

Abstract
Peptidases are classical objects of enzymology and structural studies. However, a few protein families with experimentally characterized proteolytic activity, but unknown catalytic mechanism and three-dimensional structures, still exist. Using comparative sequence analysis, we deduce spatial structure for one of such families, namely, U40, which contains just one P5 protein from bacteriophage phi-6. We show that this singleton sequence possesses conserved sequence motifs characteristic of lysozymes and is a distant homolog of lytic transglycosylases that cleave bacterial peptidoglycan. The structure of the P5 protein is therefore predicted to adopt the lysozyme-like fold shared by T4, λ, C-type, G-type lysozymes, and lytic transglycosylases. Since previous biochemical experiments with P5 of phi-6 have indicated that the purified enzyme possesses endopeptidase activity and not glycosidase activity, our results point to the possibility of a newly evolved molecular function and call for further experimental characterization of this unusual P5 protein.
Keywords: structure prediction, bacteriophage phi-6, peptidase, lytic transglycosylase, lysozyme, comparative phage genomics
Peptidases are ubiquitous enzymes that cleave peptide bonds. The proteolytic reaction can be carried out by a variety of enzymes with different folds and catalytic mechanisms. A comprehensive structural classification of peptidases is available from the MEROPS database (Rawlings et al. 2004), where evolutionarily related peptidases form clans and each clan is divided into families based on close homology. Due to the importance of peptidase activity, many peptidase families have been well studied experimentally. However, a few peptidase families with no information about three-dimensional structure or catalytic mechanism remain and are classified as “peptidases with unknown mechanism” in MEROPS. We have applied comparative sequence and structural analysis to predict the structure and catalytic mechanism of several such unknown-type peptidase families and to identify novel peptidase families (Pei and Grishin 2001, 2002, 2003; Cheng et al. 2004; Ginalski et al. 2004a,b). Here we report identification of remote homologs and structure prediction for yet another peptidase family with unknown mechanism, U40.
In MEROPS, the U40 family contains a single sequence, the P5 protein from bacteriophage phi-6. Bacteriophage phi-6 is a double-stranded RNA virus that infects Pseudomonae (Semancik et al. 1973). The P5 protein has been shown to be a lytic enzyme that cleaves the bacterial peptidoglycan (murein) layer to facilitate membrane fusion during infection and is also responsible for cell lysis in late infection (Mindich and Lehman 1979; Bamford and Palva 1980). Caldentey and Bamford (1992) purified and characterized P5 for substrate specificity. The protein was shown to be active against cell walls of various Gram-negative species. The samples of cell walls treated with P5 reacted positively with 2,4-dinitrophenol, indicating that amino groups were liberated by the enzyme. In contrast, neither muramicitol nor glucosaminitol was detected. Based on these observations, it was suggested that P5 cleaves a peptide bond and is not a glycosidase (Caldentey and Bamford 1992). The investigators of the MEROPS database have thus classified the P5 protein as a peptidase and assigned a function of “murein endopeptidase.” However, the catalytic mechanism and active site residues have not been identified for this protein.
CDD (Marchler-Bauer et al. 2002), PFAM (Bateman et al. 2004), and SMART (Letunic et al. 2004) database searches using the P5 protein sequence did not yield significant hits to known domains. A PSI-BLAST (Altschul et al. 1997) search with the P5 protein (gene identification [gi] no. 20330562, 220 residues) did not detect any other sequences with significant e-values (<0.02). The 3D-JURY fold recognition META-server (Ginalski et al. 2003) also did not yield any significant hits to existing structures. The P5 protein is apparently a singleton sequence (Pei and Grishin 2002; Siew et al. 2004) without close homologs in the current protein database (the nr database, October 2004, 2,082,196 sequences; 699,810,385 total letters). However, a sequence (gene identification no. 14422162, local alignment from 10–185 out of a length of 247 residues) from bacteriophage phi-12 was found as the best BLAST hit of P5 with an e-value of 0.045.
Bacteriophage phi-12 (Gottlieb et al. 2002a,b) belongs to the same genus (Cystovirus) as bacteriophage phi-6, and they are considered to be closely related evolutionarily. Both phages have three segments of chromosomal double-stranded RNA. This weak hit from bacteriophage phi-12, annotated as a muramidase, has the same chromosomal location (Gottlieb et al. 2002b) as P5 in bacteriophage phi-6, and therefore is likely to be a homolog of P5 protein. A PSI-BLAST search using this muramidase as a query finds the P5 protein with an e-value of 0.017 in the first round as the best hit, and further iterations find significant hits to lytic transglycosylases (Koraimann 2003) that belong to the lysozyme superfamily. For example, the lytic transglycosylase from Bartonella henselae was found with an e-value of 3e - 11 in the fourth iteration. To further verify the homology relationship between the P5 proteins and lysozymes, we started transitive PSI-BLAST searches from the lysozyme domain of the lytic transglycosylase with known structure (Protein Data Bank [PDB] ID 1SLY, residues 451–618; Thunnissen et al. 1994) (see Materials and Methods). Indeed, P5 proteins from both phages phi-6 and phi-12 were found during the course of these extensive searches. For instance, PSI-BLAST with the query gi|48869064 (a soluble lytic murein transglycosylase) found the P5 protein from bacteriophage phi-6 with an e-value of 0.01 in the third iteration.
Additional evidence for the existence of viral homologs is provided by the P5 protein of bacteriophage phi-13, which was found in transitive PSI-BLAST iterations and contained a domain closely related to lytic transglycosylases, as indicated by CDD or PFAM searches. Bacteriophage phi-13 (Qiao et al. 2000) also belongs to the genus of Cystovirus with three segments of double-stranded RNA and has the same chromosomal location of the P5 protein. A complete survey of existing Cystovirus proteins at the National Center for Biotechnology Information (NCBI) Web site did not reveal other proteins with a lytic transglycosylase domain. Complete genome sequence is available for only one other Cystovirus member: bacteriophage phi-8 (Hoogstraten et al. 2000). However, no phi-8 proteins were found to be close homologs of the P5 proteins from phi-6, phi-12, and phi-13. Unlike bacteriophages phi-12 and phi-13, the bacteriophage phi-8 is very distantly related to phi-6, displaying marked differences in gene structure and sequence (Hoogstraten et al. 2000).
The lytic transglycosylase (PFAM: SLT; COG0741) has the same fold as the well-studied hen egg-white lysozyme (HEWL) and T4 lysozyme (Strynadka and James 1996). In the SCOP database (version 1.65) (Murzin et al. 1995), they all belong to the “Lysozyme-like” fold, which consists of one superfamily and seven families. Extensive PSI-BLAST iterations for the lytic transglycosylases (bacterial muramidase, catalytic domain in SCOP) found ~2000 homologs and linked the eukaryotic families of G-type (goose-type) lysozymes and C-type (chicken-type) lysozymes. Other lysozyme families in the SCOP database, such as the families of “Phage T4 lysozymes” and “Chitosanase,” are not found using the lytic transglycosylases as queries. A multiple sequence alignment was constructed by using program PCMA (Pei et al. 2003) for the representative sequences of the lytic transglycosylases, G-type lysozymes, C-type lysozymes, and the P5 proteins from bacteriophages phi-6, phi-12, and phi-13. The alignment was manually adjusted guided by the structural superposition of known structures. To reflect similarities among the sequences, a distance diagram was made by using the Euclidean distance mapping method (Fig. 1 ▶; Grishin and Grishin 2002). Close homologs of lytic transglycosylases, G-type lysozymes, and C-type lysozymes form three well-separated clusters. The P5 protein from bacteriophage phi-13 belongs to the cluster of lytic transglycosylases, although it is relatively far from the cluster center. The P5 proteins from bacteriophages phi-6 and phi-12 are far from the rest of the sequences, suggesting an elevated substitution rate and rapid evolution of these bacteriophage sequences. Consistent with the difficulty of homology inference, the phi-6 P5 protein is the most distant from the rest of the transglycosylase sequences.
Figure 1.

Plot of Euclidian distances between lysozyme sequences. The plot is a three-dimensional projection of a multidimensional Euclidian space (Grishin and Grishin 2002). The three P5 proteins and the three families of lysozyme homologs are represented by symbols with different shapes and filling effects. Distances between symbols reflect evolutionary divergence between corresponding sequences.
Irrespective of their evolutionary divergence, all P5 proteins contain three conserved motifs that are present in lytic transglycosylases and their lysozyme homologs (Thunnissen et al. 1995; Mushegian et al. 1996). These conserved motifs are functionally and structurally important signatures for HEWL-like lysozymes. Regions of the multiple sequence alignment displaying the three motifs for representative lysozyme homologs are shown in Figure 2A ▶. The secondary structural elements containing the three conserved motifs are highlighted in the structural diagram of a lytic transglycosylase domain in Figure 2B ▶. The first motif is an N-terminal α-helix that contains the conserved catalytic residue glutamate. This α-helix is mostly buried and consists of mainly hydrophobic residues. The catalytic glutamate is situated at the end of the α-helix. The position right after the catalytic residue is also highly conserved and usually contains a serine residue that makes a critical hydrogen bond to the β-sheet in the structure. In the P5 protein from bacteriophage phi-6, this position is occupied by an asparigine. The second conserved motif has a sequence signature of GXXQ, where X is often a hydrophobic residue. This motif corresponds to the second turn of a three-stranded β-sheet in the active site. The glycine is highly conserved because of its unique backbone conformation. The glutamine makes critical interactions with a few backbone polar atoms. The third conserved motif has a sequence signature of φp, where φ is an aromatic residue and p stands for a polar residue. The motif is located at the end of an α-helix that points toward the active site in lytic transglycosylases and G-type lysozymes (α2 in Fig. 2B ▶). The polar residue p, usually an asparagine, is important for substrate binding in lytic transglycosylases and G-type lysozymes. In the C-type lysozymes, the α2 helix is almost deteriorated and the polar residue is not conserved.
Figure 2.
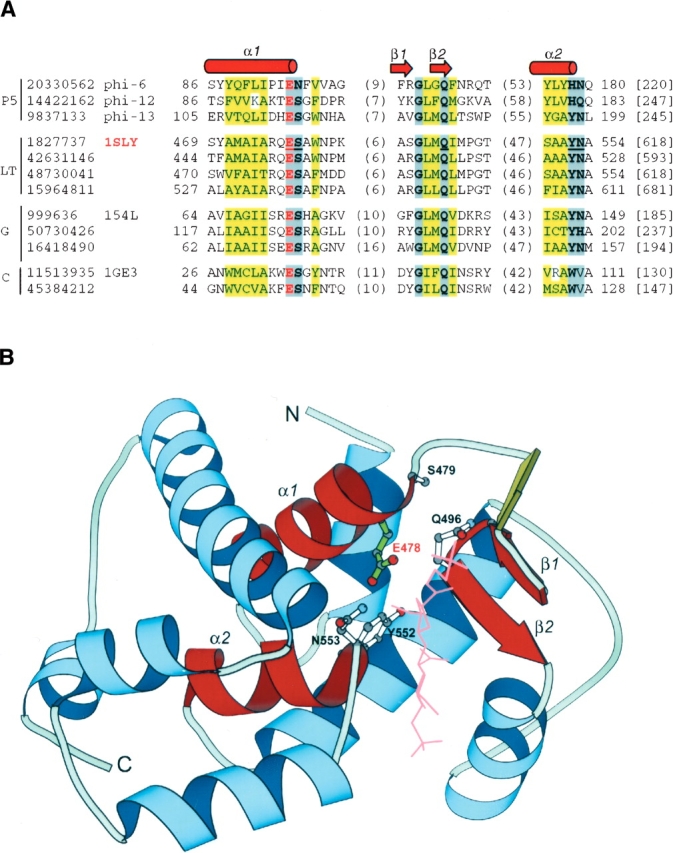
(A) A multiple sequence alignment of the motifs in P5 proteins and lysozyme homologs. The sequence identifiers on the left are NCBI gene identification (gi) numbers. The first residue number and the sequence length are shown. The numbers of residues between the motifs are shown in parentheses. The catalytic glutamate residue is in red letters, and conserved residues are shaded in gray. Positions occupied by mainly hydrophobic residues are shaded in yellow. Family abbreviations are shown to the left of sequence identifiers: P5, bacteriophage P5 proteins; LT, lytic transglycosylases; G, G-type lysozymes; and C, C-type lysozymes. The bacteriophage names and the representative PDB (Berman et al. 2002) structures are shown after the gi numbers. α-Helices and β-strands are shown as cylinders and arrows, respectively. (B) The structure of the soluble lytic transglycosylase catalytic domain from Escherichia coli. The PDB code is 1SLY, and the residue range is from 451–618. The secondary structural elements containing the three conserved motifs are highlighted in red and labeled according to A. The side-chains of a few conserved residues (underlined letters in A) in these motifs are shown in ball-and-stick representation and labeled; the catalytic residue is highlighted in green and labeled in red. The ligand (bulgecin A) is displayed in pink lines. The structure was drawn using the program BOBSCRIPT (Esnouf 1997).
Our analysis of sequence similarity searches and Cystovirus phage genomes suggests that the P5 protein from bacteriophage phi-6, as the only member of peptidase family U40, is homologous to the lytic transglycosylases and has a lysozyme-like fold. This prediction is consistent with the lytic function of this protein. The only experimental study of the P5 protein indicates that this enzyme cleaves the peptide bond in peptidoglycan and is not a glycosidase (Caldentey and Bamford 1992). The experiment to test glycosidase activity was designed to detect the reducing sugars resulting from glycosidase cleavage using NaB3H4 (Caldentey and Bamford 1992). However, transglycosylases produce a nonreducing 1,6-anhydro-bond in the N-acetylmuramic acid moiety (Holtje et al. 1975). Therefore, the negative result of reducing sugar generation cannot rule out possible transglycosylase activity of the P5 protein. Although unlikely, it is possible that the P5 protein has evolved an endopeptidase activity to maintain the same cellular function. Interestingly, a similar scenario has been proposed for some invertebrate lysozymes (Bachali et al. 2002), such as the ones from the medicinal leech Hirudo medicinalis (Zavalova et al. 1996, 2000) and the marine bivalve Tapes japonica (Takeshita et al. 2003), which possibly have both lysozyme and isopeptidase activities. Necessary caution is advised in interpreting these results since a subsequent report on H. medicinalis lysozymes (Baskova et al. 2001) described apparent separation of isopeptidase and lysozyme activities.
It is well known that many viruses evolve rapidly (Iyer et al. 2001) and new molecular functions could appear as a result of significant sequence divergence (Todd et al. 2001, 2002; Bartlett et al. 2002). The new peptidase active site could have emerged at a location different from the transglycosylase active site, for instance, in the peptide-binding groove. The experimental results suggested that the peptidase activity was directed at the peptide bond between diaminopimelic acid and d-alanine, which is quite far from the glycosidic bond targeted by lysozymes. This scenario has been suggested for the bifunctional invertebrate lysozyme from T. japonica, for which experimental studies have indicated that the isopeptidase activity and chitinase activity are housed at different active sites (Takeshita et al. 2003). Alternatively, the experimental results based on the phi-6 P5 protein purified to apparent homogeneity from disrupted viral particles (Caldentey and Bamford 1992) might not reflect its in vivo activity; or there could be a slight amount of contamination by other endopeptidases in the purified P5 protein. In addition, some membrane-bound proteases that maintain bacterial cell wall might have not been removed during substrate preparation in Caldentey and Bamford’s experiments. Preservation of conserved motifs in the P5 protein strongly suggests that it maintains transglycosylase activity. Further experimental studies are required to clarify the in vivo enzymatic activity of the P5 protein from bacteriophage phi-6 and other similar bacteriophages. Our analysis and predictions structurally annotate the U40 peptidase family and offer testable hypotheses about its potential active site residues.
Materials and methods
The PSI-BLAST program (Altschul et al. 1997) was used to search for homologs of the P5 proteins and the lytic transglycosylases against the NCBI non-redundant database (October 2004, 2,082,196 sequences; 699,810,385 total letters). The e-value threshold was 0.01 for inclusion of sequences into a profile. The other parameters were default. To ensure full coverage, found homologs were grouped by single-linkage clustering (1 bit per site threshold, ~50% sequence identity), and representative sequences from each group were used as queries for further PSI-BLAST iterations, as scripted by using the SEALS package (Walker and Koonin 1997).
A multiple sequence alignment was constructed by using the PCMA program (Pei et al. 2003) for representative sequences of C-type lysozymes, G-type lysozymes, bacterial lytic transglycosylases, and the P5 proteins. Manual adjustment of the multiple sequence alignment was made with guidance from available structures. Euclidean distance plot was made from the alignment by using the EESG program (Grishin and Grishin 2002).
Acknowledgments
We thank Lisa Kinch for critical reading of the manuscript and anonymous reviewer for helpful comments and suggestions. This work was supported by NIH grant GM67165 to N.V.G.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041250005.
References
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachali, S., Jager, M., Hassanin, A., Schoentgen, F., Jolles, P., Fiala-Medioni, A., and Deutsch, J.S. 2002. Phylogenetic analysis of invertebrate lysozymes and the evolution of lysozyme function. J. Mol. Evol. 54 652–664. [DOI] [PubMed] [Google Scholar]
- Bamford, D.H. and Palva, E.T. 1980. Structure of the lipid-containing bacteriophage phi 6: Disruption by Triton X-100 treatment. Biochim. Biophys. Acta 601 245–259. [DOI] [PubMed] [Google Scholar]
- Bartlett, G.J., Porter, C.T., Borkakoti, N., and Thornton, J.M. 2002. Analysis of catalytic residues in enzyme active sites. J. Mol. Biol. 324 105–121. [DOI] [PubMed] [Google Scholar]
- Baskova, I.P., Zavalova, L.L., Basanova, A.V., and Sass, A.V. 2001. Separation of monomerizing and lysozyme activities of destabilase from medicinal leech salivary gland secretion. Biochemistry 66 1368–1373. [DOI] [PubMed] [Google Scholar]
- Bateman, A., Coin, L., Durbin, R., Finn, R.D., Hollich, V., Griffiths-Jones, S., Khanna, A., Marshall, M., Moxon, S., Sonnhammer, E.L., et al. 2004. The Pfam protein families database. Nucleic Acids Res. 32 D138–D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman, H.M., Battistuz, T., Bhat, T.N., Bluhm, W.F., Bourne, P.E., Burkhardt, K., Feng, Z., Gilliland, G.L., Iype, L., Jain, S., et al. 2002. The Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr. 58 899–907. [DOI] [PubMed] [Google Scholar]
- Caldentey, J. and Bamford, D.H. 1992. The lytic enzyme of the Pseudomonas phage phi 6: Purification and biochemical characterization. Biochim. Biophys. Acta 1159 44–50. [DOI] [PubMed] [Google Scholar]
- Cheng, H., Shen, N., Pei, J., and Grishin, N.V. 2004. Double-stranded DNA bacteriophage prohead protease is homologous to herpesvirus protease. Protein Sci. 13 2260–2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esnouf, R.M. 1997. An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J. Mol. Graph. Model. 15 133–138. [DOI] [PubMed] [Google Scholar]
- Ginalski, K., Elofsson, A., Fischer, D., and Rychlewski, L. 2003. 3D-Jury: A simple approach to improve protein structure predictions. Bioinformatics 19 1015–1018. [DOI] [PubMed] [Google Scholar]
- Ginalski, K., Kinch, L., Rychlewski, L., and Grishin, N.V. 2004a. BTLCP proteins: A novel family of bacterial transglutaminase-like cysteine proteinases. Trends Biochem. Sci. 29 392–395. [DOI] [PubMed] [Google Scholar]
- ———. 2004b. Raptor protein contains a caspase-like domain. Trends Biochem. Sci. 29 522–524. [DOI] [PubMed] [Google Scholar]
- Gottlieb, P., Potgieter, C., Wei, H., and Toporovsky, I. 2002a. Characterization of phi12, a bacteriophage related to phi6: Nucleotide sequence of the large double-stranded RNA. Virology 295 266–271. [DOI] [PubMed] [Google Scholar]
- Gottlieb, P., Wei, H., Potgieter, C., and Toporovsky, I. 2002b. Characterization of phi 12, a bacteriophage related to phi 6: Nucleotide sequence of the small and middle double-stranded RNA. Virology 293 118–124. [DOI] [PubMed] [Google Scholar]
- Grishin, V.N. and Grishin, N.V. 2002. Euclidian space and grouping of biological objects. Bioinformatics 18 1523–1534. [DOI] [PubMed] [Google Scholar]
- Holtje, J.V., Mirelman, D., Sharon, N., and Schwarz, U. 1975. Novel type of murein transglycosylase in Escherichia coli. J. Bacteriol. 124 1067–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogstraten, D., Qiao, X., Sun, Y., Hu, A., Onodera, S., and Mindich, L. 2000. Characterization of phi8, a bacteriophage containing three double-stranded RNA genomic segments and distantly related to Phi6. Virology 272 218–224. [DOI] [PubMed] [Google Scholar]
- Iyer, L.M., Aravind, L., and Koonin, E.V. 2001. Common origin of four diverse families of large eukaryotic DNA viruses. J. Virol. 75 11720–11734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koraimann, G. 2003. Lytic transglycosylases in macromolecular transport systems of Gram-negative bacteria. Cell. Mol. Life Sci. 60 2371–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic, I., Copley, R.R., Schmidt, S., Ciccarelli, F.D., Doerks, T., Schultz, J., Ponting, C.P., and Bork, P. 2004. SMART 4.0: Towards genomic data integration. Nucleic Acids Res. 32 D142–D144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer, A., Panchenko, A.R., Shoemaker, B.A., Thiessen, P.A., Geer, L.Y., and Bryant, S.H. 2002. CDD: A database of conserved domain alignments with links to domain three-dimensional structure. Nucleic Acids Res. 30 281–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mindich, L. and Lehman, J. 1979. Cell wall lysin as a component of the bacteriophage phi 6 virion. J. Virol. 30 489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzin, A.G., Brenner, S.E., Hubbard, T., and Chothia, C. 1995. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247 536–540. [DOI] [PubMed] [Google Scholar]
- Mushegian, A.R., Fullner, K.J., Koonin, E.V., and Nester, E.W. 1996. A family of lysozyme-like virulence factors in bacterial pathogens of plants and animals. Proc. Natl. Acad. Sci. 93 7321–7326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei, J. and Grishin, N.V. 2001. Type II CAAX prenyl endopeptidases belong to a novel superfamily of putative membrane-bound metalloproteases. Trends Biochem. Sci. 26 275–277. [DOI] [PubMed] [Google Scholar]
- ———. 2002. Breaking the singleton of germination protease. Protein Sci. 11 691–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2003. Peptidase family U34 belongs to the superfamily of N-terminal nucleophile hydrolases. Protein Sci. 12 1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei, J., Sadreyev, R., and Grishin, N.V. 2003. PCMA: Fast and accurate multiple sequence alignment based on profile consistency. Bioinformatics 19 427–428. [DOI] [PubMed] [Google Scholar]
- Qiao, X., Qiao, J., Onodera, S., and Mindich, L. 2000. Characterization of phi 13, a bacteriophage related to phi 6 and containing three dsRNA genomic segments. Virology 275 218–224. [DOI] [PubMed] [Google Scholar]
- Rawlings, N.D., Tolle, D.P., and Barrett, A.J. 2004. MEROPS: The peptidase database. Nucleic Acids Res. 32 D160–D164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semancik, J.S., Vidaver, A.K., and Van Etten, J.L. 1973. Characterization of segmented double-helical RNA from bacteriophage phi6. J. Mol. Biol. 78 617–625. [DOI] [PubMed] [Google Scholar]
- Siew, N., Azaria, Y., and Fischer, D. 2004. The ORFanage: An ORFan database. Nucleic Acids Res. 32 D281–D283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strynadka, N.C. and James, M.N. 1996. Lysozyme: A model enzyme in protein crystallography. Exs 75 185–222. [DOI] [PubMed] [Google Scholar]
- Takeshita, K., Hashimoto, Y., Ueda, T., and Imoto, T. 2003. A small chimerically bifunctional monomeric protein: Tapes japonica lysozyme. Cell. Mol. Life Sci. 60 1944–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thunnissen, A.M., Dijkstra, A.J., Kalk, K.H., Rozeboom, H.J., Engel, H., Keck, W., and Dijkstra, B.W. 1994. Doughnut-shaped structure of a bacterial muramidase revealed by X-ray crystallography. Nature 367 750–753. [DOI] [PubMed] [Google Scholar]
- Thunnissen, A.M., Isaacs, N.W., and Dijkstra, B.W. 1995. The catalytic domain of a bacterial lytic transglycosylase defines a novel class of lysozymes. Proteins 22 245–258. [DOI] [PubMed] [Google Scholar]
- Todd, A.E., Orengo, C.A., and Thornton, J.M. 2001. Evolution of function in protein superfamilies, from a structural perspective. J. Mol. Biol. 307 1113–1143. [DOI] [PubMed] [Google Scholar]
- ———. 2002. Plasticity of enzyme active sites. Trends Biochem. Sci. 27 419–426. [DOI] [PubMed] [Google Scholar]
- Walker, D.R. and Koonin, E.V. 1997. SEALS: A system for easy analysis of lots of sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 5 333–339. [PubMed] [Google Scholar]
- Zavalova, L., Lukyanov, S., Baskova, I., Snezhkov, E., Akopov, S., Berezhnoy, S., Bogdanova, E., Barsova, E., and Sverdlov, E.D. 1996. Genes from the medicinal leech (Hirudo medicinalis) coding for unusual enzymes that specifically cleave endo-epsilon (γ-Glu)-Lys isopeptide bonds and help to dissolve blood clots. Mol. Gen. Genet. 253 20–25. [DOI] [PubMed] [Google Scholar]
- Zavalova, L.L., Baskova, I.P., Lukyanov, S.A., Sass, A.V., Snezhkov, E.V., Akopov, S.B., Artamonova, II, Archipova, V.S., Nesmeyanov, V.A., Kozlov, D.G., et al. 2000. Destabilase from the medicinal leech is a representative of a novel family of lysozymes. Biochim. Biophys. Acta 1478 69–77. [DOI] [PubMed] [Google Scholar]