. E.E. Tuomanen et al.) pp. 75–88. ASM Press, Washington, DC.
- Pepys, M.B. and Hirschfield, G.M. 2003. C-reactive protein: A critical update. J. Clin. Inv. 111 1808–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasia, R.M. and Vila, A.J. 2002. Exploring the role of the binding affinity of a second zinc equivalent in B. cereus metallo-β-lactamase. Biochemistry 41 1853–1860. [DOI] [PubMed] [Google Scholar]
- Rasia, R.M., Ceolín, M., and Vila, A.J. 2003. Grafting a new metal ligand in cocatalytic site of B. cereus metallo-β-lactamase: Structural flexibility without loss of activity. Protein Sci. 12 1538– 1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenow, C., Ryan, P., Weiser, J.N., Johnson, S., Fontan, P., Ortqvist, A., and Masure, H.R. 1997. Contribution of novel choline-binding proteins to adherence, colonization and immunogenicity of Streptococcus pneumoniae. Mol. Microbiol. 25 819–829. [DOI] [PubMed] [Google Scholar]
- Sánchez-Beato, A.R., López, R., and García, J.L. 1998. Molecular characterization of PcpA: A novel choline-binding protein of Streptococcus pneumoniae. FEMS Lett. 164 207–214. [DOI] [PubMed] [Google Scholar]
- Schilling, O., Wenzel, N., Naylor, N., Vogel, A., Crowder, M., Makaroff, C., and Meyer-Klaucke, W. 2003. Flexible metal binding of the metallo-β-lactamase domain: Glyoxalase II incorporates iron, manganese and zinc in vivo. Biochemistry 42 11777–11786. [DOI] [PubMed] [Google Scholar]
- Swiatlo, E., McDaniel, L.S., and Briles, D.E. 2004. Choline-binding proteins. In The pneumococcus (eds. E.I. Tuomanen et al.), pp. 49–60. ASM Press, Washington, DC.
- Tipton, B.F. and Dixon, H.B. 1979. The effect of pH on enzymes. Methods Enzymol. 63 183–234. [DOI] [PubMed] [Google Scholar]
- Tomasz, A. 1967. Choline in the cell wall of a bacterium: Novel type of polymer-linked choline in pneumococcus. Science 157 694–697. [DOI] [PubMed] [Google Scholar]
- Tu, A.H., Fulgham, R.L., McCroy, M.A., Briles, D.E., and Szalai, A.J. 1999. Pneumococcal surface protein A inhibits complement activation by Streptococcus pneumoniae. Infect. Immun. 67 4720–4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel, A., Schilling, O., Niecke, M., Bettmer, J., and Meyer-Klaucke, W. 2002. ElaC encodes a novel binuclear zinc phosphodiesterase. J. Biol. Chem. 277 29078–29085. [DOI] [PubMed] [Google Scholar]
- Vollmer, W. and Tomasz, A. 2001. Identification of the teichoic acid phosphorylcholine esterase in Streptococcus pneumoniae. Mol. Microbiol. 39 1610–1622. [DOI] [PubMed] [Google Scholar]
- Weiser, J. and Kapoor, M. 1999. Effect of intra-strain variation in the amount of capsular polysaccharide on genetic transformation of Streptococcus pneumoniae: Implications for virulence studies of encapsulated strains. Infect. Immun. 67 3690–3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox, D.E. 1996. Binuclear metallohydrolases. Chem. Rev. 96 2435–2458. [DOI] [PubMed] [Google Scholar]
- Yamaguchi, Y., Kuroki, T., Yasuzawa, H., Higashi, T., Jin, W., Kawanami, A., Yamagata, Y., Arakawa, Y., Got, M., and Kurosaki, H. 2005. Probing the role of Asp-120(81) of metallo-β-lactamase (IMP-1) by site-directed mutagenesis, kinetic studies and X-ray crystallography. J. Biol. Chem. 280 20824–20832. [DOI] [PubMed] [Google Scholar]
- Yother, J.J., Leopold, K., White, J., and Fischer, W. 1988. Generation and properties of a Streptococcus pneumoniae mutant which does not require choline or analogs for growth. J. Bacteriol. 180 2093–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang, T.M., Hollman, D.A., Crawford, P.A., Crowder, M.W., and Makaroff, C.A. 2001. Arabidopsis glyoxalase II contains a zinc/iron binuclear metal center that is essential for substrate binding and catalysis. J. Biol. Chem. 276 4788–4795. [DOI] [PubMed] [Google Scholar]