

Abstract
The Methanococcus jannaschii tRNATyr/TyrRS pair has been engineered to incorporate unnatural amino acids into proteins in E. coli. To reveal the structural basis for the altered specificity of mutant TyrRS for O-methyl-l-tyrosine (OMeTyr), the crystal structures for the apo wild-type and mutant M. jannaschii TyrRS were determined at 2.66 and 3.0 Å, respectively, for comparison with the published structure of TyrRS complexed with tRNATyr and substrate tyrosine. A large conformational change was found for the anticodon recognition loop 257–263 of wild-type TyrRS upon tRNA binding in order to facilitate recognition of G34 of the anticodon loop through π-stacking and hydrogen bonding interactions. Loop 133–143, which is close to the tRNA acceptor stem-binding site, also appears to be stabilized by interaction with the tRNATyr. Binding of the substrate tyrosine results in subtle and cooperative movements of the side chains within the tyrosine-binding pocket. In the OMeTyr-specific mutant synthetase structure, the signature motif KMSKS loop and acceptor stem-binding loop 133–143 were surprisingly ordered in the absence of bound ATP and tRNA. The active-site mutations result in altered hydrogen bonding and steric interactions which favor binding of OMeTyr over l-tyrosine. The structure of the mutant and wild-type TyrRS now provide a basis for generating new active-site libraries to evolve synthetases specific for other unnatural amino acids.
Keywords: tyrosyl-tRNA synthetase, crystallography, unnatural amino acid, protein engineering
A method was developed which allows unnatural amino acids to be genetically encoded in cells (Wang et al. 2001; Wang and Schultz 2002). The approach involves addition of a new tRNA-codon pair and a cognate aminoacyl-tRNA synthetase (hereafter referred to as synthetase) to the biosynthetic machinery, which function orthogonally to the endogenous tRNAs and synthetases in the host. The orthogonal synthetase aminoacylates only the orthogonal tRNA with the desired unnatural amino acid (and no endogenous amino acid), and this acylated tRNA delivers this amino acid to the ribosome only in response to a noncoding codon.
Specifically, it was found that the heterologous tRNATyr/TyrRS pair from archaea Methanococcus jannaschii (M. jannaschii) is poorly recognized by Escherichia coli tRNAs and aminoacyl-tRNA synthetases (Wang et al. 2000). This lack of significant recognition likely results from differences in the acceptor stem and anticodon recognition domain between prokaryotic and archaeal tRNATyr/TyrRS. The anticodon of the M. jannaschii tRNATyr was mutated to CUA to recognize the amber nonsense codon TAG, and this suppressor tRNA was further modified (yielding mutRNACUATyr ) to improve its orthogonality toward endogenous E. coli synthetases (Wang and Schultz 2001). A mutant TyrRS library in which five active-site residues of M. jannaschii TyrRS were randomly mutated was probed to select a mutant synthetase specific for the unnatural amino acid O-methyl-l-tyrosine (OMeTyr) (Wang et al. 2001). This synthetase together with the mutRNACUA Tyr was used to incorporate OMeTyr into proteins with high fidelity and efficiency in response to the amber nonsense codon.
More than 25 unnatural amino acids have now been incorporated into proteins in E. coli by this approach, including amino acids with heavy atoms, keto and alkyne side chains, photocrosslinking and redox active amino acids, and glycosylated amino acids (Zhang et al. 2004). More recently, orthogonal tRNA/aminoacyl-tRNA synthetase pairs have been generated which can be used to incorporate unnatural amino acids into mammalian cells and yeast, as well as to incorporate unnatural amino acids in response to frameshift codons.
To understand the molecular basis for the specificity of the wild-type and mutant orthogonal synthetases, we determined the crystal structure of the apo wild-type M. jannaschii TyrRS and a mutant synthetase specific for OMe-Tyr. A crystal structure of M. jannaschii TyrRS complexed with tRNATyr and substrate tyrosine was previously determined (Kobayashi et al. 2003). A comparison of these structures reveals significant conformational changes upon binding of tyrosine and tRNATyr to TyrRS, and provides a structural basis to interpret the specificity determinants of the OMeTyr-specific enzyme. These and additional structures should allow the generation of improved primary and secondary libraries of TyrRS mutants to further evolve the specificity and activity of these enzymes.
Results and Discussion
X-ray structure determination
Molecular replacement (MR) of the apo wild-type M. jannaschii TyrRS was first attempted using TyrRS homologs as search models. Bacillus stearothermophilus TyrRS (with an identity of 18% to M. jannaschii) (PDB code 2ts1; Brick et al. 1988), Thermus thermophilus enzyme (with 22% identity) (PDB code 1h3f; Yaremchuk et al. 2002), human TyrRS catalytic domain (with 37% identity) (PDB code 1n3l; Yang et al. 2002), and the predicted structure for M. jannaschii TyrRS (Zhang et al. 2002) were all used as MR models, but none provided a solution. During the preparation of Se-Met M. jannaschii TyrRS, the crystal structure of M. jannaschii TyrRS in complex with tRNATyr and tyrosine was published (PDB code 1j1u; Kobayashi et al. 2003). The coordinates from this structure, excluding the tRNATyr and tyrosine ligands, were then successfully used in MR to locate both TyrRS molecules (A, B) in the crystal asymmetric unit. The OMeTyr mutant structure was determined by MR with the apo wild-type TyrRS as the search model. The unit cell of the mutant crystal is very similar to the apo wild type with the same space group. The data collection and refinement statistics are summarized in Table 1.
Table 1.
Data collection and refinement statistics for wt M. jannaschii TyrRS and the OMeTyr-specific mutant synthetase
| wt TyrRS | OMeTyr-specific mutant | |
| Space group | P212121 | P212121 |
| No. of molecules per a.u. | 2 | 2 |
| Resolution (Å) | 48-2.66 (2.72-2.66)d | 43-3.0 (3.11-3.0) |
| Unit cell | ||
| a, b, c (Å) | 45.1, 185.3, 95.5 | 45.4, 185.0, 93.8 |
| No. of observations | 92,914 | 48,673 |
| No. of unique reflections | 23,837 | 15,817 |
| Multiplicity | 3.5 | 3.1 |
| Completeness (%) | 99.5 (96.9) | 95.4 (89.0) |
| Rsym (%)a | 6.0 (53.7) | 14.8 (51.7) |
| I/σ | 21.5 (2.1) | 7.0 (1.7) |
| Refinement: | ||
| Rcryst (%)b | 22.6 | 22.8 |
| Rfree (%)c | 27.8 | 30.8 |
| No. of protein atoms | 4838 | 4889 |
| No. of waters | 37 | 0 |
| Average B-value (Å2) | 58.7 | 38.4 |
| Average solvent B-value (Å2) | 42.2 | N/A |
| R.m.s. deviations from ideals: | ||
| Bond lengths (Å) | 0.0076 | 0.0086 |
| Bond angles (°) | 1.30 | 1.44 |
| Ramachandran plot (%): | ||
| Most favored | 90.1 | 84.2 |
| Additionally allowed | 8.7 | 13.6 |
| Generously allowed | 1.2 | 2.0e |
| Disallowed | 0.0 | 0.2e |
a Rmerge = [∑h ∑i | Ii(h) - 〈I(h) 〉 | / ∑h ∑i Ii(h)] × 100, where 〈I(h) 〉 is the mean of the I(h) observation of reflection.
b Rcryst = ∑h||Fo|-|Fc||/∑h|Fo| where Fo and Fc are the observed and calculated structure factor amplitudes.
c Rfree (%) is the same as Rcryst but for 5% of the data randomly omitted from refinement (10% for O-methyl mutant data).
d Numbers in parentheses refer to the highest-resolution shell.
e The Lys209 outlier is located in a highly flexible loop.
Overall structure
The final model for apo wild-type M. jannaschii TyrRS (Fig. 1A ▶) includes residues 1–306 for molecule A, except for two loops, residues 134–141 and 203–208, for which no interpretable electron density was found. The same loops were also untraceable in molecule B; molecule B contains residues 2–306 with loops 132–143 and 204–209 disordered. In the complex structure (with bound tRNA and tyrosine), loop 133–143 is ordered, probably due to its association with tRNA (Kobayashi et al. 2003), whereas loop 202–209 containing the KMSKS signature motif essential for ATP binding was disordered, as in the apoenzyme. In the absence of ATP or ATP analogs, this loop is also disordered in several other TyrRS structures (1j1u and 1n3l). However, the refined structure for apo OMeTyr-specific mutant synthetase includes all residues (1–306). In contrast to the wild-type apo TyrRS structure, the 133–143 and 202–209 loops are both ordered, although with higher B-values than the average for the protein. This structure thus represents the first TyrRS-like synthetase in which the entire molecule is ordered in the absence of ligand(s).
Figure 1.
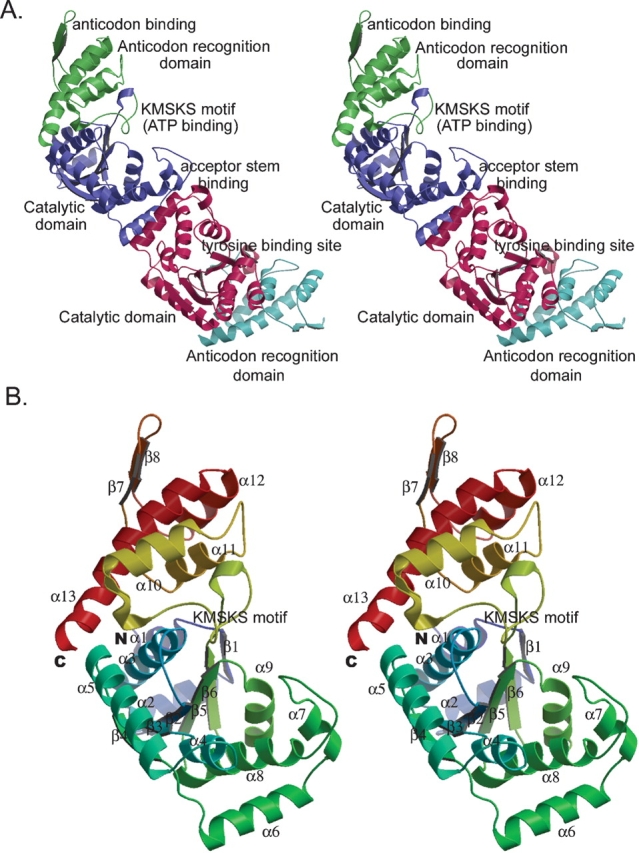
Crystal structure of apo wild-type M. jannaschii TyrRS. (A) Overall structure of apo M. jannaschii TyrRS homodimer. The N-terminal catalytic domain (blue and red) is connected via the signature KMSKS loop to the C-terminal anticodon recognition domain (green and cyan). (B) Secondary structure of M. jannaschii TyrRS. Stereo-ribbon diagram representing the overall topology and secondary structure of unliganded M. jannaschii TyrRS. Helices are shown as coils and β-strands as extended arrows. Color-coded from N (blue) to C termini (red).
The two molecules in each asymmetric unit of the apo wild-type TyrRS form a homodimer (Fig. 1A ▶), consistent with all other known TyrRS’s in solution and in the crystal (Calendar and Berg 1966; Koch 1974; Brick et al. 1988; Kobayashi et al. 2003). The dimer is the biologically active form of the synthetase with the tRNA spanning the two subunits (Kobayashi et al. 2003). The buried surface of the dimer is 1169 Å2, which represents 8.5% of the total surface area of the molecule. The surface complementarity (Sc) (Lawrence and Colman 1993) is 0.66, indicating reasonable fit of the dimer interface.
The overall structure of the apo TyrRS is similar to that of other class I aminoacyl-tRNA synthetases, with two domains based on function and topology (Fig. 1A ▶). The N-terminal catalytic domain contains residues 1–202 and is directly involved in tRNA acceptor stem binding, tyrosine binding, and charging of amino acid to tRNA. This domain consists of a central β-sheet of six twisted β-strands surrounded by eight α-helices (Fig. 1B ▶). The Rossmann-fold (β2–β6) in this domain is conserved within all class-I aminoacyl-tRNA synthetases, and forms part of the tyrosine-binding pocket and the reaction center. The signature KMSKS loop (residues 202–209), which is usually disordered in the absence of ATP, connects the N- and C-terminal domains (residues 203–306); the latter domain is referred to either as the anticodon recognition domain from its function or as the α-helical domain from its topology. This domain consists of four helices with two anti-parallel β-strands that form a β-hairpin (residues 250–269).
The thermal factor (B-value) distribution (Fig. 2A ▶) is similar for both wild-type and OMeTyr mutant apo synthetases, although the average B-value in the wild type is higher (58.0 Å2 in wild type vs. 38.9 Å2 in mutant) despite the better resolution (2.66 Å for wild type vs. 3.0 Å for mutant). When comparing the apo and complexed (PDB code 1j1u) wild-type M. jannaschii TyrRS structures, the regions where tRNA binds have greatly reduced local B-values, indicating decreased flexibility upon tRNA binding (Fig. 2 ▶).
Figure 2.

Comparison of wild-type apo and bound M. jannaschii TyrRS’s. (A) Stereo view of the B-value distribution of the apo wild-type M. jannaschii TyrRS structure. The structure trace is colored by B-values with a gradient ranging from low (25 Å2, blue) through medium (average B-value 59 Å2, yellow) to high (100 Å2, red), which highlights the more disordered regions of the structure (red). (B) Stereo view of the superimposition of apo wild-type M. jannaschii TyrRS and the bound structure (PDB code 1j1u;Kobayashi et al. 2003) of M. jannaschii TyrRS with tRNA (blue). Complexed M. jannaschii TyrRS is colored by its rms deviation (Cα) from the apo wild-type TyrRS structure (gray) with a gradient ranging from low (0.2 Å, green) via medium (1.8 Å, yellow) to high (>4.0 Å, red).
Examination of eight TyrRS primary sequences from different organisms suggests that archaea and eukarya TyrRS share common features, whereas eubacteria and mitochondria TyrRS possess more or less distinct “prokaryote-like” signatures (Shiba et al. 1997). Consistent with this analysis, the structure of apo M. jannaschii TyrRS is quite similar to its corresponding human homolog (PDB code 1n3l) with an rms deviation (Cα) of ~2.0 Å. The only significant difference is in the hinge area (residues 190–210), in which a small β-strand is inserted into the flexible loop that leads to a shift of ~20° in domain orientation. The differences between M. jannaschii and B. stearothermophilus or T. thermophilus TyrRS’s are more substantial with an overall rms deviation (Cα) >5 Å.
A comparison of apo and bound wild-type M. jannaschii TyrRS
The overall fold of the apo M. jannaschii TyrRS is identical to its counterpart complexed with tRNA and tyrosine (PDB code 1j1u; Kobayashi et al. 2003), with an overall rms deviation (Cα) of 1.50 Å. This rms deviation is surprisingly high for the same protein in the free and bound form, and does not appear to be due to different crystallization conditions, since these differences are localized to a few sites, most of which are involved in ligand binding (Fig. 2B ▶) and not in crystal packing. The regions with high rms deviation also have higher B-values (Fig. 2A ▶), consistent with their flexible nature. Moreover, the rms deviations are not evenly distributed between the two domains; the lower 0.66 Å deviation (Cα) in the C-terminal anticodon recognition domain is countered by a higher 1.66 Å rms deviation (Cα) in the catalytic domain. These superimpositions were conducted for each domain individually to eliminate differences caused by the observed interdomain rearrangement. The regions in which the two structures differ are outlined below and include the anticodon recognition region, loop 133–143, which is close to the acceptor stem-binding site, and the tyrosine-binding site. These regions are characterized by higher flexibility in the unbound structure, which are fixed upon ligand binding.
Changes in the anticodon recognition domain
The overall rms deviation before and after tRNA binding in the anticodon recognition domain is not large (Fig. 2B ▶), but is confined mainly to the tip of the recognition finger with a large rms deviation (3.2 Å vs. 0.66 Å average for this domain for Cα) in the turn (residues 257–263). This finger consists of two anti-parallel β-strands, β7 and β8, connected by a hairpin turn and is essential for anticodon recognition (Kobayashi et al. 2003). The residues composing this tip are mostly hydrophilic (Lys256, Arg257, Glu259, and Lys260). This finger region also has significantly higher B-values (Fig. 2A ▶). The average B-value at the finger tip (88.7 Å2) is more than double the average of the whole molecule (38.9 Å2). However, upon anticodon binding, the B-value of this finger tip is comparable to that of the overall molecule (40.7 Å2 vs. 34.7 Å2 overall). This suggests that the binding of the tRNA anticodon loop greatly stabilizes the conformation of the finger region.
The finger of TyrRS forms the upper surface of the binding cleft for recognition of G34 of the tRNA anticodon loop; helices α12 and α13 form the bottom surface (Fig. 3A ▶). Upon binding of tRNA, the finger closes down on the tRNA with the largest shift at Lys260 [rms deviation (Cα) of 4.2 Å]. The purine ring of G34 hydrogen bonds with Asp286 of the anticodon recognition domain of TyrRS (Kobayashi et al. 2003). The phenyl ring of Phe261 and the imidazole ring of His283 are oriented differently in the apo and bound structures (Fig. 3B ▶). When the purine ring of G34 binds between these two residues, the benzene ring of Phe261 rotates 90° and becomes parallel to G34 (Fig. 3B ▶), and His283 rotates almost 90° about χ1, resulting in a classical π stacking of these three rings (Fig. 3B ▶). π-Stacking interactions are relatively common in tRNA-synthetase recognition. For example, in the case of yeast arginyl-tRNA synthetase, Trp569 inserts between I34 and C35, forming a π-stacking sandwich (Delagoutte et al. 2000). In the case of T. thermophilus phenylalanyl-tRNA synthetase, base stacking between G34 and A35 is disrupted and G34 stacks with Tyr731 (Goldgur et al. 1997) of the synthetase. The side-chain orientation of Asp 286 in M. jannaschii TyrRS is also altered upon tRNA binding (Fig. 3B ▶). The side chain is rotated around χ1 almost 90° to align with N1 and N2 of purine ring of G34, so that two hydrogen bonds can be formed (2.6 Å and 2.7 Å) (Kobayashi et al. 2003). The other two anticodon bases, U35 and A36, also form hydrogen bonds with the synthetase, but all of these interactions are not residue-specific, since their interactions are with main-chain carbonyl groups (Kobayashi et al. 2003).
Figure 3.

M. jannaschii TyrRS conformational changes upon tRNA binding. (A) Anticodon G34 binds to a cleft formed by turns between β7 and β8 and between α12 and α13. The surface of M. jannaschii TyrRS is colored gold. The tRNA anticodon is represented by ball-and-stick; the oxygen atoms are colored red, nitrogen atoms are blue, and bonds/carbon atoms are gray (PDB code 1j1u; Kobayashi et al. 2003). (B) Superimposition of tRNA free and bound M. jannaschii TyrRS’s. The tRNA complex structure (PDB code 1j1u; Kobayashi et al. 2003) is colored blue with the apo enzyme in gray. The local region around the anticodon is visualized here. The conformation of side chains of Phe261 and His283 is altered to form a π-stacking sandwich. The side chain of Asp286 forms two hydrogen bonds with G34 upon tRNA binding. (C) Superimposition of the tRNA acceptor stem-binding site in the wild-type TyrRS complex with tRNATyr and tyrosine (PDB code 1j1u; Kobayashi et al. 2003) (purple) and apo wild-type TyrRS structure (gray). Interacting residues and tRNA are illustrated in ball-and-stick with the oxygen atoms colored red, nitrogen atoms blue, and bonds/carbon atoms in tRNA yellow. Potential hydrogen bonds are represented by a dashed line, with distances labeled in Å.
Changes in the tRNA acceptor-binding region
A comparison of the tRNATyr-Tyr bound (PDB code 1j1u) and unbound M. jannaschii TyrRS structures reveals that residues involved in tRNA acceptor stem-binding (Arg132, Arg174, and Lys175), which are highly conserved throughout different species, undergo only minor conformational changes (Fig. 3C ▶), in contrast to the large rms deviations observed at the other ligand-binding sites (tyrosine substrate-binding pocket, anticodon-binding site). For example, in both bound and free structures, Arg174 is positioned to form two hydrogen bonds with the tRNA identity element C1 (2.6 Å and 3.3 Å). The Lys175 side chain is also unchanged with a possible hydrogen bond (3.4 Å) to G73 (Fig. 3C ▶). The only change is for Arg132, which upon binding of tRNA forms a hydrogen bond (3.1 Å) to the base of G72 (Fig. 3C ▶). However, the B-value of the acceptor stem-binding site (helix α7) is greatly reduced upon tRNA binding, suggesting that binding of the tRNA rigidifies the enzyme structure.
Changes in the tyrosine-binding site
The tyrosine-binding site (Fig. 4A ▶) is located in a deep and narrow pocket (7.5 Å wide and 10.5 Å deep) that is negatively charged with Asp158 at the bottom of the pocket, and Glu36 and Glu172 at the entrance (Fig. 4B ▶). This hydrophilic pocket prevents binding of the structurally similar phenylalanine (note that the binding pocket of the phenylalanyl-tRNA synthetase is dominated by hydrophobic residues) (Goldgur et al. 1997). A structural comparison of all class I aminoacyl RS’s reveals that, despite the conserved Rossmann fold, each enzyme has its own solution for the binding of the main-chain atoms of amino acid substrate with no strictly conserved residues. However, a set of residues are conserved among TyrRS’s from various species, which correspond to Tyr32, Tyr151, Gln155, Asp158, and Gln173 in M. jannaschii TyrRS.
Figure 4.

Tyrosine-binding site in apo M. jannaschii TyrRS. (A) Electron density around the tyrosine-binding pocket in apo M. jannaschii TyrRS. The density shown is from a 2Fo-Fc map contoured at 1.8σ. Only a slice through the active-site region is shown for clarity. (B) Electrostatic distribution around the tyrosine-binding site. Positive potential is blue (≥10 mV), neutral potential (0 mV) is white, and negative potential (≤ −10 mV) is red. (C) Superimposition of tyrosine bound (cyan) and unbound (gray) M. jannaschii TyrRS. The ligand tyrosine is represented by ball and stick with the oxygen atoms colored red, nitrogen atoms blue, and bonds/carbon atoms yellow.
When the apo and complexed structures of wild-type M. jannaschii TyrRS are superimposed (Fig. 4C ▶), no substantial conformational changes are observed in the tyrosine-binding site. Instead almost every helix and β-strand forming the walls of the deep tyrosine-binding pocket undergoes small changes that allow optimal hydrogen-bonding interactions with the ligand. A comparison of the free and bound enzymes shows that all residues involved in tyrosine binding would be out of the hydrogen bonding range with substrate, if their positions remained unchanged between the apo and bound form (Fig. 5A ▶). For example, the side chain of Gln173 repositions ~2.0 Å upon substrate binding to make two hydrogen bonds (2.7 Å and 2.9 Å, respectively) with substrate (Kobayashi et al. 2003). An overall movement of 1.8 Å in helix α8 positions Asp158 within hydrogen bonding distance (2.6 Å) of the bound tyrosine. Similarly, this same helix movement also positions Gln155 to form a hydrogen bond (2.8 Å) with substrate. Finally, the aromatic ring of Tyr151 changes its χ2 by 60° to facilitate binding of tyrosine. These individual conformational changes reveal an overall structural plasticity of the active site that allows the enzyme to optimize interaction of the hydrogen-bonding network with bound tyrosine.
Figure 5.

Amino acid-binding site in wild-type TyrRS and OMeTyr-specific mutant. (A) Hydrophobic interactions and hydrogen bonds between substrate tyrosine and M. jannaschii TyrRS. Hydrogen bonds are represented with dashed lines and labeled with distances in Å, and hydrophobic interactions are indicated by red spiked arcs. The distances in parentheses in red are the distances between the apo enzyme superimposed on the tyrosine ligand position from the bound structure (Kobayashi et al. 2003). (B) Stereo view of the superimposition of wild-type M. jannaschii TyrRS and OMeTyr-specific mutant synthetase to illustrate conformational differences. Apo mutant M. jannaschii TyrRS is colored by its rms deviation (Cα) from the apo wild-type TyrRS structure (gray) with a gradient ranging from low (0.2 Å, green) via medium (1.8 Å, yellow) to high (>4.0 Å, red). (C) Stereo view of OMeTyr-specific mutant synthetase superimposed with wild-type apo M. jannaschii TyrRS (gray) to highlight mutated residues. The mutant enzyme is colored with cyan helices and pink β-strands. The four residues subjected to mutation are represented by ball-and-stick with their oxygen atoms colored red. Bonds/carbon atoms are colored green in the mutant enzyme but blue in wild-type.
Other regions of the protein adjacent to the tyrosine-binding site also undergo conformational changes upon binding of tyrosine. For example, loop 73–84, which is located at the entrance of the tyrosine-binding site, undergoes a substantial conformational change (3.7 Å) when substrate binds. This loop, which connects helix α4 and helix α5, provides a hydrophobic lid over the tyrosine-binding pocket. The conformational change of this loop results from a change in the orientation of helix α4 by ~20° (Fig. 4C ▶). This conformational change may play a role in sequestering the activated amino acid from water during the catalytic reaction. Finally, helix α9 (residues 172–183), which includes Glu172, Gln173, Arg174, Lys175, and His177, has significantly higher B-values in the free versus bound structure. In the bound structure on the inside face of the helix, the side-chain carbonyl of Gln173 forms a hydrogen bond with the amino group of the substrate tyrosine (Kobayashi et al. 2003), whereas residues on the outside face of the helix, Arg174 and Lys175, bind to the tRNA acceptor stem. The special disposition of helix α9, which is highly conserved from M. jannaschii to human, suggests that it may mediate communication between binding of the tRNA to the synthetase and in the positioning of active-site residues to charge tRNA with the amino acid.
The OMeTyr-specific mutant synthetase
The initial TyrRS library used to evolve synthetases with specificity for unnatural amino acids was based on the B. stearothermophilus TyrRS crystal structure (the M. jannaschii structure was not available). Five residues (Tyr34, Asn123, Asp176, Phe177, and Leu180) in the B. stearothermophilus TyrRS active site were identified that were in close proximity to the phenyl ring of substrate tyrosine (L. Wang and P.G. Schultz, unpubl.). Based on sequence homology alignment, corresponding residues in the M. jannaschii TyrRS residues (Tyr32, Glu107, Asp158, Ile159, and Leu162) were randomized to generate the mutant synthetase library. From this library, a synthetase was evolved that was able to incorporate OMeTyr efficiently and selectively (>99%) into proteins. The value of kcat/KM of the mutant synthetase for OMeTyr is ~100-fold higher than that for tyrosine (Wang et al. 2001). This OMeTyr mutant synthetase has the following mutations in the tyrosine binding pocket: Tyr32Gln, Asp158Ala, Leu162Pro, and Glu107Thr.
The overall structures of the apo wild-type M. jannaschii TyrRS and apo OMeTyr-specific mutant synthetase are very similar with only a 0.66 Å rms deviation in Cα (Fig. 5B ▶). The only significant changes are in the KMSKS motif loop 202–209 and in loop 133–143, which is close to the acceptor stem-binding site. Both of these loops are disordered in the apo wild-type structure, but surprisingly traceable in the mutant structure even at the lower resolution of that structure.
KMSKS loop
The KMSKS signature motif is conserved in all class I tRNA synthetases (for review, see Mechulam et al. 1995) and is part of a mobile loop that is involved in ATP binding, initial binding of tRNATyr, and catalysis of aminoacyl-adenylate formation (First and Fersht 1995; Xin et al. 2000).
This motif is located in the middle of loop 202–209, which links the two domains. Loop 202–209 is disordered in most TyrRS structures in the absence of ATP or ATP analogs (Brick et al. 1988; Yang et al. 2002; Kobayashi et al. 2003). Consistent with this observation, the loop is disordered in both apo and complexed wild-type M. jannaschii TyrRS. However, the density for loop 202–209 is traceable in the OMeTyr mutant. A conserved mechanism of aminoacyl adenylation among TyrRS (Yaremchuk et al. 2002), TrpRS (Ilyin et al. 2000), and LeuRS (Cusack et al. 2000) involves a conformational rearrangement in the KMSKS loop, resulting in the repositioning of the essential catalytic lysine (Lys204 in M. jannaschii TyrRS). The conformation of the loop 202–209 in the mutant is very similar to that of the corresponding loop in the T. thermophilus apo structure (PDB code 1h3f) (loop 230–237 in T. thermophilus), which is presumably in a nonproductive conformation. This suggests that the conformation in the apo mutant M. jannaschii structure also represents an inactive conformation.
Loop 133–143
Loop 133–143, which directly follows the acceptor stem-binding site, is ordered in the OMeTyr-specific mutant synthetase, but not in the apo wild-type TyrRS structure. Although not directly involved in tRNA binding (but close to the tRNA binding site), the loop undergoes a large rms deviation (3.1 Å in Cα) upon binding of tRNA. In the OMe-Tyr mutant structure, this loop is ordered, but with higher B-values (61.9 Å2 vs. 38.9 Å2 of the overall protein), suggesting greater flexibility. The catalytic role of this conformational isomerism is unclear, as no potential hydrogen bonds are formed between this loop and the tRNA acceptor stem. A conformational change in this loop could possibly mediate communication between α7, which binds to the tRNA acceptor stem, and α8, which binds to the substrate tyrosine via Tyr151, Gln155, and Asp158.
Active-site mutations
Although four residues in the tyrosine-binding pocket were mutated to change specificity, no large backbone movements were observed. The four active-site mutations are clearly defined from the X-ray crystallographic data of the apo OMeTyr-specific mutant synthetase at 3.0 Å resolution (Fig. 5C ▶). The overall shape of the binding pocket is not significantly changed upon mutation (Fig. 5C ▶). In the wild-type enzyme, Tyr32 and Asp158 are both directly involved in hydrogen bond interactions with the side-chain hydroxyl of the tyrosine ligand (Fig. 5A ▶). Mutation of these two residues in the mutant synthetase results in a loss of these hydrogen bonds. Mutation of Asp158 to the smaller amino acid, Ala, relieves a steric clash of active-site residues with the methyl group of O-methyl tyrosine. Leu162 is also close to the tyrosine side chain (4.3 Å). When this residue is mutated to Pro, the wall of the tyrosine-binding pocket retreats by 3.8 Å, leaving ample space for the additional methyl group of the unnatural amino acid, OMeTyr. This mutation also causes helix α8 to terminate at Tyr161 rather than at Gly163 in the wild type. The side chain of Glu107, however, faces away from the tyrosine-binding site and is ~12.5 Å from the para position of the phenyl ring, which differs from that of its counterpart Asn 123 in B. stearothermophilus. Therefore, mutation of Glu107 to Thr probably plays a minor role if at all in the specific incorporation of OMeTyr. Nevertheless, this outcome could not have been predicted ahead of time from the B. stearothermophilus TyrRS structure. Thus, three of the four mutations in the active site favor binding of the new substrate OMeTyr and disfavor binding of tyrosine. This conclusion is consistent with the selection scheme that was used to evolve this synthetase, which included rounds of positive selection for OMeTyr and negative selection against endogenous amino acids.
Conclusions
Structural studies of tRNA synthetases have revealed diverse effects induced by ligand binding. For some tRNA/ synthetase complexes, these changes are global. For example, in glutamyl- and aspartyl-tRNA synthetases, the anticodon and substrate-binding domains are both rigid. However, the hinge region connecting these two domains twists slightly, which results in the reorientation (~10°) of the domains with respect to each other such that they tilt toward the tRNA upon binding (Rees et al. 2000; Sekine et al. 2003). On the other hand, in other synthetases, ligand binding leads only to local conformational changes. These changes are restricted to areas in close proximity to the ligand-binding sites, yet they can still be dramatic. Yeast arginyl-tRNA synthetase has an rms deviation of 3.6 Å (Cα) upon tRNA binding, but these changes are primarily localized within the catalytic and anticodon-binding regions (Delagoutte et al. 2000).
A comparison of the apo M. jannaschii TyrRS structure with that of TyrRS complexed with tRNATyr and tyrosine reveals that several regions of the synthetase become ordered or change orientation upon ligand binding. These include π-stacking of Phe261 and His283 with G34 of the anticodon, repositioning of Asp286 to hydrogen bond with G43, and cooperative movement of Tyr151, Gln155, Gln173, Asp158, and Tyr32 to optimally bind tyrosine. In addition, it is noteworthy that some regions of the synthetase, which are disordered in wild-type TyrRS, are more ordered in the OMeTyr-specific mutant synthetase. These include the signature KMSKS loop involved in ATP binding and catalysis, and the connecting loop (133–143) between helix α7 and α8. Since only residues in the active site were targeted for mutation in the mutant synthetase, the mobility differences between the wild-type and mutant TyrRS’s suggest that the different domains of the wild-type TyrRS communicate with each other.
Comparison of the wild-type and mutant TyrRS’s also reveals the structural basis whereby the active-site mutations specifically accommodate OMeTyr and disfavor binding of tyrosine. These include eliminating bulky residues to avoid stereochemical clashes with the larger substrate, as well as disruption of hydrogen bonds to the hydroxyl group of tyrosine. Many mutant synthetases have now been evolved to incorporate various tyrosine analogs into proteins. It will be of considerable interest to visualize how a single protein framework can be mutated to accommodate such a variety of ligands. Moreover, structural analysis of these mutant synthetases should lead to improvements in library design for evolving synthetases with new specificity and enhanced activities.
Materials and methods
Materials
Luria broth, 2YT broth, and agar were obtained from Life Technologies. All common buffers and reagents were purchased from Sigma-Aldrich.
Expression and purification
The expression of apo M. jannaschii TyrRS was described previously (Wang et al. 2001). The protein was initially purified by a heat denaturation protocol (Steer and Schimmel 1999). The resulting protein was further purified with a monoQ column (Amersham Pharmacia Biotech) using a gradient of 50 mM to 1 M NaCl at pH 8.0 in 25 mM Tris buffer, followed by chromatography on a Superdex 200 HR 10/30 size-exclusion column (Amersham Pharmacia Biotech). The purity of protein was >96%, as analyzed by silver-stained sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The purified protein was then tested to confirm activity. The plasmid expressing the OMeTyr-specific mutant synthetase was inserted into a pBR322-derived vector (Wang et al. 2001) and transformed into BL21(DE3) Gold cells (Retrogene). The bacterial culture was grown to an absorbance of 0.6 at 595 nm from a freshly streaked plate at 37°C, and then induced with 1 mM IPTG at 37°C for 3 h. Cells were disrupted by sonication in a buffer of 0.5 M NaCl, 50 mM HEPES (pH 7.9), 10% glycerol, 0.1% Triton-100, and the lysate clarified by centrifugation at 20,000g for 50 min. The supernatant was then incubated with 10 mL of nickel affinity beads (QIAGEN) for 3 h. The beads were washed extensively with 0.5 M NaCl, 10 mM imidazole, 25 mM HEPES (pH 7.9), until the base line for the absorbance remained constant. Protein was eluted with a 10 mM to 250 mM imidazole gradient in 0.5 M NaCl, 25 mM HEPES (pH 7.9). Fractions containing protein were detected by SDS-PAGE and pooled. Any contaminants were then removed with an ion-exchange monoQ column using a gradient of 50 mM to 1M NaCl at pH 8.0 in 25 mM Tris buffer. The purity of the protein was assessed by SDS-PAGE and was greater than 95%. The purified mutant protein was dialyzed into a buffer containing 50 mM NaCl and 25 mM Tris (pH 8.0).
Crystallization
The M. jannaschii TyrRS was concentrated to 16 mg/mL with Millipore Ultrafree 15, and crystal screening trays were set up by the vapor diffusion method (Ducruix and Giege 1999) in 2-μL sitting drops. Several conditions produced crystals, but most did not diffract beyond 5 Å. However, structure-quality crystals were eventually grown from 12%–15% PEG4000, 0.2M ammonium sulfate, 0.2M sodium acetate (pH 4.6–5.6), at 4°C after 6 wk as needle clusters. Crystals for the OMeTyr mutant synthetase were grown under similar conditions.
Data collection
An apo wild-type crystal was cryoprotected with 25% glycerol prior to diffraction data collection on a CCD 2 × 2 detector at −179°C at beam line 5.0.2 of the Advanced Light Source (ALS). The data were processed with HKL2000 (Otwinowski and Minor 1997). The crystal was indexed in space group P212121 with two molecules per asymmetric unit, with unit cell dimensions a = 45.12 Å, b = 185.29 Å, and c = 95.48 Å. The crystal is estimated to have a solvent content of 59%, based on a Matthews’ coefficient (Matthews 1968) of 3.0 Å3 Da−1. The Rsym is 6.0% with a data completeness of 99.5% to 2.66 Å resolution. The OMeTyr-specific mutant TyrRS data from a similarly cryoprotected crystal were collected on a CCD 3 × 3 detector at ALS beam line 5.0.2 to 3.0 Å resolution. However, due to the small size of the crystal, the diffraction was weak with a higher Rsym of 14%. The statistics for the data collection and processing for both data sets are summarized in Table 1.
Structure solution and refinement
The structures of apo M. jannaschii TyrRS and the OMeTyr-specific mutant synthetase were determined by MR using the program AMoRe from the CCP4 package (Navaza 1994). TyrRS’s from B. stearothermophilus (PDB code 2ts1; Brick et al. 1988), T. thermophilus (PDB code 1h3f; Yaremchuk et al. 2002), and the catalytic domain of human TyrRS (PDB code 1n3l; Yang et al. 2002) were first used as search models, but no solution could be found by MR. Finally, an MR solution was obtained using the complexed TyrRS (PDB code 1j1u; Kobayashi et al. 2003) as the initial model, excluding the coordinates for tRNA and substrate. Refinement was carried out using CNS (Brünger et al. 1998), with a 5% test set (1097 reflections) excluded for Rfree cross validation (Brünger 1992). Electron density maps (2Fo-Fc and Fo-Fc) were calculated after each cycle of refinement of simulated annealing or individual B-value refinement with CNS. Poorly defined regions of the structure were omitted and then manually rebuilt in O (Jones et al. 1991). Since there are two molecules per asymmetric unit, noncrystallographic symmetry (NCS) was used to restrain the refinement and improve the quality of the maps. NCS restraints were released in the refinement for the more disordered regions of the structure (N and C termini, 133–143 and 254–286, KMSKS motif 202–209). During the last round of refinement, all NCS restraints were released. The refined model for apo M. jannaschii TyrRS was then used in the structure determination for the OMeTyr-specific mutant synthetase. The geometries of the two models were evaluated by Procheck (Laskowski et al. 1993). Lys209 falls into a disallowed region, but is located in the highly flexible loop containing the KMSKS motif. The statistics for the refinement are summarized in Table 1.
The figures presented in this paper were created with Bobscript (Esnouf 1997) and rendered with Raster3D (Merritt and Murphy 1994), except that Figure 3A ▶ was generated with PMV (Sanner 1999) and Figure 5A ▶ with LIGPLOT (Wallace et al. 1995). The coordinates and structure factors for the structures have been deposited in the Protein Data Bank (Berman et al. 2000) with PDB codes 1u7d (wild type) and 1u7x (mutant).
Acknowledgments
This work was supported by NIH grants GM62159 (P.G.S.) and GM38273 (I.A.W.) and a Skaggs predoctoral fellowship (Y.Z.). We also thank the staff of ALS Beamline 5.0.2 for technical support in X-ray data collection.
Abbreviations
TyrRS, tyrosyl-tRNA synthetase
OMeTyr, O-methyl-l-tyrosine
ALS, Advanced Light Source
MR, molecular replacement
CCP4, Collaborative Computational Project 4
rms deviation, root-mean-square deviation
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041239305.
References
- Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., and Bourne, P.E. 2000. The Protein Data Bank. Nucleic Acids Res. 28 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brick, P., Bhat, T.N., and Blow, D. 1988. Structure of tyrosyl-tRNA synthetase refined at 2.3 Å resolution. Interaction of the enzyme with the tyrosyl adenylate intermediate. J. Mol. Biol. 208 83–98. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T. 1992. Free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 355 472–475. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallographic and NMR system: A new software suite for macro-molecular structure determination. Acta Crystallogr. D 54 905–921. [DOI] [PubMed] [Google Scholar]
- Calendar, R. and Berg, P. 1966. Purification and physical characterization of tyrosyl ribonucleic acid synthetases from Escherichia coli and Bacillus subtilis. Biochemistry 5 1681–1690. [DOI] [PubMed] [Google Scholar]
- Cusack, S., Yaremchuk, A., and Tukalo, M. 2000. The 2 Å crystal structure of leucyl-tRNA synthetase and its complex with a leucyl-adenylate analogue. EMBO J. 19 2351–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delagoutte, B., Moras, D., and Cavarelli, J. 2000. tRNA aminoacylation by arginyl-tRNA synthetase: Induced conformations during substrates binding. EMBO J. 19 5599–5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducruix, A. and Giege, R. 1999. Crystallization of nucleic acids and proteins: A practical approach, 2d ed., pp. 130–137. Oxford University Press, New York.
- Esnouf, R.M. 1997. An extensively modified version of Molscript that includes greatly enhanced coloring capabilities. J. Mol. Graph. Model. 15 132–134. [DOI] [PubMed] [Google Scholar]
- First, E.A. and Fersht, A.R. 1995. Analysis of the role of the KMSKS loop in the catalytic mechanism of the tyrosyl-tRNA synthetase using multimutant cycles. Biochemistry 34 5030–5043. [DOI] [PubMed] [Google Scholar]
- Goldgur, Y., Mosyak, L., Reshetnikova, L., Ankilova, V., Lavrik, O., Khodyreva, S., and Safro, M. 1997. The crystal structure of phenylalanyl-tRNA synthetase from Thermus thermophilus complexed with cognate tRNAPhe. Structure 5 59–68. [DOI] [PubMed] [Google Scholar]
- Ilyin, V.A., Temple, B., Hu, M., Li, G., Yin, Y., Vachette, P., and Carter Jr., C.W. 2000. 2.9 Å crystal structure of ligand-free tryptophanyl-tRNA synthetase: Domain movements fragment the adenine nucleotide binding site. Protein Sci. 9 218–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Kobayashi, T., Nureki, O., Ishitani, R., Yaremchuk, A., Tukalo, M., Cusack, S., Sakamoto, K., and Yokoyama, S. 2003. Structural basis for orthogonal tRNA specificities of tyrosyl-tRNA synthetases for genetic code expansion. Nat. Struct. Biol. 10 425–432. [DOI] [PubMed] [Google Scholar]
- Koch, G.L. 1974. Tyrosyl transfer ribonucleic acid synthetase from Bacillus stearothermophilus. Preparation and properties of the crystallizable enzyme. Biochemistry 13 2307–2312. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26 283–291. [Google Scholar]
- Lawrence, M.C. and Colman, P.M. 1993. Shape complementarity at protein/ protein interfaces. J. Mol. Biol. 234 946–950. [DOI] [PubMed] [Google Scholar]
- Matthews, B.W. 1968. Solvent content of protein crystals. J. Mol. Biol. 33 491–497. [DOI] [PubMed] [Google Scholar]
- Mechulam, Y., Meinnel, T., and Blanquet, S. 1995. A family of RNA-binding enzymes. the aminoacyl-tRNA synthetases. Subcell. Biochem. 24 323–376. [DOI] [PubMed] [Google Scholar]
- Merritt, E.A. and Murphy, M.E.P. 1994. Raster3D Version 2.0—A program for photorealistic graphics. Acta Crystallogr. D 50 869–873. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. HKL: Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Rees, B., Webster, G., Delarue, M., Boeglin, M., and Moras, D. 2000. Aspartyl tRNA-synthetase from Escherichia coli: Flexibility and adaptability to the substrates. J. Mol. Biol. 299 1157–1164. [DOI] [PubMed] [Google Scholar]
- Sanner, M.F. 1999. Python: A programming language for software integration and development. J. Mol. Graph. Model. 17 57–61. [PubMed] [Google Scholar]
- Sekine, S., Nureki, O., Dubois, D.Y., Bernier, S., Chenevert, R., Lapointe, J., Vassylyev, D.G., and Yokoyama, S. 2003. ATP binding by glutamyl-tRNA synthetase is switched to the productive mode by tRNA binding. EMBO J. 22 676–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba, K., Motegi, H., and Schimmel, P. 1997. Maintaining genetic code through adaptations of tRNA synthetases to taxonomic domains. Trends Biochem. Sci. 22 453–457. [DOI] [PubMed] [Google Scholar]
- Steer, B.A. and Schimmel, P. 1999. Major anticodon-binding region missing from an archaebacterial tRNA synthetase. J. Biol. Chem. 274 35601–35606. [DOI] [PubMed] [Google Scholar]
- Wallace, A.C., Laskowski, R.A., and Thornton, J.M. 1995. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 8 127–134. [DOI] [PubMed] [Google Scholar]
- Wang, L. and Schultz, P.G. 2001. A general approach for the generation of orthogonal tRNAs. Chem. Biol. 8 883–890. [DOI] [PubMed] [Google Scholar]
- ———. 2002. Expanding the genetic code. Chem. Commun. (Camb). 7 1–11. [DOI] [PubMed] [Google Scholar]
- Wang, L., Magliery, T.J., Liu, D.R., and Schultz, P.G. 2000. A new functional suppressor tRNA/aminoacyl-tRNA synthetase pair for the in vivo incorporation of unnatural amino acids into proteins. J. Am. Chem. Soc. 122 5010–5011. [Google Scholar]
- Wang, L., Brock, A., Herberich, B., and Schultz, P.G. 2001. Expanding the genetic code of Escherichia coli. Science 292 498–500. [DOI] [PubMed] [Google Scholar]
- Xin, Y., Li, W., and First, E.A. 2000. The “KMSKS” motif in tyrosyl-tRNA synthetase participates in the initial binding of tRNA(Tyr). Biochemistry 39 340–347. [DOI] [PubMed] [Google Scholar]
- Yang, X.-L., Skene, R.J., McRee, D.E., and Schimmel, P. 2002. Crystal structure of a human aminoacyl-tRNA synthetase cytokine. Proc. Natl. Acad. Sci. 99 15369–15374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaremchuk, A., Kriklivyi, I., Tukalo, M., and Cusack, S. 2002. Class I tyrosyl-tRNA synthetase has a class II mode of cognate tRNA recognition. EMBO J. 21 3829–3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, D., Vaidehi, N., Goddard, W.A., Danzer, J.F., and Debe, D. 2002. Structure-based design of mutant Methanococcus jannaschii tyrosyl-tRNA synthetase for incorporation of O-methyl-L-tyrosine. Proc. Natl. Acad. Sci. 99 6579–6584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z., Gildersleeve, J., Yang, Y.-Y., Xu, R., Loo, J.A., Uryu, S., Wong, C.-H., and Schultz, P.G. 2004. A new strategy for the synthesis of glyco-proteins. Science 303 371–373. [DOI] [PubMed] [Google Scholar]