

Abstract
Modeling a protein structure based on a homologous structure is a standard method in structural biology today. In this process an alignment of a target protein sequence onto the structure of a template(s) is used as input to a program that constructs a 3D model. It has been shown that the most important factor in this process is the correctness of the alignment and the choice of the best template structure(s), while it is generally believed that there are no major differences between the best modeling programs. Therefore, a large number of studies to benchmark the alignment qualities and the selection process have been performed. However, to our knowledge no large-scale benchmark has been performed to evaluate the programs used to transform the alignment to a 3D model. In this study, a benchmark of six different homology modeling programs— Modeller, SegMod/ENCAD, SWISS-MODEL, 3D-JIGSAW, nest, and Builder—is presented. The performance of these programs is evaluated using physiochemical correctness and structural similarity to the correct structure. From our analysis it can be concluded that no single modeling program outperform the others in all tests. However, it is quite clear that three modeling programs, Modeller, nest, and SegMod/ ENCAD, perform better than the others. Interestingly, the fastest and oldest modeling program, SegMod/ ENCAD, performs very well, although it was written more than 10 years ago and has not undergone any development since. It can also be observed that none of the homology modeling programs builds side chains as well as a specialized program (SCWRL), and therefore there should be room for improvement.
Keywords: homology modeling, structure quality, alignment quality
Knowledge of the three-dimensional structure of a protein can often provide invaluable information. The structure can provide hints about functional and evolutionary features of the protein, and in addition structural information are useful in drug design efforts. The structure of a protein can, in theory, be obtained by three methods, either by the use of experimental information, normally from X-ray crystallography or NMR spectroscopy, by purely theoretical methods, or by the use of homology modeling. In spite of great progress within the structural genomics efforts, it is still unreasonable to believe that the structure of more than a tiny fraction of all the billions of proteins in the world will be studied by experimental methods in the foreseeable future. Purely theoretical methods do not yet seem to be able to provide high-resolution information for the majority of proteins. Hence, for the vast majority of proteins the only way to get structural information is through the use of homology modeling methods.
Homology modeling methods use the fact that evolutionary related proteins share a similar structure. Therefore, models of a protein with unknown structure (target) can be built based on an alignment of a protein of known structure (template). This typically involves four steps (Sánchez and Sali 1997; Marti-Renom et al. 2000): (1) identification of homologs that can by used as template(s) for modeling; (2) alignment of the target sequence to the template(s); (3) building a model for the target based on the information from the alignment(s); and (4) evaluation of the model. Finally, all four steps can be repeated until a satisfactory model is obtained.
History of molecular modeling
The first approaches to modeling by homology were done by Browne et al. (1969) using wire and plastic models of bonds and atoms. A model of α-lactalbumin was constructed by taking the coordinates of a hen’s egg-white lysozyme and modifying, by hand, those amino acids that did not match the structure. The sequence identity between these two proteins was 39%. Since then, many different homology modeling packages have been developed (Marti-Renom et al. 2000). In principle, they can be grouped into three different groups: rigid-body assembly, segment matching, or modeling by satisfaction of spatial restraints.
The first modeling programs were based on rigid-body assembly methods, where a model is assembled from a small number of rigid bodies obtained from the core of the aligned regions (Blundell et al. 1987; Greer 1990). The assembly involves fitting the rigid bodies onto the framework and rebuilding the nonconserved parts, i.e., loops and side chains. Here, we test four programs using a rigid-body assembly method: SWISS-MODEL (Schwede et al. 2004), nest (Petrey et al. 2003), 3D-JIGSAW (Bates et al. 2001), and Builder (Koehl and Delarue 1994, 1995). The main difference between the rigid-body assembly programs lies in how side chains and loops are built. Nest (Petrey et al. 2003) uses a stepwise approach, changing one evolutionary event from the template at a time, while 3D-JIGSAW and Builder use mean-field minimization methods (Koehl and Delarue 1996).
The segment-matching approach uses a subset of atomic positions, derived from the alignment as a guide to find matching segments in a representative database of all known protein structures (Jones and Thirup 1986; Claessens et al. 1989; Levitt 1992). The database contains short segments of protein structure that are selected using energy or geometry rules, or a combination of these criteria. Here, we have studied one of the first segment based methods SegMod/ENCAD (Levitt 1992).
The methods using “modeling by satisfaction of spatial restraints” use a set of restraints derived from the alignment, and the model is then obtained by minimizing the violations to these restraints. One of the most frequently used modeling programs, Modeller (Sali and Blundell 1993), uses this approach.
Predictors participating in CASP (Moult et al. 2003) have used different programs to build 3D coordinates from the alignment. During the first CASPs a wide variety of programs were used. However, in the last two CASPs the clearly most popular package has been Modeller (Sali and Blundell 1993). In addition SWISS-MODEL (Schwede et al. 2004) has been used by some groups and several groups have used their own programs, such as nest (Petrey et al. 2003) and 3D-JIGSAW (Bates et al. 2001) or commercial packages such as ICM (Cardozo et al. 1995), Insight (Accelrys, http://www.accelrys.com/insight/), or Quanta (Accelrys, http://www.accelrys.com/quanta/). One advantage of Modeller and SWISS-MODEL are that they both are quite fast and that they are free for academic use. It has been reported that SWISS-MODEL is better for the core and Modeller for the rest (Kosinski et al. 2003), but it is believed that the accuracy for the different modeling approaches are similar when used optimally (Marti-Renom et al. 2000).
Homology modeling benchmark
Despite the importance of homology modeling very few large-scale assessments of homology modeling approaches have been performed. This is in sharp contrast to the fold recognition field where dozens of different benchmarking strategies have been reported (Godzik et al. 1992; Jones et al. 1992; Fischer and Eisenberg 1996 Fischer and Eisenberg 1999; Abagyan and Batalov 1997; Brenner et al. 1998; Park et al. 1998; Jaroszewski et al. 2002; Wallner et al. 2002; Fischer and Rychlewski 2003; Moult et al. 2003; Rychlewski and Fischer 2005). The reason for this is probably that it is generally believed that the most important part of homology modeling is the alignment and the ability to detect the structural similarities based on the amino acid sequence (Chothia and Lesk 1986) and not the homology modeling procedure itself (Tramontano et al. 2001; Tramontano and Morea 2003). For closely related protein sequences with identities over 40%, the alignment is most often close to optimal. As the sequence similarity decreases, the alignment becomes more difficult and will contain an increasingly large number of gaps and alignment errors (Rost 1999; Marti-Renom et al. 2000; Elofsson 2002).
One example of what kind of models an alignment error can give rise to is illustrated in Figure 1 ▶. The modeled protein is the N-terminal domain of ribosomal protein L2 from Bacillus stearothermophilus (d1rl2a2) belonging to the Cold shock DNA-binding domain-like SCOP family (b.40.4.5) and is modeled onto a template (d1jj2a2) of the same protein but from another organism (Archaeon Haloarcula marismortui). The sequence identity is 42% and the alignment contains an incorrect gap of 25 residues at the N-terminal part of the protein, while the alignment otherwise is gap-less. The gap puts two adjacent residues 40 Å apart in space, and the final model will depend on how the modeling programs balance the restraints from the alignment with chemical restraints. Models built by Modeller are almost unaffected since the gap just adds a few additional spatial restraints to the final optimization procedure. However, the programs that use rigid-body assembly force the N-terminal part to be separated from the structure (see Fig. 1B ▶). This shows that at least when using nonoptimal alignments it may matter which modeling program is used to build the final model.
Figure 1.

Example of models produced with an alignment containing an error. (A) SWISS-MODEL model, (B) Backbone model, (C) Modeller model, (D) Native structure. The N-terminal helix in the backbone model is clearly wrong, since the distance between two adjacent residues is 40 Å. For the Modeller model this has no great impact (it is just one of many restraints); however, for the SWISS-MODEL model the error is enough to break the sheet in order to include the helix in the model. Figures were made using MOLSCRIPT (Kraulis 1991).
One additional conclusion from the comparative modeling category in CASP4 (Tramontano et al. 2001) and CASP5 (Tramontano and Morea 2003) were that the final models rarely are closer to the native structure than is the template structure. Indicating that model building in general does not refine the models. However, since predictors at CASP use different alignments and different modeling programs it is difficult to evaluate performance of individual modeling programs.
Here, we have tested alignments between protein domains from the same family using six homology modeling programs: Modeller (Sali and Blundell 1993), SegMod/ ENCAD (Levitt 1992), SWISS-MODEL (Schwede et al. 2004), 3D-JIGSAW (Bates et al. 2001), nest (Petrey et al. 2003) within the JACKAL modeling package (http:// trantor.bioc.columbia.edu/programs/jackal/index.html), and Builder (Koehl and Delarue 1994, 1995). As a further reference SCWRL3 (Canutescu et al. 2003) was used to build side chains on models with backbone coordinated copied from the templates.
In this study we try to answer two questions: Does any significant difference between the performance of different homology modeling programs exist or is the quality of the final model only dependent on the alignment? If there are differences is there a way to select the best model and would that procedure provide better models? Evaluation of the models was performed using both physiochemical criterion and structural similarity to the correct structure. In addition, the ability to converge and produce a model was evaluated.
Results
In the first part of this study we have compared the different homology modeling programs described in Table 1 by using 1037 alignments of protein pairs from the same SCOP family, with sequence identities ranging from 30% to 100%. For each alignment and program, a model is generated and evaluated. We have tried to evaluate several aspects of the programs, including (1) the reliability, i.e., the ability to produce coordinates for all residues in the alignment; (2) the speed by which the programs produce models; (3) the similarity to the correct structure; and (4) the physiochemical correctness of the models. Although most of the comparisons are straightforward, there are problems caused by the fact that all modeling programs do not create coordinates for all residues in all models; some programs crash for some targets, while other modeling programs do not model some residues, mainly loops. These differences in the models cause problems when the quality of the models from the different modeling programs is compared to each other. If a modeling program excludes all “difficult” residues the per residue quality will be higher than for a modeling program that includes these residues. Therefore, in the comparisons below we have only included the subset of residues that are produced by all modeling programs. However, for measures of the overall quality of a model (root-mean-square deviation [RMSD], MaxSub, acceptable models) we have included all residues that are produced by a modeling program. The RMSD measure actually favors shorter models, but this should to some degree be compensated by the MaxSub measure that favors longer models.
Table 1.
Description of the homology modeling programs used in this study
| Modeling program | Description |
| Modeller6v2 | Modeling by satisfying spatial restraints |
| Modeller6v2–10 | For each query 10 models created by Modeller6v2 and the one with the closest RMSD to the target structure is chosen |
| Modeller7v7 | Updated version of Modeller6v2 |
| SegMod/ENCAD | Segment matching followed by molecular dynamics refinement |
| SWISS-MODEL | Web server using rigid-body assembly with loop modeling |
| 3D-JIGSAW | Web server using rigid-body assembly with loop modeling using a mean-field minimization methods |
| nest | Rigid-body assembly with loop modeling using an artificial evolution method |
| Builder | Self-Consistent Mean Field Approach |
| SCWRL3 | State-of-the-art prediction of protein side-chain conformations; the backbone is copied from the alignment |
| SCWRL-CONS | State-of-the-art prediction of protein side-chain conformations, the backbone is copied from the alignment. Side-chain conformations of conserved residues are not changed. |
Reliability
The reliability of a homology modeling program is the ability to produce coordinates, that look like a protein, for all residues in the alignment. There are two types of reliability problems: missing coordinates and problems with convergence. The first type is easy to assess, since it is known what residues the model should contain. The second type is slightly more difficult, but since problems with convergence frequently are manifested by large extended fragments. Consequently, to find the models with extended parts, all models were compared to its simple backbone model created by copying the aligned coordinates from the template. Large changes in RMSD (>3 Å) between the model and the simple backbone model were taken as an indicator of a model that had failed to converge. In theory, it is possible that some of these large deviations could have moved the models closer to the native structure. However, none of these large changes from template backbone made the models better; in fact, none of the models with an RMSD larger than 3 Å to the simple backbone model was closer than 3 Å to the native structure.
The most severe case of missing residues is caused by programs that fail to produce a model at all. However, all modeling programs, except SWISS-MODEL, produced a model for more than 99% of the alignments. SWISS-MODEL failed to produce a model for 10% of the alignments (see Table 2). The reported reasons for the failures were either too long loops or problems in finding the right loop in the loop library. This means that more difficult alignments with more loops will probably crash more often. Indeed, the alignments with sequence identity below 50% crash four times more frequently than alignments with more than 70% sequence identity.
Table 2.
Overview of the different modeling programs used
| Modeling program | No. of alignments | No. of models | No. of crashed | No. of RMSD >3 Å from backbone | Average time (batch of 〈10〉) |
| Modeller6v2 | 1037 | 1037 | 0 (0.0%) | 51 (4.9%) | 〈43s〉 |
| Modeller7v7 | 1037 | 1035 | 1 (0.1%) | 55 (5.3%) | 〈90s〉 |
| Modeller6v2–10 | 1037 | 1037 | 0 (0.0%) | 20 (1.9%) | 〈430s〉 |
| SegMod/ENCAD | 1037 | 1036 | 1 (0.1%) | 21 (2.0%) | 〈6s〉 |
| SWISS-MODEL | 1037 | 932 | 105 (10.1%) | 48 (4.6%) | 〈165s〉 (〈22s〉) |
| 3D-JIGSAW | 1037 | 1032 | 5 (0.5%) | 13 (1.3%) | 〈1322s〉 (〈482s〉) |
| nest | 1037 | 1029 | 8 (0.8%) | 19 (1.8%) | 〈17s〉 |
| Builder | 1037 | 1030 | 7 (0.7%) | 46 (4.4%) | 〈19s〉 |
| SCWRL3 | 1037 | 1036 | 1 (0.1%) | 0 (0.0%) | 〈2s〉 |
| SCWRL-CONS | 1037 | 1037 | 0 (0.0%) | 0 (0.0%) | 〈2s〉 |
Number of alignments, number of models produces, number of crashes, number of models more than 3 Å RMSD from the backbone model, and average time to make a model; for the Web servers, the average time for a batch of 10 models is also included.
In addition, three of the programs—SWISS-MODEL, 3D-JIGSAW, and Builder—sometimes create models with missing coordinates (see Fig. 2 ▶). SWISS-MODEL lost residues in only 71 models (7.6%), while Builder and 3D-JIGSAW had missing residues in more than two-thirds of the models. In contrast, less than half of the models contain gaps, i.e., 3D-JIGSAW and Builder did not produce coordinates for all residues that are aligned to the template. For 3D-JIGSAW, this is due to a bug in the code, and will be updated in the next version (P. Fitzjohn, pers. comm.). Fortunately, most frequently the residues missing are few, since only 5% of the models contain more than 20 missing residues. SWISS-MODEL and Builder only miss residues at the N or C terminus, while 3D-JIGSAW also deleted residues in the middle of the target sequence.
Figure 2.

Histogram over the number of models that contain missing residues, i.e., where the program for some reason does not model all residues in the target sequence. SCWRL3 does not attempt to model loops; therefore, this number represents the alignments containing gaps.
Three programs—Modeller, SWISS-MODEL, and Builder—produce more models that do not converge compared to the other programs (see Table 2). There is a correlation between models that crash using some programs with models that do not converge using another program. The models that fail to converge can, in most cases, be detected and then sometimes corrected by rerunning the same program using alternative parameters. As seen by Modeller6v2–10, more than half of the alignments with convergence problems could be overcome by rerunning the same program 10 times using different random seeds.
How fast can the prosgrams build models?
Another important factor when modeling a protein sequence is speed. A representative set of 50 alignments was selected from the initial 1037 alignments and the time it took to produce the models using a standard PC (1.4 GHz AMD XP processor) was monitored. SegMod/ENCAD was the fastest modeling program, producing a model in 6 sec; in fact, all locally run programs were quite fast, producing a model in less than a minute. As expected the Web-based programs were slowest, but the situation could be improved by submitting several alignments at the same time (see Table 2).
Structural similarity to the correct structure
The similarity between a model and the correct structure was assessed by CA-RMSD and MaxSub (see Fig. 3 ▶). In agreement with earlier observation, it is clear that no improvement over a simple model with copied coordinates (SCWRL) can be seen. One should bear in mind that the modeling programs that do not model all residues are favored using the RMSD measure. Therefore, another common measure for protein model quality, MaxSub (Siew et al. 2000), was also used. The MaxSub score is related to the fraction of CA atoms in a model that can be superimposed with the correct structure with <3.5 Å RMSD. Hence, a model with 10% missing residues cannot receive a MaxSub score higher than 0.9; i.e., models with removed residues are penalized. As expected, Builder and 3D-JIGSAW, which remove the highest number of residues, performed slightly worse than the other programs, while no difference can be found between the other programs.
Figure 3.

Different measures used to assess the quality of the protein models. (A) RMSD values transformed using 1/(1 + RMSD) to avoid problem with high values. (B) MaxSub. (C) Backbone quality. (D) Side-chain quality as measured by fraction of correct side-chain torsion angles (χ1 and χ2). Error bars are constructed using standard error.
Backbone dihedral angles
Another measure of the overall structure can be obtained by analyzing how well the backbone dihedral angles (φ/ψ) agree with the correct ones. As for the RMSD and MaxSub measures, no modeling program performs better than the backbone models with coordinates from the template (see Fig. 3C ▶). However, the three Modeller programs all perform as well as the backbone models, while the other programs are slightly worse. SegMod/ENCAD, at high sequence identities, and Builder perform worse than the others methods. For SegMod/ENCAD, this is a result of the energy minimization step using ENCAD, since the models before the minimization do not show this decrease (data not shown).
Side-chain quality
The side-chain quality can be analyzed by RMSD for all atoms or by detecting the fraction of correct rotamers found. The latter measure is a more specific measure of side-chain quality, and subtle differences are more easily observed. In fact, using RMSD for all atoms it is difficult to detect any difference between the homology modeling programs (see Fig. 3A ▶). However, if the fraction of correct rotamers are used it is obvious that SCWRL-CONS builds better side chains than the modeling programs (see Fig. 3D ▶). In addition, at low sequence identities (<50%) it is possible to distinguish three groups: SCWRL3 and SCWRL-CONS perform best, followed by SegMod/ENCAD, SWISS-MODEL, Builder, and nest, while Modeller and 3D-JIGSAW are the worst programs, with only 30% correct residues. At higher sequence identities Builder and SCWRL3 drop in performance compared to the other programs. SCWRL3 drops in performance at high sequence identity because information about conserved rotamers is not used, while this information is used by SCWRL-CONS and apparently also somehow by nest and SWISS-MODEL, which both perform on par with SCWRL-CONS at high sequence identities. It can also be noted that the side-chain prediction problem faced here is much more difficult than if the side chains were built on the native backbone, where SCWRL3 creates more than 70% correct side chains.
Stereochemistry
Stereochemistry was assessed by WHAT_CHECK (Hooft et al. 1996). The output from WHAT_CHECK is, in principle, a list of residues that have “bad” stereochemistry using different measures such as bond lengths, bond angles, side-chain planarity, torsion angles, or contacts. “Bad” is defined as a significant number of standard deviations from what is observed in native structures. In addition to checking the chemistry for all models, the native structure was also assessed using the same tests. This provided an estimate on what could be considered as “good” chemistry, under the assumption that the native structure has good chemistry. Indeed according to WHAT_CHECK, the native structures had only 2% bad residues, most of them coming from “bad” bond angles.
In general, all modeling programs performed well for most of the checks; no model contained van der Waals overlap or residues in disallowed regions of the Ramachandran map, and only a few models had side chains with bad rotamers. However, differences were observed for bond lengths, bond angles, and side-chain planarity (see Fig. 4A ▶). 3D-JIGSAW, Builder, and SWISS-MODEL created more residues with bad chemistry for difficult targets, while the other modeling programs showed a fairly constant number of bad residues at all sequence identities.
Figure 4.

(A) Fraction of residues with “bad” bond angles, bond length side-chain planarity according to WHAT_CHECK for each method and also for the native structure. For a residue to be part of the any category it has to be classified as “bad” for any of the categories above. (B) The sequence identity dependence for the residues from the any “bad” category above. (C) Models with MaxSub score >0.6. (D) Acceptable model are models that have a MaxSub score of at least 0.6 and not more than 10% of its residues missing or with bad chemistry. 3D-JIGSAW and Builder have a significantly lower number of acceptable models and were removed for clarity.
SegMod/ENCAD produced slightly less bad residues than contained in the native structure, while all other programs produced more. The good stereochemistry is a result of the energy minimization step using ENCAD. Therefore, we applied energy minimization on the Modeller and nest models to investigate if their stereochemistry could be improved. Indeed, using ENCAD improved the stereochemistry significantly; however, using GROMACS it got worse. Both minimization methods distorted the backbone conformation, resulting in a less correct backbone. This demonstrates the difficulties involved in the refinement of a protein model, but also shows that it might be possible to improve the current protocols.
Discussion
Improvement over the template
It has been shown in several studies and also at CASP5 (Tramontano and Morea 2003) that a model only rarely is closer to the native structure than is the template it was built on. This is also true for most cases in this benchmark (see Fig. 5 ▶). MaxSub was calculated for the template structure and for the model, and a difference of 0.02 was assumed to be significant. For sequence identities below 40% all modeling programs manage to bridge some gaps and build some loops correctly; therefore, some models are better than the template. In this region the Modeller programs, nest, SegMod/ENCAD, and SWISS-MODEL, improved 20% of the models. In the same region SWISS-MODEL deteriorated 10% of the models, while the three other programs only deteriorated 5% of the models. At higher sequence identities, the number of improved models is decreased, while the fraction of models that get deteriorated remain fairly constant. All improvements are mainly due to the inclusion of “trivially” placed loop residues, but this still shows that molecular modeling approaches sometimes adds value over simply copying the template coordinates. Overall, nest only rarely made the models worse, while all other programs deteriorated at least 5% of the models (see Fig. 5B ▶). In addition, we found a few examples of significant improvements describe below (see Fig. 6 ▶). In the first example, the HCV helicase from Human hepatitis C virus (HCV) belonging to RNA helicase family (SCOP code: c.37.1.14) was modeled on a template (d8ohm_2) from a different isolate of the same domain with Modeller7v7. The sequence identity between the target and template is 91%, and the alignment contains no gaps. The RMSD is significantly reduced from 2.06 Å between the template and native structure to 1.40 Å for the model. This improvement is impressive, especially since no other modeling program improved this target at all. The reason for the improvement is not that a few loops are built correctly; it is rather so that the whole structure has moved closer to the native one. In the second example, colicin E7 belonging to colicin E immunity protein family (SCOP code: a.28.2.1), was modeled on colicin E9 (d1emva_) using 3D-JIGSAW. The sequence identity between the two proteins is 54%, and the alignment contains only one single residue gap. The RMSD is reduced from 2.05 Å between the template and native structure to 1.30 Å between the model and the native structure. This improvement is mainly due to one eight-residue loop that only 3D-JIGSAW built correctly.
Figure 5.

Improvement over template as measured by (A) the difference between the fraction of models that gets significantly (ΔMX > 0.02) improved, fimp, and the fraction that gets significantly deteriorated, fdet, or by (B) the average fraction of models that gets improved, 〈fimp〉, and deteriorated, 〈fdet〉. 3D-JIGSAW and Builder were removed for clarity.
Figure 6.

Two examples of a model that is improved upon modeling: in red, the template structure model is shown; in green, the final model; in blue, the native structure. (A) Modeller7v7 model of domain d1heia2 with d8ohm 2 as a template. The alignment contains no gaps, and the sequence identity between the target and template sequence is 91%. The RMSD between the template and the native structure is 2.06 Å, and for the Modeller7v7 model only 1.40 Å. The MaxSub score is also improved from 0.75 to 0.87. (B) 3D-JIGSAW model of domain d1unka with d1emva as a template. The alignment contains a single residue gap, and the sequence identity between target and template sequence is 58%. The RMSD between the template and the native structure is 2.05 Å, and for the 3D-JIGSAW model 1.30 Å.The MaxSub score is improved from 0.80 to 0.87. Figures were made using MOLSCRIPT (Kraulis 1991).
Even though improvements over the template are rare, these examples show that sometimes a model can be significantly improved. Basically, all improvements are observed in the region below 40% sequence identity and overall nest is the only program that makes more models better than worse.
Acceptable models
By using global measures such as RMSD it is difficult to detect any significant difference between the homology modeling programs. However, by looking at more detailed measures there are clear differences. It is clear that some programs are very reliable and always produce a model, some models contain large extended parts as a result of poor convergence, some models have missing residues, and some programs sacrifice the stereochemistry for a more correct backbone or vice versa. To get an estimate of how often the different programs produced an “acceptable model,” two criteria were used. First, an acceptable model should have good stereochemistry and few missing residues, therefore only models with <10% bad stereochemistry or missing residues were accepted. Second, an acceptable model should have a MaxSub score higher than 0.6.
Using these two criteria, SegMod/ENCAD produced the highest fraction of acceptable models over all sequence identity levels (91.5%) (see Fig. 4D ▶). This is a remarkable good performance, since even as many as 7% of the native structures had more than 10% residues with bad chemistry, giving an acceptance rate for the native structures of 93%. The different Modeller programs performed equally well as SegMod/ENCAD at high sequence identity but worse at lower identities, due to bad convergence for some models; the performance of SWISS-MODEL dropped at low sequence identities because it frequently failed to produce a model; nest produced the same number of acceptable models as Modeller at low sequence identities, but dropped together with SCWRL at higher sequence identities due to a few models with bad chemistry. As expected, 3D-JIGSAW and Builder, which did not model all residues, produce a significantly lower fraction of acceptable models compared to the other modeling programs (<40%), and were therefore removed from Figure 4D ▶ for clarity.
Selecting the best model
One possibility to produce the best possible model would be to produce many different models, and than try to select the best of them using some scoring function. Usually, this is done by generating many different alternative alignments and then using one homology modeling program to create models followed by some quality assessment and a selection process. This is also the basis for consensus methods that have been shown to be very successful in protein structure predictions (Lundström et al. 2001; Wallner et al. 2003). In contrast, here we use one alignment but many homology modeling programs to create alternative models.
We only included the modeling programs that produced an acceptable number of correct models, and only the best Modeller program in this final selection, i.e., Modeller6v2–10, SegMod/ENCAD, SWISS-MODEL, nest, and SCWRL-CONS. This means that for each alignment we have at most five different alternative models.
To assess if it was possible to select the best model for each target using a suitable scoring function, the following scoring functions were applied to each model: ProsaII (Sippl 1993), Errat (Colovos and Yeates 1993), ProQ (Wallner and Elofsson 2003), GROMACS energy calculations (Lindahl et al. 2001), and RMSD to the backbone model. In addition, the average RANK was also used as a scoring function. This measure is simply the average ranking of the model based on all scoring functions. The selection was evaluated based on the fraction of models that were among the best (“among best”) and number of acceptable models (see Materials and Methods for details).
One striking feature from Table 3 is that the different scoring functions seem to favor or disfavor different modeling programs. Prosa likes SegMod/ENCAD but not nest, Errat does not like Modeller but likes SWISS-MODEL and nest, GROMACS really likes SegMod/ENCAD and Builder but not Modeller, and 3D-JIGSAW and ProQ favor Modeller and nest but disfavor SegMod/ENCAD. In some cases it is easy to understand why a certain modeling program is preferred over others, i.e., that GROMACS favors SegMod/ ENCAD is due to the fact that they both use molecular mechanics energy functions, while ProQ is trained on Modeller models, and this could be the reason why ProQ favors Modeller models. However, in other cases no obvious explanations are found.
Table 3.
Selection of the best possible model using a number of different scoring functions
| Modeling program | 〈MX〉 | Among best | Accept | Prosa | Errat | Gromacs | ProQ | RMSD CA | RANK |
| Modeller6v2–10 | 0.834 | 904 (87.2) | 86.6% | 204 (19.7) | 14 (1.4) | 27 (2.6) | 281 (27.1) | 394 (38.0) | 48 (4.6) |
| SegMod/ENCAD | 0.834 | 887 (85.6) | 91.5% | 395 (38.1) | 208 (20.1) | 553 (53.4) | 34 (3.3) | 218 (21.0) | 220 (21.2) |
| SWISS-MODEL | 0.836 | 763 (81.8) | 78.0% | 173 (18.5) | 315 (33.8) | 79 (8.5) | 133 (14.3) | 86 (9.2) | 232 (24.9) |
| nest | 0.836 | 940 (91.4) | 81.9% | 108 (10.5) | 294 (28.6) | 262 (25.5) | 287 (27.9) | 339 (32.9) | 350 (34.0) |
| SCWRL-CONS | 0.835 | 892 (86.0) | 80.0% | 157 (15.1) | 206 (19.9) | 116 (11.2) | 302 (29.1) | —b | 187 (18.0) |
| 〈MX〉 | 0.844a | — | — | 0.835 | 0.836 | 0.836 | 0.836 | 0.837 | 0.837 |
| Among best | — | 1037a | — | 927 (89.4) | 912 (87.9) | 927 (89.4) | 917 (88.4) | 925 (89.2) | 933 (90.0) |
| Accept | — | — | 92.6%a | 85.5% | 87.2% | 88.3% | 82.4% | 86.0% | 85.1% |
Among best is the number of models that have a MaxSub score significantly close to the best possible choice (0.02). Acceptable models are models with <10% residues with bad chemistry and missing residues and a MaxSub score >0.6. Also, the preference for different scoring functions to select models for certain methods are shown. RANK is the average ranking of the models based on all scoring functions. A random pick corresponds to 〈MX〉 = 0.833, 897 (86.5%) models among best and 85.8% acceptable models.
a Best possible choice.
b All SCWRL-CONS models have RMSD-CA equal to zero and were therefore excluded from this selection.
It is clear that no single modeling program produce both the highest number of models (“among best”) and acceptable models. Nest makes few mistakes and has many of its models “among best,” i.e., selecting a model from nest is almost always a good choice if the objective is to get a model with a good MaxSub score. However, since nest creates some models with bad chemistry the number of “acceptable models” gets quite low. A selection of models that with many models among the best and a high number of acceptable models would be ideal. This can, to some extent, be achieved by selecting models based on GROMACS energy calculations, which selects 89.4% models among the best and 88.3% among the acceptable models, not as good as the best single modeling program: nest, 91.4% among best, and SegMod/ENCAD with 91.5% acceptable models, but still the best tradeoff between the two measures. The RANK might be slightly better than GROMACS for “among best” but the number of acceptable models is lower. Overall, most of the scoring functions select a higher number of acceptable models than compared to random. ProQ seems to select the least number of acceptable models of all scoring functions, since it has a bias not to select SegMod/ ENCAD.
Conclusions
It is obvious from the analysis above that no single modeling program performs best in all different test. All programs have its pros and cons (see Table 4 for a summary of them). It is clear that three modeling programs—Modeller, nest, and SegMod/ENCAD—perform better than the others, partly because they reliable produce a chemically correct model. These three programs are also quite fast producing a model in less than a minute on a 1.4-GHz AMD XP processor, which make any of them suitable for large-scale studies.
Table 4.
Pros and cons for the different modeling programs
| Modeling program | Pros | Cons |
| Modeller6v2 | reliable | convergence problem, bad side chains |
| Modeller7v7 | reliable | convergence problem, bad side chains |
| Modeller6v2–10 | reliable | bad side chains |
| SegMod/ENCAD | fast, good stereochemistry | bad backbone conformation |
| SWISS-MODEL | good stereochemistry | unreliable, many crashes, convergence problem |
| 3D-JIGSAW | — | missing residues, bad side chains, bad stereochemistry |
| nest | reliable, rarely deteriorate the models compared to template | bad stereochemistry |
| Builder | — | missing residues, bad backbone, bad stereochemistry, convergence problem |
| SCWRL3 | good side chains | no real modeling |
SegMod/ENCAD performs very well in all tests except for backbone conformation, nest very rarely makes the models worse than the template, but the chemistry is not as good as for SegMod/ENCAD, while Modeller is in general good, except for a few examples of poor convergence and suboptimal side-chains positioning. The convergence problem can be solved by rerunning Modeller 10 times with different random and select the model with the lowest RMSD to the template (Modeller6v2–10) and the side chains can be improved by rebuilding them using SCWRL3. None of the homology modeling programs builds side chains as well as SCWRL3, and, therefore, there should be room for improvement within this area.
Finally, we examined the possibility to select the best out of a set of models built using the different homology modeling programs. We found that several evaluation methods selects models that were better than the average model, but that no selection method performed significantly better than the best homology modeling method.
Materials and methods
Data set
Our data set consists of alignments between protein sequences with known 3D structure belonging to the same family according to SCOP (Murzin et al. 1995). The structures should have a resolution better than 3 Å and an R-factor <0.25. In order to not bias the set toward a particular family the number of alignments for one family was restricted to five. The alignments were constructed using the Needleman-Wunsch (Needleman and Wunsch 1970) global alignment algorithm. The main reason for using Needleman-Wunsch was to get alignments that behaved like a real modeling problem with errors. The errors will, however, be rare, since the alignments in most cases are trivial. The final alignment set consisted of 1037 alignments that covered the whole spectrum of sequence identity from 30% to 100%.
Programs used in the benchmark
All homology modeling programs used here use as their input an alignment between a target sequence and a template sequence. Based on this alignment and the known structure, the coordinates for the heavy atoms of query sequence are built. The difference between the programs is how the information contained in the alignment is used to build a 3D model. Below, follow a short overview of the programs used in this study.
Modeller
Modeller (Sali and Blundell 1993) is perhaps the most frequently used homology modeling program. It is one of the first fully automated programs, and it is also relatively fast, making it suitable for whole-genome modeling (Marti-Renom et al. 2000; Pieper et al. 2004). Models are obtained by satisfying spatial restraints derived from the alignment and expressed as probability density functions (pdfs) for the different types of restraints. The pdfs restrain CA–CA and backbone N–O distances, and backbone and side-chain dihedral angles for different residue types. The generated model violates these restraints as little as possible. A new version of Modeller was recently released, and both the new 7v7 and the old 6v2 have been tested here. In addition, a third Modeller version, Modeller6v2–10, was also tested. Here, 10 models are created for each alignment using different initial random seeds, and the model with the lowest RMSD to the template structure is chosen. The reason for including this program was that Modeller sometimes has a problem with convergence, i.e., producing models with extended structures. Modeller is available from http://salilab.org/modeller/.
SegMod/ENCAD
SegMod/ENCAD is a combination of a segment-matching routine (SegMod) (Levitt 1992) and a molecular dynamics simulation program (ENCAD) (Levitt 1983). The SegMod program is based on a database of known protein structures. First, the aligned coordinates are copied and then it tries to bridge the gaps by breaking down the target structure into a set of short segments and search the database for segments that match the framework of the target structure. The matching is based on three criteria: sequence similarity, conformational similarity, and compatibility with the target structure using van der Waals’ interactions. The final model is then energy minimized using ENCAD. SegMod/ENCAD is available upon request from michael.levitt@stanford.edu.
SWISS-MODEL
SWISS-MODEL (Schwede et al. 2004) is a Web-based homology modeling server (http://swissmodel.expasy.org/). Models are generated from the alignment in a stepwise manner. First, backbone coordinates for aligned positions are extracted from the template. Second, regions of insertions and deletions in the alignment are modeled by either searching a loop library or by a search in conformational space using constraint space programming. The best loop is selected using a scoring scheme, which accounts for force field energy, steric hindrance, and favorable interactions such as hydrogen bond formation. Third, side-chain conformations are selected from a backbone-dependent rotamer library using a scoring function assessing favorable interactions (hydrogen bonds, disulfide bridges) and unfavorably close contacts.
3D-JIGSAW
3D-JIGSAW (Bates et al. 2001) is a Web-based homology modeling server (http://www.bmm.icnet.uk/servers/3djigsaw). Models are created by extracting coordinates from aligned positions. Obvious gaps in the structures, elements between secondary structures, and backbone angles incompatible with the target sequence are modeled by database fragment searches. A complete backbone is selected from an ensemble of secondary structure elements and connecting loops using a self-consistent mean field approach (Koehl and Delarue 1995). Side chains are built using rotamers from the template structure and a side-chain rotamer library together with a second mean field calculation (Koehl and Delarue 1994). Loops are trimmed by adjusting torsion angles within each loop to give good geometry. Finally, to remove steric clashes, 100 steps of steepest descents energy minimization are run by using CHARMM (Brooks et al. 1983).
nest
Nest (Petrey et al. 2003) is the core program within the JACKAL Modeling Package. The model building is based on an artificial evolution method. In this modeling program changes from the template structure such as residue mutation, insertion, and deletions are made one at a time. After each change a torsion energy minimizer is applied and an energy is calculated based on a simplified potential function that includes van der Waals, hydrophobic, electrostatic, torsion angle, and hydrogen bond terms. The change that produces the most favorable change in energy is accepted, and the process is repeated until the target sequence is completely modeled. The Jackal Package can be downloaded from http://trantor.bioc.columbia.edu/programs/jackal/.
Builder
Builder (Koehl and Delarue 1994, 1995) is based on the Self Consistent Mean-Field theory (SCMF) (Koehl and Delarue 1996), both for loop modeling and for side-chain conformation prediction. SCMF is based on a multicopy sampling in conformation space: the protein is replaced by an effective system containing the framework, multiple copies of the backbone in the gap regions, and multiple copies of all side chains on all backbones. Each side-chain copy k on a backbone copy j of a residue i is given a weight P(i, j, k), while the backbone copy j is given the weight B (i, j). The weights are initialized to follow a uniform distribution (1/N). The effective energies for all backbone copies and side-chain copies are then computed using the Mean Field Theory (Koehl and Delarue 1995). These energy values are used to update the probabilities following a Boltzmann-like law. The whole procedure is iterated until the total energy of the system does not change, i.e., when it has reached self-consistency. The side chains and backbone conformations of residue i are then chosen to be the copies with the highest converged probabilities. Builder is available upon request from koehl@cs.ucdavis.edu.
SCWRL
SCWRL (Canutescu et al. 2003) is a program that, given a backbone, builds the side chains on it. SCWRL uses a backbone-dependent rotamer library (Dunbrack 2002) and an energy function based on the log probabilities of these rotamers and a simple repulsive steric energy term. Here, SCWRL was used to build side chains on the backbone models obtained by copying the aligned template coordinates, in two ways: rebuilding all side chains or rebuilding only nonconserved side chains (SCWRL-CONS). SCWRL is available from http://dunbrack.fccc.edu/SCWRL3.php.
Quality tests
A number of different tests were performed to assess the quality of the models from the different homology modeling programs, including stereochemistry, RMSD, MaxSub, backbone dihedral angles, and side chain quality (see below). All tests except stereochemistry were done by comparing the model to the correct structure. The final assessment was complicated by the fact that all modeling programs did not produce coordinates for all residues in the alignments. This forced us to exclude models and residues that did not exist for all modeling programs. The RMSD and MaxSub checks were performed for all models that existed for all modeling programs, while all the other checks were performed for all residues that existed in all models from all modeling programs.
Stereochemistry
The stereochemistry of each model was evaluated by running WHAT_CHECK (Hooft et al. 1996). WHAT_CHECK performs a number of checks including bond lengths, bond angles, side chain planarity, torsion angles, van der Waals overlap, and backbone dihedrals. Each check gives a Z-score for each residue, which is the number of standard deviations each residue deviates from what is expected from observations in real X-ray structures. Based on this Z-score, residues are defined by WHAT_CHECK to be “ok,” “poor,” or “bad,” depending on how large the deviation is. Here, we have only considered the residues with “bad” Z-score in our analysis.
RMSD
RMSD is the most common check to measure structural similarity between a model and a correct structure. The RMSD between two structures is simply the square root of the average squared distances between equivalent atoms after an optimal superposition. RMSD was calculated for all atoms and for CA atoms only.
MaxSub
MaxSub (Siew et al. 2000) is a measure developed to overcome the drawbacks of RMSD, especially for models with high RMSD values. It aims at detecting segments in common between the model and the correct structure. Based on these segments a structural comparison score, Sstr, is calculated
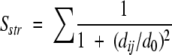 |
where the sum is taken over all residues in the segments, dij, is the distance between the CA atom in residue i in the model and j in the correct structure and d0, a distance threshold (normally set to 5 Å). For a perfect model dij will be 0 and Sstr will be equal to the length of the model, for a completely wrong model Sstr will be zero. MaxSub is the Sstr averaged over the whole structure
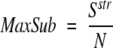 |
where N is the number of residues in the correct structure.
Backbone dihedral angles
The quality of the backbone was assessed by calculating the backbone dihedral angles, φ and ψ for the model and comparing them to correct values from the native structure. A residue in the model was considered to have correct backbone dihedral angles if they occupied the same “core” region (Morris et al. 1992) in the Ramachandran plot as the native structure or if the distance from the native (φ/ψ) in the Ramachandran plot was <15°.
Side-chain quality
The side-chain quality was assessed by RMSD for all atoms and by calculating the fraction of side-chain dihedral angles, χ1 and χ2, that where within 30° from the angles in the correct structure.
Acceptable models
It is difficult to use one single quality measure that captures all properties of a protein model. A model might look good using one measure and poor using another. Therefore, many different tests were used to evaluate the final models. However, it would also be desirable to have an estimate on how often the different programs produce an “acceptable model,” i.e., a model that might not be perfect but is still ok. It seems reasonable that an acceptable model should contain coordinates for all residues in the alignment, and that the stereochemistry should not be too bad. The model should also be reasonably close to the correct structures. Here, we used two criteria summarized below to define an acceptable model:
Less than 10% of its residues with bad stereochemistry or missing
MaxSub >0.6.
Acknowledgments
This work was supported by grants from the Graduate Research School in Genomics and Bioinformatics to B.W. and Swedish Foundation for Strategic Research and the Swedish Research Council to A.E.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041253405.
References
- Abagyan, R.A. and Batalov, S. 1997. Do aligned sequences share the same fold? J. Mol. Biol. 273 355–368. [DOI] [PubMed] [Google Scholar]
- Bates, P.A., Kelley, L.A., MacCallum, R.M., and Sternberg, M.J. 2001. Enhancement of protein modeling by human intervention in applying the automatic programs 3D-JIGSAW and 3D-PSSM. Proteins 45 (Suppl. 5): 39–46. [DOI] [PubMed] [Google Scholar]
- Blundell, T.L., Sibanda, B.L., Sternberg, M.J., and Thornton, J.M. 1987. Knowledge-based prediction of protein structures and the design of novel molecules. Nature 326 347–352. [DOI] [PubMed] [Google Scholar]
- Brenner, S.E., Chothia, C., and Hubbard, T. 1998. Assessing sequence comparison methods with reliable structurally identified evolutionary relationships. Proc. Natl. Acad. Sci. 95 6073–6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks, B.R., Bruccoleri, R.E., Olafson, B.D., States, D.J., Swaminathan, S., and Karplus, M. 1983. CHARMM: A program for macromolecular energy minimization and dynamics calculations. J. Comput. Chem. 4 187–217. [Google Scholar]
- Browne, W.J., North, A.C., Phillips, D.C., Brew, K., Vanaman, T.C., and Hill, R.L. 1969. A possible three-dimensional structure of bovine α-lactalbumin based on that of hen’s egg-white lysozyme. J. Mol. Biol. 42 65–86. [DOI] [PubMed] [Google Scholar]
- Canutescu, A.A., Shelenkov, A.A., and Dunbrack Jr., R.L. 2003. A graphtheory algorithm for rapid protein side-chain prediction. Protein Sci. 12 2001–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardozo, T., Totrov, M., and Abagyan, R. 1995. Homology modeling by the ICM method. Proteins 23 403–414. [DOI] [PubMed] [Google Scholar]
- Chothia, C. and Lesk, A.M. 1986. The relationship between the divergence of sequence and structure in proteins. EMBO J. 5 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claessens, M., Van Cutsem, E., Lasters, I., and Wodak, S. 1989. Modelling the polypeptide backbone with ”spare parts” from known protein structures. Protein Eng. 2 335–345. [DOI] [PubMed] [Google Scholar]
- Colovos, C. and Yeates, T.O. 1993. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 2 1511–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbrack Jr., R.L. 2002. Rotamer libraries in the 21st century. Curr. Opin. Struct. Biol. 12 431–440. [DOI] [PubMed] [Google Scholar]
- Elofsson, A. 2002. A study on how to best align protein sequences. Proteins 15 330–339. [Google Scholar]
- Fischer, D. and Eisenberg, D. 1996. Protein fold recognition using sequence-derived predictions. Protein Sci. 5 947–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— 1999. Predicting structures for genome proteins. Curr. Opin. Struct. Biol. 9 208–211. [DOI] [PubMed] [Google Scholar]
- Fischer, D. and Rychlewski, L. 2003. The 2002 Olympic games of protein structure prediction. Protein Eng. 16 157–160. [DOI] [PubMed] [Google Scholar]
- Godzik, A., Kolinski, A., and Skolnick, J. 1992. Topology fingerprint approach to the inverse protein folding problem. J. Mol. Biol. 227 227–238. [DOI] [PubMed] [Google Scholar]
- Greer, J. 1990. Comparative modeling methods: Application to the family of the mammalian serine proteases. Proteins 7 317–334. [DOI] [PubMed] [Google Scholar]
- Hooft, R.W., Vriend, G., Sander, C., and Abola, E.E. 1996. Errors in protein structures. Nature 381 272. [DOI] [PubMed] [Google Scholar]
- Jaroszewski, L., Li, W., and Godzik, A. 2002. In search for more accurate alignments in the twilight zone. Protein Sci. 11 1702–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, T.A. and Thirup, S. 1986. Using known substructures in protein model building and crystallography. EMBO J. 5 819–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, D.T., Taylor, W.R., and Thornton, J.M. 1992. A new approach to protein fold recognition. Nature 358 86–89. [DOI] [PubMed] [Google Scholar]
- Koehl, P. and Delarue, M. 1994. Application of a self-consistent mean field theory to predict protein side-chains conformation and estimate their conformational entropy. J. Mol. Biol. 239 249–275. [DOI] [PubMed] [Google Scholar]
- ———. 1995. A self consistent mean field approach to simultaneous gap closure and side-chain positioning in homology modelling. Nat. Struct. Biol. 2 163–170. [DOI] [PubMed] [Google Scholar]
- ———. 1996. Mean-field minimization methods for biological macromolecules. Curr. Opin. Struct. Biol. 6 222–226. [DOI] [PubMed] [Google Scholar]
- Kosinski, J., Cymerman, I.A., Feder, M., Kurowski, M.A., Sasin, J.M., and Bujnicki, J.M. 2003. A “FRankenstein’s monster” approach to comparative modeling: Merging the finest fragments of fold-recognition models and iterative model refinement aided by 3D structure evaluation. Proteins 53 (Suppl. 6): 369–379. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Levitt, M. 1983. Molecular dynamics of native protein. I. Computer simulation of trajectories. J. Mol. Biol. 168 595–617. [DOI] [PubMed] [Google Scholar]
- ——— 1992. Accurate modeling of protein conformation by automatic segment matching. J. Mol. Biol. 226 507–533. [DOI] [PubMed] [Google Scholar]
- Lindahl, E., Hess, B., and van der Spoel, D. 2001. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Model. 7 306–317. [Google Scholar]
- Lundström, J., Rychlewski, L., Bujnicki, J., and Elofsson, A. 2001. Pcons: A neural-network-based consensus predictor that improves fold recognition. Protein Sci. 10 2354–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti-Renom, M.A., Stuart, A.C., Fiser, A., Sánchez, R., Melo, F., and Sali, A. 2000. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 29 291–325. [DOI] [PubMed] [Google Scholar]
- Morris, A.L., MacArthur, M.W., Hutchinson, E.G., and Thornton, J.M. 1992. Stereochemical quality of protein structure coordinates. Proteins 12 345–364. [DOI] [PubMed] [Google Scholar]
- Moult, J., Fidelis, K., Zemla, A., and Hubbard, T. 2003. Critical assessment of methods of protein structure prediction (CASP)-round V. Proteins 53 (Suppl. 6): 334–339. [DOI] [PubMed] [Google Scholar]
- Murzin, A.G., Brenner, S.E., Hubbard, T., and Chothia, C. 1995. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247 536–540. [DOI] [PubMed] [Google Scholar]
- Needleman, S.B. and Wunsch, C.D. 1970. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 48 443–453. [DOI] [PubMed] [Google Scholar]
- Park, J., Karplus, K., Barrett, C., Hughey, R., Haussler, D., Hubbard, T., and Chothia, C. 1998. Sequence comparisons using multiple sequences detect three times as many remote homologues as pairwise methods. J. Mol. Biol. 284 1201–1210. [DOI] [PubMed] [Google Scholar]
- Petrey, D., Xiang, Z., Tang, C.L., Xie, L., Gimpelev, M., Mitros, T., Soto, C.S., Goldsmith-Fischman, S., Kernytsky, A., Schlessinger, A., et al. 2003. Using multiple structure alignments, fast model building, and energetic analysis in fold recognition and homology modeling. Proteins 53 (Suppl. 6): 430–435. [DOI] [PubMed] [Google Scholar]
- Pieper, U., Eswar, N., Braberg, H., Madhusudhan, M.S., Davis, F.P., Stuart, A.C., Mirkovic, N., Rossi, A., Marti-Renom, M.A., Fiser, A., et al. 2004. MODBASE, a database of annotated comparative protein structure models, and associated resources. Nucleic Acids Res. 32 D217–D222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost, B. 1999. Twilight zone of protein sequence alignments. Protein Eng. 12 85–94. [DOI] [PubMed] [Google Scholar]
- Rychlewski, L. and Fischer, D. 2005. LiveBench–8: The large-scale, continuous assessment of automated protein structure prediction. Protein Sci. 14 240–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali, A. and Blundell, T.L. 1993. Comparative modelling by statisfaction of spatial restraints. J. Mol. Biol. 234 779–815. [DOI] [PubMed] [Google Scholar]
- Sánchez, R. and Sali, A. 1997. Advances in comparative protein-structure modeling. Curr. Opin. Struct. Biol. 7 206–214. [DOI] [PubMed] [Google Scholar]
- Schwede, T., Kopp, J., Guex, N., and Peitsch, M.C. 2004. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 31 3381–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siew, N., Elofsson, A., Rychlewski, L., and Fischer, D. 2000. Maxsub: An automated measure to assess the quality of protein structure predictions. Bioinformatics 16 776–785. [DOI] [PubMed] [Google Scholar]
- Sippl, M.J. 1993. Recognition of errors in three-dimensional structures of proteins. Proteins 17 355–362. [DOI] [PubMed] [Google Scholar]
- Tramontano, A. and Morea, V. 2003. Assessment of homology-based predictions in CASP5. Proteins 53 (Suppl. 6): 352–368. [DOI] [PubMed] [Google Scholar]
- Tramontano, A., Leplae, R., and Morea, V. 2001. Analysis and assessment of comparative modeling predictions in CASP4. Proteins 45 (Suppl.) 5: 22–38. [DOI] [PubMed] [Google Scholar]
- Wallner, B. and Elofsson, A. 2003. Can correct protein models be identified? Protein Sci. 12 1073–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallner, B., Fang, H., and Elofsson, A. 2002. Using evolutionary information for the query and target improves fold recognition. Proteins 54 342–350. [DOI] [PubMed] [Google Scholar]
- ———. 2003. Automatic consensus-based fold recognition using Pcons, Proq, and Pmodeller. Proteins 53 (Suppl. 6): 534–541. [DOI] [PubMed] [Google Scholar]