

Abstract
The His46Arg (H46R) mutant of human copper-zinc superoxide dismutase (SOD1) is associated with an unusual, slowly progressing form of familial amyotrophic lateral sclerosis (FALS). Here we describe in detail the crystal structures of pathogenic H46R SOD1 in the Zn-loaded (Zn-H46R) and metal-free (apo-H46R) forms. The Zn-H46R structure demonstrates a novel zinc coordination that involves only three of the usual four liganding residues, His 63, His 80, and Asp 83 together with a water molecule. In addition, the Asp 124 “secondary bridge” between the copper- and zinc-binding sites is disrupted, and the “electrostatic loop” and “zinc loop” elements are largely disordered. The apo-H46R structure exhibits partial disorder in the electrostatic and zinc loop elements in three of the four dimers in the asymmetric unit, while the fourth has ordered loops due to crystal packing interactions. In both structures, nonnative SOD1–SOD1 interactions lead to the formation of higher-order filamentous arrays. The disordered loop elements may increase the likelihood of protein aggregation in vivo, either with other H46R molecules or with other critical cellular components. Importantly, the binding of zinc is not sufficient to prevent the formation of nonnative interactions between pathogenic H46R molecules. The increased tendency to aggregate, even in the presence of Zn, arising from the loss of the secondary bridge is consistent with the observation of an increased abundance of hyaline inclusions in spinal motor neurons and supporting cells in H46R SOD1 transgenic rats.
Keywords: copper-zinc superoxide dismutase, familial amyotrophic lateral sclerosis, ALS, amyloid, SOD1, H46R
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder characterized by the progressive loss of motor neurons in the brain and spinal cord leading to paralysis and eventual death, typically within 5 yr of symptom onset (Haverkamp et al. 1995). Approximately 10% of cases are familial (FALS), and ~20% of these are associated with autosomal dominant mutations in the gene encoding the 32-kDa homodimeric anti-oxidant enzyme copper-zinc superoxide dismutase (SOD1) (Deng et al. 1993; Rosen et al. 1993; Cleveland and Rothstein 2001). SOD1 catalyses the disproportionation of superoxide (O2−) radical to dioxygen and hydrogen peroxide via the cyclic reduction and reoxidation of its bound copper ion (Fridovich 1975).
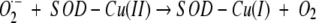 |
(1) |
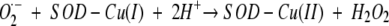 |
(2) |
To date, >100 FALS-inducing point mutations have been identified that map to all regions of the SOD1 molecule, including the dimer interface, β-barrel, loop regions, disulfide bond residues, and both the copper- and zinc-binding sites (a list can be found at http://www.alsod.org). FALS SOD1 mutations were first thought to reduce SOD1 enzymatic activity and thereby cause increased oxidative damage to motor neurons (Deng et al. 1993). However, the majority of FALS mutant SOD1 proteins display SOD1 activity comparable to that of the wild-type enzyme (Borchelt et al. 1995; Rabizadeh et al. 1995), and SOD1 knockout mice do not develop ALS (Reaume et al. 1996). In contrast, transgenic mice overexpressing human FALS mutant SOD1 in addition to their own fully functional SOD1 develop symptoms characteristic of ALS, while those overexpressing the wild-type human enzyme do not (Gurney 1997). Taken together, these results strongly suggest that the pathogenic mechanism of SOD1-linked FALS involves the gain of a toxic property by the mutant SOD1 enzyme, rather than a loss of function.
H46R SOD1-mediated FALS has been described as a milder subtype of the disease compared with the majority of FALS cases, where the mean age of onset is 42.8 yr and the mean survival after diagnosis is 4.8 yr (Ceroni et al. 2001). The H46R SOD1 mutation was first observed in four Japanese families (Aoki et al. 1994) that displayed an unusual FALS pathology. The patients initially presented with weakness in the legs, while upper extremity symptoms were delayed by >5 yr and bulbar symptoms by >8 yr. The mean age of onset was 49.6 yr, and mean survival was 17.3 yr. These findings were expanded in a more recent, larger study of 17 Japanese patients that showed an earlier mean age of onset (44.3 yr) and a mean disease duration of ~12 yr (Arisato et al. 2003). This stands in sharp contrast to the A4V and I113T mutants that are associated with rapid disease progression, and for which crystal structures have recently been determined (Cardoso et al. 2002; Hough et al. 2004).
Transgenic rats expressing H46R SOD1 develop motor neuron disease (Nagai et al. 2001), and neural tissues from these rats contain intracytoplasmic Lewy-like inclusion bodies similar to those observed in spinal cord tissue of human ALS patients. G93A rats examined in the same study have neural tissues with fewer aggregates and an obvious vacuolar pathology. Although the H46R rats in this study express over twice the amount of mutant SOD1 as do the G93A rats, motor neuron disease progressed more slowly for the animals harboring H46R (24 d) compared with those expressing G93A (8 d). This finding correlates with the lengthened disease course in human patients with the H46R mutation.
Each subunit of the SOD1 homodimer binds one copper and one zinc ion and folds as an eight-stranded Greek-key β-barrel that is stabilized by an intra-subunit disulfide bond near the active site (Tainer et al. 1982). Protruding from the β-barrel are two major structural elements termed the “electrostatic” and “zinc” loops. The electrostatic loop (loop VII, residues 121–144) contains charged residues that help guide the negatively charged superoxide substrate toward the catalytic copper site. The zinc loop (loop IV, residues 49–84) contains two distinct substructures. The first consists of residues 49–62 and is termed the “disulfide loop,” which is anchored to the β-barrel through the bond formed between Cys 57 and Cys 146, the latter of which is located on the C-terminal β-strand (strand 8) of the β-barrel. The second consists of residues 63–84 and includes all four zinc ligands (His 63, His 71, His 80, and Asp 83) and forms the zinc-binding site. Particularly important interactions between the electrostatic and zinc loops in the wild-type enzyme come from the side chain of Asp 124 of the electrostatic loop, which accepts a hydrogen bond simultaneously from the nonliganding nitrogen atom of zinc ligand His 71 and from the nonliganding nitrogen atom of copper ligand His 46. This Asp 124–mediated linkage between the copper- and zinc-binding sites and between the electrostatic and zinc loop elements is termed the “secondary bridge.” The “primary bridge” between the copper- and zinc-binding sites is also known as the “bridging imidazolate” and comes from the side chain of His 63, which binds to the two metals simultaneously in the Cu(II) form of the enzyme. Together, the electrostatic and zinc loop elements in their wild-type conformation form the substrate channel leading to the active site.
Histidine 46 is one of the four copper ligands in the SOD1 active site. As described above, Asp 124 makes simultaneous hydrogen bonds to the nonliganding imidazole nitrogen atoms of the copper ligand His 46 and zinc ligand His 71 to form the secondary bridge between the metal-binding sites that helps to stabilize and orient correctly the electrostatic and zinc loop elements. Because the pathogenic mutation occurs at the copper-binding site, H46R SOD1 has a substantially reduced ability to bind copper in vitro and possesses dramatically lower activity as compared to wild-type and other pathogenic SOD1 molecules (Carri et al. 1994).
Even when both copper and zinc are available in the growth medium, recombinant H46R SOD1 expressed in insect cells shows little detectable SOD activity and is severely metal deficient in comparison to wild-type SOD1 expressed and purified in the same manner (Hayward et al. 2002). Moreover, H46R SOD1 expressed in yeast fails to rescue the oxygen-sensitive phenotype of ySOD1Δ yeast, unlike other FALS mutant SOD1s with mutations outside of the metal-binding region, for example, G37R (Ratovitski et al. 1999). This metal-deficient H46R SOD1 also exhibits reduced thermal stability (as measured by differential scanning calorimetry) in comparison to recombinant metal-replete native SOD1 (Rodriguez et al. 2002). The reduction in stability relative to the wild-type protein is due primarily to metal deficiency of the mutant protein. In addition, cultured neuroblastoma cells expressing H46R SOD1 are more sensitive to paraquat than are those expressing human wild-type SODl (Gabbianelli et al. 1999).
To understand better the structural determinants of mutant SOD1 toxicity, we have determined several X-ray crystal structures of FALS SOD1 protein molecules. As part of this endeavor, we present here the structures of H46R SOD1 in the zinc-loaded and apo forms at 2.15 Å and 2.50 Å resolution, respectively. Previously, we described the modes of filamentous assembly by H46R proteins (Elam et al. 2003b). Here, we present a detailed analysis of the structures, and we report (1) that binding of zinc at the zinc site does not cause the zinc loop and secondary bridge to adopt the conformation characteristic of wild-type SOD1 that has been observed in the other metal-replete FALS mutants structurally characterized to date, and (2) that binding of zinc to the zinc site of H46R SOD1 does not prevent the formation of nonnative interactions between the protein dimers. The copper and zinc sites themselves show novel spatial arrangements that appear to compensate partially for the lack of metals and/or loop disorder. These results are discussed in light of the distinct ALS phenotype associated with H46R SOD1.
Results
Overall molecular structure
The X-ray crystal structures of human pathogenic SOD1 mutant H46R in its zinc-bound and apo forms were determined to resolutions of 2.15 Å and 2.5 Å, respectively, as described previously (Elam et al. 2003b). The structure of Zn-H46R contains two SOD1 dimers in the crystallographic asymmetric unit, designated as W/X and Y/Z, where the slash (/) represents the naturally occurring homodimer interface. Each subunit has zinc bound in the zinc-binding site but no metal bound in the copper-binding site. Analysis by inductively coupled mass spectrometry (ICP) revealed that Zn-H46R in solution contains 0.23 equivalents of copper and 1.9 equivalents of zinc per dimer. The overall β-barrel structure of Zn-H46R is quite similar to that of wild-type SOD1. However, large stretches of the electrostatic and zinc-binding loop elements are disordered and are not visible in the electron density maps (Fig. 1 ▶; Table 1). This comparison reveals significant differences between the Zn-H46R and native structures in residues immediately before or after the disordered loops. The zinc ligand His 71 falls within the disordered region and is absent from the electron density. Part of the zinc loop, residues 78–81 immediately following the disordered region, forms the interface to an adjacent dimer in the “helical filament” packing motif (see below) (Elam et al. 2003b).
Figure 1.

Superposition of a monomer of holo-wtSOD1 (red), apo-H46R (yellow), and Zn-H46R (blue). The apo-H46R monomer (G) is disordered in residues 68–77 (zinc loop) and 128–140 (electrostatic loop). The Zn-H46R structure is disordered in residues 66–77 and 126–141. The largest structural shifts between Zn-H46R and wtSOD1 occur in residues 118–125 and 78–80. In the latter case, the first residue after the break in the electron density Glu 78 is >7 Å away from its position in wtSOD1. In the case of the truncations of the model at residues 65 and 142, structural changes are limited to the final modeled residue. Superposition of the Zn-H46R dimers onto the best-defined dimer of the native hSOD structure yields a root mean square deviation (RMSD) for Cα atoms of 0.97 Å (not including those residues absent from the Zn-H46R model). Comparison of apo-H46R with the wild-type SOD1 dimer yields a RMSD of ~0.35 Å for all atoms shared between the two structures. The dimer interface is not altered by the H46R mutation in either structure.
Table 1.
Disordered regions
| Monomer | Disordered regions | RMS to wild-type |
| Zn-H46R | 0.97 Å | |
| W | 66–77, 126–141 | |
| X | 66–77, 126–141 | |
| Y | 66–77, 126–141 | |
| Z | 66–77, 126–141 | |
| Apo-H46R | 0.35 Å | |
| G | 68–77, 128–140 | |
| H | 68–78, 133–139 | |
| I | 68–77, 133–140 | |
| J | 70–77, 132–141 | |
| K | 68–77, 125–141 | |
| L | 67–78, 127–137 | |
| M | — | |
| N | — |
The apo-H46R structure has four dimers in the crystallographic asymmetric unit with subunits designated as G/H, I/J, K/L, and M/N. Structural differences between the dimers are limited to specific interactions between residues at the vacant metal sites and in the degree of disorder in the electrostatic and zinc loops. All metal sites in apo-H46R are completely devoid of both copper and zinc ions, consistent with ICP analysis that indicated 0.0 equivalents of copper and 0.1 equivalents of zinc per dimer. In all apo-H46R monomers, residues 65–80 in the zinc loop demonstrate some degree of disorder, and in almost all cases, residues 68–77 had no electron density visible for chain tracing (Table 1). Exceptions were subunits M and N that, despite high thermal parameters in this region (in the range 46–100 Å2), displayed enough electron density to trace the backbone fully, resulting in a dimer in which the conformations of these loop elements resembles those of holo wild-type SOD1. The amount of disorder in the electrostatic loop of each monomer was dependent on the exact nature of interactions occurring at the metal sites and on crystal packing interactions with symmetry-related molecules. A general trend was observed in that the more tenuous the interactions between Asp 124, Arg 46 (which replaces His 46), and His 71 that link the copper and zinc sites (see below), the more disorder was observed in the electrostatic loop adjacent to Asp 124. Despite the differences at the metal sites and the localized disorder, the overall structure of apo-H46R is similar to that of Zn-H46R and wtSOD1.
Mutation site and copper center
The copper-binding site of Zn-H46R is devoid of metal ions, and the side-chain positions are altered relative to those of wtSOD1 (Fig. 2A ▶). A superposition of the Zn-H46R copper site with that of holo- and apo-wtSOD1 is shown in Figure 3A ▶. The altered side-chain position of His 120 is associated with a 0.4 Å shift in the position of its Cα atom, part of a concerted movement of residues Val 118–Asp 125, the latter of which is the final ordered residue in the electrostatic loop. His 63, which bridges between the copper and zinc ions in Cu(II)-wtSOD1, adopts a different conformation in the absence of copper. The new position of the His 63 imidazole ring allows a bond to form between its Nɛ2 atom and a sulfate anion, while at the same time its conformation is sterically constrained by the close approach of Arg 46, whose Cδ atom comes within 3.17 Å of the His Nɛ2 atom. Sulfate occupies a position in the active site channel similar to that previously observed in the structures of native and D125H SOD1 (Elam et al. 2003a; Strange et al. 2003) such that its oxygen atoms form a number of hydrogen bonds to the protein (Fig. 2B ▶; Table 2). Thus, the presence of sulfate in the Zn-H46R structure appears to stabilize the vacant copper site through the formation of a hydrogen-bonding network with His 120, His 63, and Arg 143.
Figure 2.
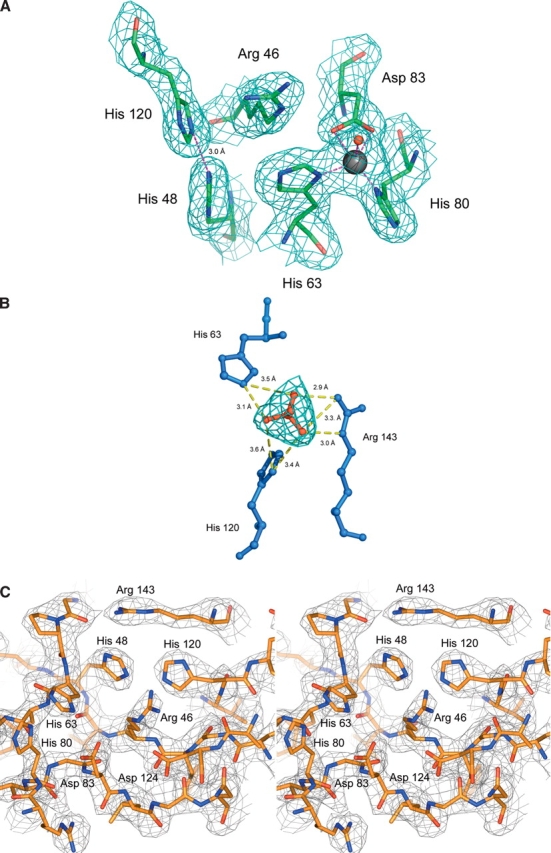
(A) 2Fobs-Fcalc electron density maps, contoured at 1σ, for the copper and zinc sites of Zn-H46R. The copper site is vacant, and the mutated residue Arg 46 is well-defined. The zinc site is fully occupied, and the zinc ion is ligated by only three residues, His 63, His 80, and Asp 83. The usual fourth zinc ligand, His 71, is disordered and not present in the electron density. A water molecule (shown as a red sphere) acts as an additional weak ligand some 2.4 Å from Zn. The copper ligands His 48 and His 120 are stabilized by a 3 Å hydrogen bond between their Nɛ2 atoms. (B) Binding of sulfate near to the vacant copper site in Zn-H46R. Sulfate forms hydrogen bonds to Arg 143 and to the copper ligands His 63 and His 120, thus stabilizing the site in the absence of metal (see Table 3). (C) Stereo image of the structure and electron density around the altered active site of the copper and zinc sites of apo-H46R SOD1 monomer G. The 2.5 Å σA-weighted electron density, with coefficients 2mFo - DFc, is contoured at 1σ. Note the lack of metal ion electron density and novel hydrogen bonding between His 48 and His 120 of the SOD1 copper site and His 63 and His 80 of the SOD1 zinc site. Asp 83 and Asp 124 (the secondary bridging residue) are within hydrogen bonding distance of mutated Arg 46, further stabilizing the site. The fourth zinc ligand, His 71, exhibits no electron density and therefore was not modeled in this monomer.
Figure 3.
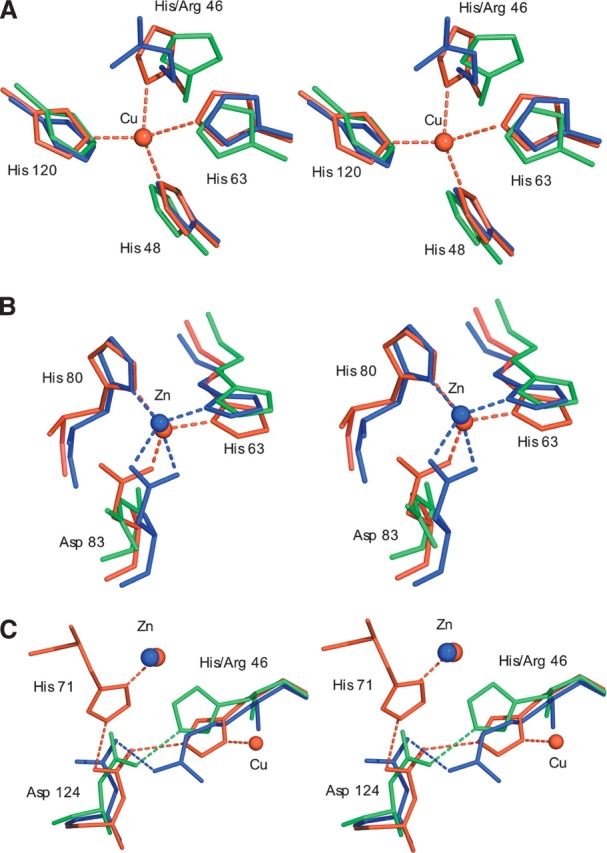
(A) Stereo figures showing the superposition of Zn-H46R (blue), wtSOD1 (red), and apo wtSOD1 (green) for the copper site. The Nη1 atom of Arg 46 occupies a position 2.8 Å from the position occupied by copper in the native hSOD structure, while the Cɛ atom resides some 2.9 Å away. The new conformation of His 120 in Zn-H46R allows the copper-ligating Nɛ2 atoms of histidines 48 and 120 to form a ~3 Å hydrogen bond. His 48 remains in a very similar conformation in the mutant and wtSOD1 structures. This preserved conformation is likely to be a consequence of the hydrogen bond between its Nδ1 atom and the O atom of Gly 61. The side chain of His 120 is shifted, occupying a position slightly closer to the vacant copper position (Nɛ2 to copper distance would be ~1.8 Å). (B) The zinc site. In Zn-H46R zinc is ligated by only three protein residues where Asp 83 acts as a bidentate ligand. His 71 is absent in Zn-H46R and is omitted from this figure for clarity, as is the water ligand observed in the Zn-H46R structure. (C) The secondary bridge. In wtSOD1, residue Asp 124 forms a hydrogen-bonded link between the copper ligand His 46 and the zinc ligand His 71. Asp 124 Oδ2 forms a strong (~2.6 Å) hydrogen bond to His46 Nɛ2, while its Oδ1 atom bonds to His71 Nɛ2. In addition, the Oδ1 atom is close to the backbone nitrogen atoms of residues 125 (3.1 Å) and 126 (2.8 Å) of the electrostatic loop, providing additional stabilization of the structure. In Zn-H46R, the mutant residue 46 maintains hydrogen bonding to Asp 124, but residue His 71 is disordered and the bridge is broken. In apo-wtSOD1, in the absence of metals His 46 hydrogen bonds to Asp 124, which adopts a different rotamer to that present in wtSOD1.
Table 2.
Metal-ligand and sulfate bond distances in Zn-H46R (Å)
| W | X | Y | Z | |
| Zn-His 63 Nɛ 1 | 2.0 | 1.9 | 2.0 | 1.8 |
| Zn-Asp 83 Oδ1 | 2.3 | 2.4 | 2.5 | 2.5 |
| Zn-Asp 83 Oδ 2 | 2.2 | 2.4 | 2.1 | 2.2 |
| Zn-His 80 Nδ 1 | 1.9 | 2.0 | 1.8 | 1.8 |
| Zn-water | 2.4 | 2.5 | 2.4 | 2.4 |
| Sulfate O1–His 63 Nɛ 2 | 3.0 | 3.1 | 3.2 | 2.9 |
| Sulfate O2–Arg 143 Nɛ | 3.1 | 2.7 | 3.0 | 2.8 |
| Sulfate O4–Arg 143 Nη2 | 3.1 | 2.8 | 2.9 | 2.8 |
In the apo-H46R structure, the conformation of the copper and zinc sites in the absence of metal varies in the eight monomers depending on the stability of the secondary bridge and consequent stability of the electrostatic loop (Table 1). One common feature in all monomers is that the copper ligands His 48 and His 120 are well-ordered and are stabilized by a hydrogen bond between their Nɛ2 atoms, adopting a similar position to that observed in Zn-H46R (see Fig. 2c ▶). No sulfate is present in the structure of apo-H46R, although it was a minor component of the crystallization buffer. Overall, in both apo and zinc-bound H46R, Arg 46 physically blocks the site where copper binds in wtSOD1. In most of the subunits, however, the rest of the site makes only slight shifts to satisfy the hydrogen bonding donor and acceptor nature of the ligands. Parts of the active site channel are absent from the Zn-H46R and apo-H46R structures due to the disordered zinc and electrostatic loops, which exposes the metal sites to solvent.
The zinc sites
The zinc-binding sites in the Zn-H46R structure are fully occupied and yet their structures are significantly different from those observed in wtSOD1 (Fig. 3B ▶; Table 2). The zinc atom is shifted some 0.7 Å from its position in wt-SOD1, and, only His 80 remains in a similar position in the mutant structure. His 80 forms a ~1.8–1.9 Å bond to zinc through its Nδ1 atom, although the imidazole ring is rotated by ~20°. The new position of His 63 maintains a bond between its Nδ1 atom and zinc at a distance of ~1.9 Å. His 71 falls in the disordered region of the H46R structure and is absent from the electron density maps. A water molecule has been modeled at ~2.5 Å from copper in the approximate direction of the missing His 71 ligand. The largest shift at the zinc site occurs in residue Asp 83. In the absence of the His 71 ligand, the side chain of Asp 83 adopts a new position such that the zinc-ligating Oδ2 atom is shifted by ~1.6 Å relative to wtSOD1. This movement, concomitant with the shift in the position of zinc, allows Asp 83 to act as a bidentate ligand to zinc (Oδ1 distance, 2.3–2.5 Å; Oδ2 distance, 2.1–2.4 Å). The zinc atom in this rearranged metal center is five coordinate, but with only three ligand residues, and an additional weak water ligand. This zinc coordination has not been observed previously in any structure of SOD1 (Table 2).
In contrast, apo-H46R has no bound metal at the zinc site. The zinc ligands display a variable amount of order as in the copper site, however, all monomers show a severely disrupted zinc site. The position and stability of individual ligand residues at the zinc site in different monomers is quite variable, and different arrangements of hydrogen bonding are present in the monomers (Table 3). Residue His 71 falls within the disordered region of the zinc loop and is therefore absent from the zinc site. It is apparent that the stability of the zinc site ligands (and this portion of the zinc loop) is correlated with the relative stability of the electrostatic loop, which is in turn influenced by the presence or absence of the secondary bridge and/or packing interactions.
Table 3.
Hydrogen bonds at the copper and zinc sites (Å)
| Monomer | W | X | Y | Z |
| Zn-H46R | ||||
| Asp 124Oδ2 Arg 46 Nɛ2 | 3.1 | 3.4 | 3.4 | 3.9 |
| His 120 Nɛ2-His 48 Nɛ2 | 3.0 | 2.9 | 3.0 | 3.0 |
| Monomer | G | H | I | J | K | L | M | N |
| a Bidentate interaction with Asp 83 Oδ1 and Oδ2. | ||||||||
| Apo-H46R | ||||||||
| Copper site | ||||||||
| His 48Nɛ2-His 120Nɛ2 | 3.0 | 3.1 | 3.1 | 3.1 | 3.1 | 3.0 | 3.1 | 3.1 |
| Arg 46Nη1-His 120 Nɛ | 3.5 | 3.3 | 3.2 | 3.2 | 3.3 | 3.4 | — | |
| Arg 46Nη2-Gly 138O | 3.1 | |||||||
| Arg 46Nη2-Thr 137O | 2.7 | |||||||
| Arg 46Nη2-His 63 Nɛ2 | 3.1 | |||||||
| Arg 46Nɛ-His 63 Nɛ2 | 3.2 | |||||||
| Zinc site | ||||||||
| His 63Nɛ2-His 80Nɛ2 | 2.7 | 2.9 | 3.0 | 2.5 | ||||
| His 80Nδ1-His 63Nδ1 | 3.2 | 2.6 | ||||||
| His 80Nɛ2-Arg 69O | 2.8 | 3.4 | ||||||
| His 63Nδ1-Asp 83Oδ1 | 3.0 | |||||||
| Involving Asp124 | ||||||||
| Asp 124-Arg 46Nη2 | 2.7 | 2.9 | 2.9 | 3.1 | 3.4 | 3.0 | 2.9 | |
| Asp 124-Arg 46Nɛ | 3.2 | |||||||
| Asp 124Oδ1-His 71 | 2.8 | |||||||
| Asp 124Oδ2-Asp 83 | 2.9 | 2.9/3.0a | ||||||
The secondary bridge
In wtSOD1, Asp 124 acts as the “secondary bridge” connecting the copper and zinc sites through its simultaneous hydrogen bonds to the copper ligand His 46 and the zinc ligand His 71 (Fig. 3c ▶). In Zn-H46R, the mutated Arg 46 forms a much weaker (~3.1 Å) hydrogen bond through its Nη2 atom to Asp 124 Oɛ2. The Asp 124 Cγ–Oδ2–Arg 46 Nη angle is ~80°–90° in Zn-H46R compared with ~130° for the Asp 124 Cγ–Oδ2–His 63 Nη2 angle in wtSOD1, indicating a conformation less preferable for hydrogen bonding. Asp 124 is shifted significantly in position. The hydrogen bond between Asp 124 Oδ1 and Asp 125 N is maintained in monomer F but broken in the remaining monomers. Residue 125 is the final residue visible in Zn-H46R prior to the disordered region of the electrostatic loop. His 71 is also disordered and not present in the Zn-H46R structure, thus the secondary bridge is broken. Interestingly, if His 71 maintained its wild-type position, it would be within ~2.0 Å of the zinc atom in Zn-H46R.
Each of the eight monomers in the apo-H46R structure exhibits some electron density for Asp 124, and in several subunits, this residue is able to make at least a tenuous hydrogen bond with the mutated residue Arg 46. The degree of disorder and the B-factor in Asp 124 for each monomer correlate with its ability to form hydrogen bonds with the backbone nitrogen atoms of Asp 125 and Leu 126.
Solvent exposed cysteine
The solvent-exposed residue Cys 111 lies in a similar position in both apo-H46R and Zn-H46R to that previously observed for wild-type SOD1. The thiol side chain of this residue from each subunit faces approximately toward the 2-fold symmetry axis at the dimer interface, such that the sulfur–sulfur separation is ~9 Å. The adjacent residues in the polypeptide, His 110 and Asp 109, face away from the interface and from Cys 111, and are therefore not in a suitable position to form a binding site for metals together with the Cys 111, as had previously been suggested by Liu et al. (2000).
Helical filament packing in Zn-H46R
The formation of helical filamentous arrays in the Zn-H46R structure and linear amyloid-like filamentous arrays in the crystal structure of apo-H46R has been described previously (Elam et al. 2003b). Each helical filament was also described as a water-filled nanotube due to the ~30 Å diameter hole in the center. Each nanotube packs side-to-side with four adjacent nanotubes (Fig. 4 ▶). The interfaces between Zn-H46R molecules in adjacent filaments consist of two salt-bridges between the Lys 36 residues of monomers W and X in one filament and the Asp 11 residues in monomers Y and Z of a second filament along with a number of more tenuous water-mediated interactions and close reciprocal hydrophobic interactions of Pro 13 (Fig. 4 ▶). This interaction is repeated for each of the four dimers forming one turn of the helical filament.
Figure 4.

(A) End-on view of a plane of helical filaments in the Zn-H46R structure. The crystallographic asymmetric unit consists of a pair of SOD1 dimers (boxed). (B) Surface representation showing the interface between monomers of Zn-H46R arising from adjacent helical filaments. The interacting residues Asp 11 and Lys 36 are shown in a ball-and stick representation. A further interaction involves reciprocal interactions of Pro 13.
Linear filament and zigzag packing interactions in apo-H46R
As previously described (Elam et al. 2003b), the disorder in the zinc and electrostatic loops of apo-H46R allows the protein to form linear amyloid-like filaments and zigzag packing interactions through the same edge strand deprotection of β-strands 5 and 6 as in Zn-H46R and another FALS-associated SOD1 mutant S134N. In the crystal, each linear filament interacts side-to-side with two others to form a plane of linear filaments (Fig. 5A ▶). Similarly, each zigzag packing arrangement of dimers interacts with two others to form a separate non-intersecting plane of zigzag arrays (Fig. 5B ▶). The two distinct planes stack in alternating layers, that lie approximately normal to the ac plane within the monoclinic crystal. A few somewhat tenuous interactions occur between these layers (Fig. 6 ▶).
Figure 5.

Sheets of filaments in the apo-H46R structure. (A) Linear filament sheet: planar view (i) and end-on view (ii). Filaments are arranged anti-parallel to each other with the axis of propagation determined by head-to-head interactions between the altered conformation electrostatic loop elements of the G/H (dark green) and I/J (purple) dimers. The G/H (bottom right) and I/J (top left) components of the asymmetric unit are colored dark pink. The sheet is connected by tenuous water-mediated side-to-side interaction between linear filaments between monomers G and H. (B) Zigzag packing interaction sheet: planar view (i) and end-on view (ii). These interactions are arranged anti-parallel to each other with the axis of propagation determined by head-to-head interactions between the altered conformation electrostatic loop elements of the K/L (blue) and M/N (light green) dimers. The K/L (right) and M/N (left) components of the asymmetric unit are in dark pink. The sheet packing is completed by side-to-side interactions between two of the zigzag filament subunits, chains L and N.
Figure 6.
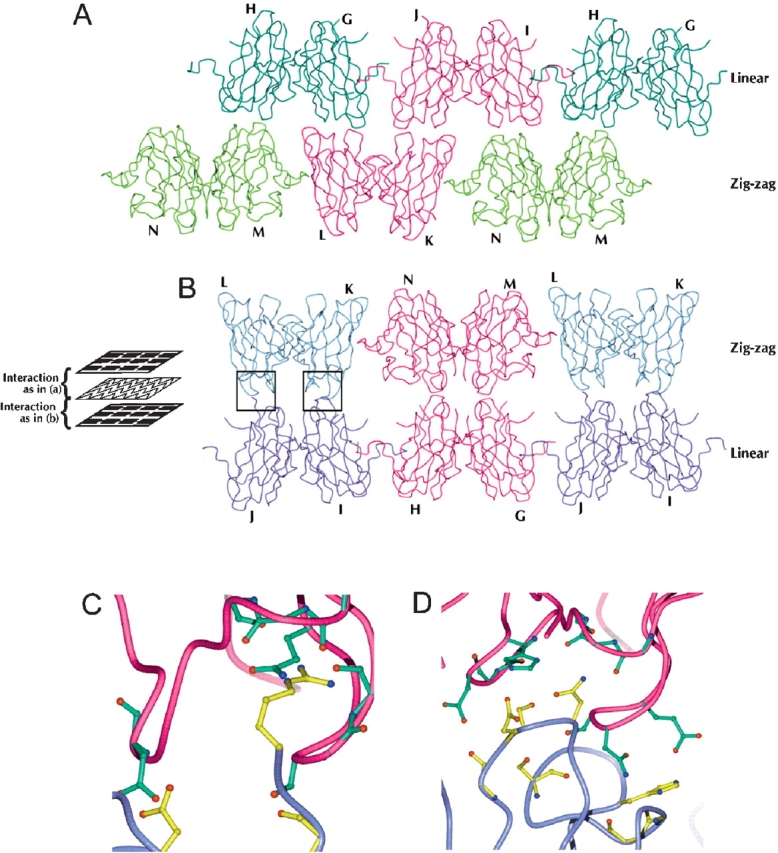
Interactions between apo-H46R filament planes. The asymmetric unit consists of dimers G/H, I/J, L/K, and M/N. The linear filament and zigzag interaction planes lie parallel to each other in space as layers. The apo-H46R molecules of each plane make interactions of two types between these layers perpendicular to the filament planes: between the bottom surface of the linear plane and the top surface of the zigzag plane (A) and between the bottom surface of the zigzag plane and the top surface of the linear plane (B) (see schematic). Dimers G/H (dark green) and I/J (purple) form the linear filament layer, and L/K (blue) and M/N (light green) dimers form the zigzag layer. The components of the asymmetric unit are in dark pink. There are three sites of interaction between the four molecules within the asymmetric unit and consequently between the two classes of filament: (1) a few hydrogen-bond, water-mediated, and van der Waals interactions between the region of chain M near Asp 109 and the area around the same residue in chain G; (2) a similar interaction involving residues around the Asp 109 residues in monomers N and H; and (3) reciprocal van der Waals interactions only between residues 12–13 and 37–39 of chains K and J. The most extensive of the interactions between the linear and zigzag sheets occurs between monomers L and J and K and I, which form a total of 30 side-chain–to–side-chain and side-chain–to–main-chain hydrogen bonds through their respective areas adjacent to and including part of the zinc loop (C,D). Another interaction involves reciprocal hydrogen bonds between the β-strand connecting loops (residues 13–15, 36–39, and 91–92) of monomers L and G. The third interaction occurs through the same area of chains I and N but is very tenuous, consisting of only one electrostatic interaction between residues Asp 92 and Lys 36 and a few van der Waals interactions. (C) Closeup of hydrogen bonding interactions between monomers L and J at the zigzag and linear plane interface (left boxed region in B). (D) Closeup of hydrogen bonding interactions between monomers K and I at the zigzag and linear plane interface (right boxed region in B). (C,D) The K/L dimer is shown in dark pink, and the I/J dimer is in purple. Residues involved in the interaction have either their side chain or backbone modeled in detail, green (K/L) or yellow (I/J), depending on which atoms of the residue are directly involved in hydrogen bonding.
Discussion
Altered metal binding to H46R SOD1
The SOD1 activity and copper loading of H46R in vivo has been the focus of much work since the mutation was first identified >10 yr ago. Activity measurements on erythrocyte lysate from H46R FALS patients showed SOD1 activity between 70% and 80% of control levels (Aoki et al. 1998). Coupled with these data, an electrospray ionization mass spectrometry study using erythrocyte lysate from H46R patients indicated that the ratio of mutant to normal SOD1 was ~0.15 (Nakanishi et al. 1998). Taken together, these results suggest that the H46R fraction was inactive and that the observed SOD1 activity in erythrocyte lysates from H46R patients in previous studies arose from wild-type SOD1 only. Ogawa and coworkers (Ogawa et al. 1997) observed abnormal copper peaks during purification of mutant H46R protein from erythrocytes of FALS patients. This was suggested to be the result of impaired copper binding by the H46R protein.
An activity assay and ESR spectroscopy study on H46R expressed in an Escherichia coli SOD1 knock-out system (Carri et al. 1994), showed no detectable SOD1 activity in cell extracts and <5% copper loading (measured by integration of the ESR spectrum). Wild-type SOD1 expressed similarly had ~80% activity and a corresponding level of copper. More recently, a combined metal binding, Raman spectroscopy and UV/visible spectroscopy study showed that Cu2+ does not bind to the copper site of apo H46R SOD1 and that Cu2+ competes with other metal ions such as Zn2+ and Co2+ for binding to the zinc site. Furthermore, Cu2+ was proposed to bind strongly to a surface residue, Cys 111, near the dimer interface of H46R SOD1, giving rise to a distinct optical absorption band at 370 nm (Liu et al. 2000).
Both the zinc-loaded and apo-crystal structures of H46R SOD1 presented here show that the copper site is completely vacant. This is consistent with measurements of copper content on the H46R protein expressed in insect cells (Hayward et al. 2002) that was used to grow the apo-H46R crystals examined in this study. The H46R SOD1 protein used to grow the Zn-H46R crystals examined here was expressed in yeast and showed a copper content of 0.23 equivalents per dimer prior to crystallization, yet the crystal structure showed no indication that copper was bound to the protein at either the copper or zinc site or near Cys 111. It is likely in the case of Zn-H46R that we are selectively crystallizing the copper-free form of the protein. The observation of a structurally different zinc site in Zn-H46R, in which only three of the usual four protein ligands participate, may indicate a higher susceptibility for metal loss at the zinc site compared with the wild-type enzyme. A decreased affinity for zinc has been determined for several FALS SOD1 mutants (Crow et al. 1997) and zinc-deficient SOD1 (either wild type or mutant) has been shown to induce apoptosis in liposomal delivery to cultured motor neurons (Estevez et al. 1999).
Several studies have shown some FALS SOD1 mutants do not properly segregate zinc and copper at their metal sites when reconstituted from their apo-forms in vitro (Lyons et al. 1996; Goto et al. 2000) and we have previously observed in crystal structures of wild-type and FALS mutant SOD1 that in the absence of sufficient copper, zinc ions will sometimes occupy the copper site (Elam et al. 2003a; S. Antonyuk, R. Strange, M. Hough, P. Doucette, J. Valentine, and S. Hasnain, in prep.). The fully occupied zinc sites in Zn-H46R suggest that ample zinc was present during protein production and crystallization, yet there was no evidence of any metal (even zinc) at the copper site in the crystal structure, consistent with the Arg side chain sterically impairing the copper site for metal binding.
The geometry of the zinc site is observed to be five coordinate, with a water ligand, His 80, His 63, and the bidentate carboxylate of Asp 83. It seems likely that Co2+ is binding in the same fashion as zinc to apo-H46R, since the visible spectrum of Co2+ H46R shows an extinction coefficient for the d-d bands near 600 nm that is somewhat lower than that of Co2+ bound to the zinc site of wt SOD1, where it is known to be four coordinate (Liu et al. 2000; Lyons et al. 2000).
Earlier studies of Cu2+ binding to apo-H46R have demonstrated the existence of a new metal-binding site on this protein, which was assigned as Cys 111 on the basis of its characteristic copper-cysteinate visible absorption and resonance Raman spectroscopic properties (Liu et al. 2000). The nature of the resonance Raman spectrum led to a tentative conclusion that both cysteines might be binding simultaneously to the copper chromophore. However, this conclusion now seems unlikely based on an examination of the crystal structure, unless a binuclear copper site is forming that bridges the two cysteines. The locations of the two Cys 111 residues are close enough, however, to explain why chemical modification using either dithiobis(2-nitro-benzoic acid) or 4-vinylpyridine gave only a 50% yield. Modification of one of the Cys 111 side chains with either reagent would be predicted to create a significant steric barrier to modification of the second Cys 111 of the protein dimer.
The secondary bridge and disordered loop regions
The disordered loop regions in the H46R crystal structures appear to be correlated with the disruption of the Asp 124 secondary bridge as a consequence of the mutation of His 46 and the subsequent disorder of the zinc ligand, His 71. In Zn-H46R, the electrostatic loop becomes disordered shortly after residue 125, adjacent to Asp 124. The loss of the bond from zinc to His 71 is related to the disorder of the zinc loop from residues 68–78 (or vice versa). In apo-H46R, the electrostatic and zinc loops are disordered in all dimers but one, in which crystal contacts appear to stabilize these loops. It is possible that the zigzag packing interaction is an intermediate to the linear, amyloid-like filament conformation, where the transition between the two could be dependent on the destabilization of the His 71-Asp 124 hydrogen bond in the secondary bridge. Overall, our results combined with other observations (Lyons et al. 2000; Banci et al. 2002; Strange et al. 2003) strengthen the idea that the hydrogen-bonding network around Asp 124 forms a crucial component in the stability and conformation of both zinc and electrostatic loops.
A significant observation is that the presence of zinc in its altered coordination environment at the zinc site in the Zn-H46R structure is not sufficient to maintain the wild type–like loop conformation. It is possible that zinc bound in this altered zinc site may be quite labile in solution, further disrupting the zinc loop as seen in apo-H46R. The observation that His 71 is ordered in apo-wtSOD1 but not in Zn-H46R is suggestive that the disruption of the secondary bridge and the zinc loop conformation is a consequence of the H46R mutation rather than of metal depletion. It is clear that the His → Arg mutation at residue 46 compromises the correct formation and function of the secondary bridge. This is particularly significant considering that pools of stable, zinc-loaded, but copper-deficient wild-type CuZnSOD have been observed in vivo (Steinkuhler et al. 1991, 1994; Petrovic et al. 1996; Schmidt et al. 2000), and presumably, FALS mutants would also be expected to exist in that form. The H46R mutant may have highly disordered electrostatic and zinc loops prior to its interaction with the SOD1 copper chaperone CCS, if it interacts at all.
H46R and FALS pathogenesis
H46R is of particular interest in the study of SOD1-mediated FALS, as the mutant protein has severely impaired copper binding at the active site while at the same time it causes a distinctive, very slowly progressing form of FALS. Several proposed mechanisms for the pathogenicity of FALS mutant proteins involve altered copper-mediated chemistry or loss of substrate specificity (for review, see Valentine et al. 1999; Valentine and Hart 2003). H46R-associated FALS is presumably not the result of these mechanisms due to its vacant copper site, but mechanisms in which oxidative stress is caused by mishandling or release of copper from mutant SOD1 have not been entirely excluded. One possibility is that SOD1 mutants without active site copper may correlate with a milder phenotype by producing toxicity through a mechanism distinct from those mutants capable of binding active site copper ion. An alternative hypothesis has been that the SOD1 mutants, particularly in their metal-deficient forms, lose the stabilizing effects of well-ordered electrostatic and zinc loops, and become susceptible either to fibrillar or nonspecific aggregation (Elam et al. 2003b). In fact, both apo- and Zn-H46R form filamentous arrays within their crystals, which suggests a mechanism by which the mutant protein becomes susceptible to aggregation. While zinc binding is still possible in the H46R mutant, the novel arrangement at the zinc-binding site as a result of the H46R mutation is not sufficient to stabilize the two loops, despite the presence of bound zinc. Therefore, if the loss of β-strand edge protection caused by loop disorder is a crucial mechanism by which SOD1 aggregates in affected tissues, then the H46R mutant protein might be expected to aggregate more readily than wild-type SOD1 or other FALS mutant SOD1 proteins that bind metals similarly to wild type. H46R SOD1 transgenic rats do in fact develop an abundance of hyaline inclusions in spinal motor neurons and supporting cells (Nagai et al. 2001). G93A transgenic rats produced from the same genetic background as the H46R rats in the same study had motor neurons with far fewer aggregates and a distinct vacuolar pathology. In spite of the increased aggregation and a 2.4-fold higher relative expression level of H46R compared to G93A SOD1, the onset and progression of motor neuron disease in H46R rats was substantially slower than for those with the G93A mutation (144 d vs. 122 d and 24 d vs. 8 d for onset and duration, respectively).
These data suggest that the distinctive H46R FALS phenotype may not result from genetic modifying factors shared by the limited number of Japanese families in which this mutation has been observed. The discrepancy that although H46R readily forms aggregates as observed from transgenic animal studies, the disease progresses much less rapidly than expected is consistent with the observation of several research groups who now believe that protofibrils rather than insoluble fibers or inclusions are toxic. The X-ray structures presented here strongly support this hypothesis because the rapid aggregation of H46R may actually reduce the amount of toxic protofibrils and hence be neuroprotective. Further support for this idea comes from recent studies showing that increasing SOD1 aggregate formation did not correlate with an increase in cell death in tissue culture (Lee et al. 2002; Takeuchi et al. 2002) or in FALS transgenic mice (Lino et al. 2002).
Conclusions
The H46R mutation in SOD1 results in an enzyme with significant structural differences relative to the wild-type enzyme. In both Zn-H46R and apo-H46R, the secondary bridge formed by His 46-Asp 124-His 71 is disrupted and the copper site is devoid of metal. Apo-H46R is also devoid of Zn, while Zn-H46R contains an altered zinc site relative to wild type due to the disordered nature of residue His71. The three remaining protein ligands form a novel five coordinate zinc environment with both Oδ1 and Oδ2 of Asp 83 coordinated to zinc. The coordination environment of zinc is completed by a weak water ligand. The electrostatic and zinc-loop regions of the enzyme are disordered in the H46R mutant, and notably, the presence of zinc in its altered site in Zn-H46R does not stabilize these loops. Zn-H46R dimers form so-called helical filaments in the crystals while apo-H46R forms linear, amyloid-like filaments and zigzag packing assemblies. The observation of these packing arrangements in the crystals is consistent with the increased tendency for this mutant to aggregate in motor neurons in the H46R SOD1 transgenic rat. SOD1-containing inclusions do not, however, appear to correlate with toxicity in FALS motor neurons since H46R rats exhibit much more aggregated protein than those of G93A rats, yet have a milder form of the disease. The observation that the disease progression of H46R rats correlates with the slowly progressive H46R phenotype in humans suggests that the distinctive FALS H46R phenotype may not be the result of protective genetic factors in the affected families, but rather due to the properties of the mutant SOD1 molecule itself.
Materials and methods
Experimental procedures
The expression, purification, and crystal structure determinations for H46R in its apo- (accession code 1OZT) and Zn-loaded (accession code 1OEZ) forms were described previously (Elam et al. 2003b). Briefly, recombinant human H46R SOD1 was produced in Saccharomyces cerevisiae (Zn-H46R) and baculovirus /Sf21 (apo-H46R) expression systems. Metal content was determined using ICP as described previously (Hayward et al. 2002). Prior to data collection, crystals of Zn-H46R were soaked for 5 min in mother liquor plus 25% ethylene glycol before transfer into a nitrogen cryostream for flash-cooling at 100 K. Crystals of apo-H46R were passed through a 50% (w/v) sorbitol-saturated mother liquor and flash frozen in liquid nitrogen. Single crystal X-ray diffraction data were collected as described (Elam et al. 2003b). The data were integrated and scaled by using HKL2000 (Zn-H46R) and the DENZO/SCALEPACK suite (apo-H46R) (Otwinowski 1993).
Structure solution and refinement
The Zn-H46R structure was determined by molecular replacement using MOLREP (Vagin and Teplyakov 1997) with the coordinates of a monomer of the 1.78 Å holo wild-type hSOD structure (Protein Data Bank [PDB] code 1 HL5; Strange et al. 2003) as the search model. Two SOD dimers were located in the crystallographic asymmetric unit. This initial model was refined by using the maximum likelihood method implemented in REFMAC5 (Murshudov et al. 1997) as part of the CCP4i program suite and rebuilt interactively by using σA-weighted electron density maps with coefficients 2mFo-DFc and mFo-DFc in the program O (Jones et al. 1991). Five percent of the data (1774 reflections) was set aside to calculate a free R-factor. Each monomer was refined independently, without the application of noncrystallographic symmetry restraints. A bulk solvent correction was applied and individual solvent molecules added using ARP/WARP. In the latter stages of refinement, TLS parameters were determined. Refinement converged to a final R-factor of 20.6% (R-free = 23.4%).
The apo-H46R structure was also determined by molecular replacement using the program EPMR (Kissinger et al. 1999) with the 1.9 Å resolution structure of FALS SOD1 mutant G37R (PDB code 1AZV; Hart et al. 1998) as the search model. Four SOD dimers were located in the crystallographic asymmetric unit. The structure was iteratively refined by using the program CNS (Brunger et al. 1998). When necessary, the molecular model was adjusted in the program O. NCS restraints were applied to the residues forming the β-barrel core only. A bulk solvent correction was applied, and water molecules were added. Refinement converged to a final R-factor of 22.1% (R-free = 26.8%). No stereo-chemical restraints on metal-ligand bond distances or angles were applied in the refinement of either structure. The final models were evaluated by using the programs WHATCHECK and PROCHECK (Laskowski et al. 1993). The final coordinates and structure factors have been deposited in the RSCB PDB (Bernstein et al. 1977), accession codes 1OEZ (Zn-H46R) and 1OZT (apo-H46R).
Acknowledgments
This work was supported by a Motor Neuron Disease Association grant (S.S.H.); NIH grants NS39112 (P.J.H.), NS44170 (L.J.H.), and GM28222 (J.S.V.); a Robert A. Welch Foundation grant AQ-1399 (P.J.H.); and grants from the ALS Association (L.J.H., J.S.V., and P.J.H.). J.S.E. was supported for this work by a grant from the American Federation on Aging Research (AFAR). We thank our colleagues in the International Consortium on SOD and ALS (ICOSA) for valuable discussions.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041256705.
Supplemental material: see www.proteinscience.org
References
- Aoki, M., Ogasawara, M., Matsubara, Y., Narisawa, K., Nakamura, S., Itoyama, Y., and Abe, K. 1994. Familial amyotrophic-lateral-sclerosis (ALS) in Japan associated with H46R mutation in Cu/Zn superoxide-dismutase gene: A possible new subtype of familial ALS. J. Neurol. Sci. 126 77–83. [DOI] [PubMed] [Google Scholar]
- Aoki, M., Abe, K., and Itoyama, Y. 1998. Molecular analyses of the Cu/Zn superoxide dismutase gene in patients with familial amyotrophic lateral sclerosis (ALS) in Japan. Cell. Mol. Neurobiol. 18 639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arisato, T.O.R., Arata, H., Abe, K., Fukada, K., Sakoda, S., Shimizu, A., Qin, X.H., Izumo, S., Osame, M., and Nakagawa, M. 2003. Clinical and pathological studies of familial amyotrophic lateral sclerosis (FALS) with SOD1 H46R mutation in large Japanese families. Acta Neuropathol. 106 561–568. [DOI] [PubMed] [Google Scholar]
- Banci, L., Felli, I.C., and Kummerle, R. 2002. Direct detection of hydrogen bonds in monomeric superoxide dismutase: Biological implications. Biochemistry 41 2913–2920. [DOI] [PubMed] [Google Scholar]
- Bernstein, F.C., Koetzle, T.F., Williams, G.J.B., Meyer Jr., F.M., Brice, M.D., Rodgers, J.R., Kennard, O., Shimanouchi, T., and Tasumi, M. 1977. The Protein Data Bank: A computer based archival file for macromolecular structures. J. Neurol. Biol. 112 535–542. [DOI] [PubMed] [Google Scholar]
- Borchelt, D.R., Guarnieri, M., Wong, P.C., Lee, M.K., Slunt, H.S., Xu, Z.S., Sisodia, S.S., Price, D.L., and Cleveland, D.W. 1995. Superoxide-dismu-tase-1 subunits with mutations linked to familial amyotrophic-lateral-sclerosis do not affect wild-type subunit function. J. Biol. Chem. 270 3234–3238. [DOI] [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography and NMR system: A new software suite for macro-molecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54 905–921. [DOI] [PubMed] [Google Scholar]
- Cardoso, R.M., Thayer, M.M., DiDonato, M., Lo, T.P., Bruns, C.K., Getzoff, E.D., and Tainer, J.A. 2002. Insights into Lou Gehrig’s disease from the structure and instability of the A4V mutant of human Cu,Zn superoxide dismutase. J. Mol. Biol. 324 247–256. [DOI] [PubMed] [Google Scholar]
- Carri, M.T., Battistoni, A., Polizio, F., Desideri, A., and Rotilio, G. 1994. Impaired copper-binding by the H46R mutant of human Cu,Zn superoxide-dismutase involved in amyotrophic-lateral-sclerosis. FEBS Lett. 356 314–316. [DOI] [PubMed] [Google Scholar]
- Ceroni, M., Curti, D., and Alimonti, D. 2001. Amyotrophic lateral sclerosis and SOD1 gene: An overview. Funct. Neurol. 16 171–180. [PubMed] [Google Scholar]
- Cleveland, D.W. and Rothstein, J.D. 2001. From Charcot to Lou Gehrig: Deciphering selective motor neuron death in ALS. Nat. Rev. Neurosci. 2 806–819. [DOI] [PubMed] [Google Scholar]
- Crow, J.P., Sampson, J.P., Zhuang, Y.X., Thompson, J.A., and Beckman, J.S. 1997. Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite. J. Neurochem. 69 1936–1944. [DOI] [PubMed] [Google Scholar]
- Deng, H.X., Hentati, A., Tainer, J.A., Iqbal, Z., Cayabyab, A., Hung, W.Y., Getzoff, E.D., Hu, P., Herzfeldt, B., Roos, R.P., et al. 1993. Amyotrophic-lateral-sclerosis and structural defects in Cu,Zn superoxide-dismutase. Science 261 1047–1051. [DOI] [PubMed] [Google Scholar]
- Elam, J.S., Malek, K., Rodriguez, J.A., Doucette, P.A., Taylor, A.B., Hayward, L.J., Cabelli, D.E., Valentine, J.S., and Hart, P.J. 2003a. An alternative mechanism of bicarbonate-mediated peroxidation by copper-zinc superoxide dismutase: Rates enhanced via proposed enzyme-associated peroxycarbonate intermediate. J. Biol. Chem. 278 21032–21039. [DOI] [PubMed] [Google Scholar]
- Elam, J.S., Taylor, A.B., Strange, R.W., Antonyuk, S.V., Doucette, P.A., Rodriguez, J.A., Hasnain, S.S., Hayward, L.J., Valentine, J.S., and Hart, P.J. 2003b. Amyloid-like filaments and water-filled nanotubes formed by SOD1 mutant proteins linked to familial amyotrophic lateral sclerosis. Nat. Struct. Biol. 10 461–467. [DOI] [PubMed] [Google Scholar]
- Estevez, A.G.C.J., Sampson, J.B., Reiter, C., Zhuang, Y.X., Richardson, G.J., Tarpey, M.M., Barbeito, L., and Beckman, J.S. 1999. Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient superoxide dismutase. Science 286 2498–2500. [DOI] [PubMed] [Google Scholar]
- Fridovich, I. 1975. Superoxide dismutase. Annu. Rev. Biochem. 44 147–159. [DOI] [PubMed] [Google Scholar]
- Gabbianelli, R., Ferri, A., Rotilio, G., and Carri, M.T. 1999. Aberrant copper chemistry as a major mediator of oxidative stress in a human cellular model of amyotrophic lateral sclerosis. J. Neurochem. 73 1175–1180. [DOI] [PubMed] [Google Scholar]
- Goto, J.J., Zhu, H.N., Sanchez, R.J., Nersissian, A., Gralla, E.B., Valentine, J.S., and Cabelli, D.E. 2000. Loss of in vitro metal ion binding specificity in mutant copper-zinc superoxide dismutases associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 275 1007–1014. [DOI] [PubMed] [Google Scholar]
- Gurney, M. 1997. Transgenic animal models of familial amyotrophic lateral sclerosis. J. Neurol. 244 S15–S20. [DOI] [PubMed] [Google Scholar]
- Hart, P.J., Liu, H., Pellegrini, M., Nersissian, A.M., Gralla, E.B., Valentine, J.S., and Eisenberg, D. 1998. Subunit asymmetry in the three dimensional structure of a human CuZnSOD mutant found in amyotrophic lateral sclerosis. Protein Sci. 7 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haverkamp, L.J., Appel, V., and Appel, S.H. 1995. Natural-history of amyotrophic-lateral-sclerosis in a database population: Validation of a scoring system and a model for survival prediction. Brain 118 707–719. [DOI] [PubMed] [Google Scholar]
- Hayward, L.J., Rodriguez, J.A., Kim, J.W., Tiwari, A., Goto, J.J., Cabelli, D.E., Valentine, J.S., and Brown, R.H. 2002. Decreased metallation and activity in subsets of mutant superoxide dismutases associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 277 15923–15931. [DOI] [PubMed] [Google Scholar]
- Hough, M.A., Grossmann, J.G., Antonyuk, S.V., Strange, R.W., Doucette, P.A., Rodriguez, J.A., Whitson, L.J., Hart, P.J., Hayward, L.J., Valentine, J.S., et al. 2004. Dimer destabilization in superoxide dismutase may result in disease-causing properties: Structures of motor neuron disease mutants. Proc. Natl. Acad. Sci. 101 5976–5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Kissinger, C.R., Gehlhaar, D.K., and Fogel, D.B. 1999. Rapid automated molecular replacement by evolutionary search. Acta Crystallogr. D Biol Crystallogr. 55 484–491. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. Procheck: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 25 283–291. [Google Scholar]
- Lee, J.P., Gerin, C., Bindokas, V.P., Miller, R., Ghadge, G., and Roos, R.P. 2002. No correlation between aggregates of Cu/Zn superoxide dismutase and cell death in familial amyotrophic lateral sclerosis. J. Neurochem. 82 1229–1238. [DOI] [PubMed] [Google Scholar]
- Lino, M.M., Schneider, C., and Caroni, P. 2002. Accumulation of SOD1 mutants in postnatal motoneurons does not cause motoneuron pathology or motoneuron disease. J. Neurosci. 22 4825–4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, H.B., Zhu, H.N., Eggers, D.K., Nersissian, A.M., Faull, K.F., Goto, J.J., Ai, J.Y., Sanders-Loehr, J., Gralla, E.B., and Valentine, J.S. 2000. Copper2+ binding to the surface residue cysteine 111 of His46Arg human copper-zinc superoxide dismutase, a familial amyotrophic lateral sclerosis mutant. Biochemistry 39 8125–8132. [DOI] [PubMed] [Google Scholar]
- Lyons, T.J., Liu, H.B., Goto, J.J., Nersissian, A., Roe, J.A., Graden, J.A., Cafe, C., Ellerby, L.M., Bredesen, D.E., Gralla, E.B., et al. 1996. Mutations in copper-zinc superoxide dismutase that cause amyotrophic lateral sclerosis alter the zinc binding site and the redox behavior of the protein. Proc. Natl. Acad. Sci. 93 12240–12244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons, T.J., Nersissian, A., Huang, H.J., Yeom, H., Nishida, C.R., Graden, J.A., Gralla, E.B., and Valentine, J.S. 2000. The metal binding properties of the zinc site of yeast copper-zinc superoxide dismutase: Implications for amyotrophic lateral sclerosis. J. Biol. Inorg. Chem. 5 189–203. [DOI] [PubMed] [Google Scholar]
- Murshudov, G.N., Vagin, A.A., and Dodson, E.J. 1997. Refinement of macro-molecular structures by the maximum likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53 240–255. [DOI] [PubMed] [Google Scholar]
- Nagai, M., Aoki, M., Miyoshi, I., Kato, M., Pasinelli, P., Kasai, N., Brown, R.H., and Itoyama, Y. 2001. Rats expressing human cytosolic copper-zinc superoxide dismutase transgenes with amyotrophic lateral sclerosis: Associated mutations develop motor neuron disease. J. Neurosci. 21 9246–9254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi, T., Kishikawa, M., Miyazaki, A., Shimizu, A., Ogawa, Y., Sakoda, S., Ohi, T., and Shoji, H. 1998. Simple and defined method to detect the SOD-1 mutants from patients with familial amyotrophic lateral sclerosis by mass spectrometry. J. Neurosci. Meth. 81 41–44. [DOI] [PubMed] [Google Scholar]
- Ogawa, Y., Kosaka, H., Nakanishi, T., Shimizu, A., Ohoi, N., Shouji, H., Yanagihara, T., and Sakoda, S. 1997. Stability of mutant superoxide dismutase-1 associated with familiar amyotrophic lateral sclerosis determines the manner of copper release and induction of thioredoxin in erythrocytes. Biochem. Biophys. Res. Comm. 241 251–257. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. 1993. Proceedings of the CCP4 Study Weekend Data Collection and Processing, 29–30 January 1993, Daresbury Laboratory, UK.
- Petrovic, N., Comi, A., and Ettinger, M.J. 1996. Identification of an apo-superoxide dismutase (Cu,Zn) pool in human lymphoblasts. J. Biol. Chem. 271 28331–28334. [DOI] [PubMed] [Google Scholar]
- Rabizadeh, S., Gralla, E.B., Borcheldt, D.R., Gwinn, R., Valentine, J.S., Sisodia, S., Wong, P., Lee, M., Hahn, H., and Breseden, D.E. 1995. Mutations associated with amyotrophic lateral sclerosis convert superoxide dismutase from an antiapoptotic gene to a proapoptotic gene: Studies in yeast and neural cells. Proc. Natl. Acad. Sci. 92 3024–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratovitski, T., Corson, L.B., Strain, J., Wong, P., Cleveland, D.W., Culotta, V.C., and Borchelt, D.R. 1999. Variation in the biochemical/biophysical properties of mutant superoxide dismutase 1 enzymes and the rate of disease progression in familial amyotrophic lateral sclerosis kindreds. Hum. Mol. Genet. 8 1451–1460. [DOI] [PubMed] [Google Scholar]
- Reaume, A.G., Elliott, J.L., Hoffman, E.K., Kowall, N.W., Ferrante, R.J., Siwek, D.F., Wilcox, H.M., Flood, D.G., Beal, M.F., Brown, R.H., et al. 1996. Motor neurons in Cu/Zn superoxide dismutase–deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat. Genet. 13 43–47. [DOI] [PubMed] [Google Scholar]
- Rodriguez, J.A., Valentine, O.S., Eggers, D.K., Roe, J.A., Tiwari, A., Brown, R.H., and Hayward, L.J. 2002. Familial amyotrophic lateral sclerosis–associated mutations decrease the thermal stability of distinctly metallated species of human copper/zinc superoxide dismutase. J. Biol. Chem. 277 15932–15937. [DOI] [PubMed] [Google Scholar]
- Rosen, D.R., Siddique, T., Patterson, D., Figlewicz, D.A., Sapp, P., Hentati, A., Donaldson, D., Goto, J., Oregan, J.P., Deng, H.X., et al. 1993. Mutations in Cu/Zn superoxide-dismutase gene are associated with familial amyotrophic-lateral-sclerosis. Nature 362 59–62. [DOI] [PubMed] [Google Scholar]
- Schmidt, P.J., Kunst, C., and Culotta, V.C. 2000. Copper activation of superoxide dismutase 1 (SOD1) in vivo: Role for protein–protein interactions with the copper chaperone for SOD1. J. Biol. Chem. 275 33771–33776. [DOI] [PubMed] [Google Scholar]
- Steinkuhler, C., Sapora, O., Carri, M.T., Nagel, W., Marcocci, L., Ciriolo, M.R., Weser, U., and Rotilio, G. 1991. Increase of Cu,Zn-superoxide dismutase activity during differentiation of human K562 cells involves activation by copper of a constantly expressed copper-deficient protein. J. Biol. Chem. 266 24580–24587. [PubMed] [Google Scholar]
- Steinkuhler, C., Carri, M.T., Micheli, G., Knoepfel, L., Weser, U., and Rotilio, G. 1994. Copper-dependent metabolism of Cu,Zn-superoxide dismutase in human K562 cells: Lack of specific transcriptional activation and accumulation of a partially inactivated enzyme. Biochem. J. 302 687–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strange, R.W., Antonyuk, S., Hough, M.A., Doucette, P.A., Rodriguez, J.A., Hart, P.J., Hayward, L.J., Valentine, J.S., and Hasnain, S.S. 2003. The structure of holo and metal-deficient wild-type human Cu, Zn superoxide dismutase and its relevance to familial amyotrophic lateral sclerosis. J. Mol. Biol. 328 877–891. [DOI] [PubMed] [Google Scholar]
- Tainer, J.A., Getzoff, E.D., Beem, K.M., Richardson, J.S., and Richardson, D.C. 1982. Determination and analysis of the 2 Å structure of copper, zinc superoxide dismutase. J. Mol. Biol. 160 181–217. [DOI] [PubMed] [Google Scholar]
- Takeuchi, H., Kobayashi, Y., Ishigaki, S., Doyu, M., and Sobue, G. 2002. Mitochondrial localization of mutant superoxide dismutase 1 triggers caspase-dependent cell death in a cellular model of familial amyotrophic lateral sclerosis. J. Biol. Chem. 277 50966–50972. [DOI] [PubMed] [Google Scholar]
- Vagin, A. and Teplyakov, A. 1997. MOLREP: An automated program for molecular replacement. J. Appl. Cryst. 30 1022–1025. [Google Scholar]
- Valentine, J.S. and Hart, P.J. 2003. Misfolded CuZnSOD and amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. 100 3617–3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentine, J.S., Gralla, E.B., Goto, J.J., Liu, H.B., and Zhu, H.I. 1999. Copper-zinc superoxide dismutase and ALS. J. Inorg. Biochem. 74 55. [DOI] [PubMed] [Google Scholar]