

Abstract
Conservation of function is the basic tenet of protein evolution. Conservation of key electrostatic properties is a frequently employed mechanism that leads to conserved function. In a previous report, we identified several conserved electrostatic properties in four protein families and one functionally diverse enzyme superfamily. In this report, we demonstrate the evolutionary and catalytic importance of electrostatic networks in three ubiquitous metabolic enzymes: triosephosphate isomerase, enolase, and transaldolase. Evolutionary importance is demonstrated using phylogenetic motifs (sequence fragments that parallel the overall familial phylogeny). Phylogenetic motifs frequently correspond to both catalytic residues and conserved interactions that fine-tune catalytic residue pKa values. Further, in the case of triosephosphate isomerase, quantitative differences in the catalytic Glu169 pKa values parallel subfamily differentiation. Finally, phylogenetic motifs are shown to structurally cluster around the active sites of eight different TIM-barrel families. Depending upon the mechanistic requisites of each reaction catalyzed, interruptions to the canonical fold may or may not be identified as phylogenetic motifs.
Keywords: protein family evolution, phylogenetic motifs, electrostatic networks, residue pKa, TIM-barrel proteins
Conservation of function is the ultimate evolutionary driving force (Gu 2003). Closely related enzymes generally catalyze the same, or very similar, reactions. Grouping closely related proteins into protein families and superfamilies is a natural extension of this observation. For example, enzymes within a given protein family generally catalyze the same reaction, whereas members of the same superfamily generally catalyze different, albeit related, reactions. From a cursory analysis, however, the similarity within superfamily reactions is not always immediately obvious. For example, members of the enolase superfamily catalyze a diverse array of reactions from throughout the metabolic chart (Babbitt et al. 1996). Despite little global similarity in the reactions catalyzed, the reactions conserve a common mechanistic strategy. All members of the enolase superfamily catalyze reactions that involve the formation of an enolic intermediate by abstraction of an α-carbon proton from a carboxylate substrate (Babbitt et al. 1995). Several other functionally diverse enzyme superfamilies have been identified after defining a common mechanistic strategy (Gerlt and Babbitt 1998).
We recently demonstrated that several conserved electrostatic properties are responsible for maintaining function across four closely related protein families and the functionally diverse enolase superfamily (Livesay et al. 2003). Using pairwise distance probability density functions, conservation within the spatial distribution of charge around active-site regions is demonstrated. In the case of the copper, zinc superoxide dismutase (CuZnSOD) family, the conserved charge distribution leads to qualitatively conserved electrostatic potential maps and quantitatively conserved calculated Brownian dynamics rate constants. Further, phylogenetic trees of only the electrostatically relevant residues within the CuZnSOD and enolase superfamily active sites are shown to reproduce the complete familial tree. This result confirms that conservation of electrostatics is an important mechanism leading to conservation of function.
Subsequently, we reversed the former scenario and later demonstrated that sequence fragments approximating the complete familial tree (termed phylogenetic motifs) represent good functional-site predictions (La et al. 2005). We briefly highlight the key results of our previous report here (see Materials and Methods for a technical description of the approach). Across a structurally and functionally diverse protein family data set, phylogenetic motifs (PMs) consistently correspond to functional sites defined by surface loops, active site clefts, and partially buried regions interacting with prosthetic groups. In all instances, the functional importance of the identified PMs is verified through structural comparisons. PMs structurally cluster around known functionality despite little overall sequence proximity. Similarity between traditional and phylogenetic motifs is generally observed. However, there are instances when PMs are not (overall) well conserved in sequence. This point is intriguing because it implies that PMs are able to functionally annotate regions where traditional motifs fail. Tree significance, especially in the PM regions, has also been demonstrated using bootstrapping. The PM approach is similar in spirit to the evolutionary trace (Lichtarge et al. 1996, 1997, 2003) method, and as expected, the results from the two methods are consistent. Ostensibly, PMs identify sequence clusters of evolutionary trace residues, which generally improves functional-site prediction. Additionally, the general use of the evolutionary trace method is to map the tree-determinant positions onto protein structure (Yao et al. 2003). However, no structural information is used in PM identification, making PMs a valuable postgenomic technology.
Defining what constitutes a functional site is not trivial. In our previous work (La et al. 2005), this determination was made simply through structural proximity to known catalytic residues and substrate binding sites. As discussed previously (La et al. 2005), the catalytic Glu of triosephosphate isomerase and all residues interacting with the substrate analog correspond to PM residues. Additionally, enolase PMs are structurally clustered at the active site, with regions from both the triosephosphate isomerase (TIM)-barrel and N-terminal domains predicted as PMs.
The aim of this report is to delve deeper into the catalytic specifics of three TIM-barrel families. We demonstrate congruence between PM predictions and calculated electrostatic interactions within active site residues. In both the triosephosphate isomerase and enolase examples, the catalytic residues and many of the electrostatic interactions responsible for maintaining functional pKa values correspond to PMs. Through stabilizing and destabilizing interactions, the electrostatic interactions fine-tune the catalytic pKa values. In the case of triosephosphate isomerase, subfamily phylogenetic differences parallel quantitative differences in the calculated pKa values. In the case of transaldolase, the catalytic Lys, which forms a Schiff base, is not identified as a PM residue. Nevertheless, four of the five strongest interactions with it are, again confirming the evolutionary importance of electrostatic networks vis-à-vis conservation of function. Further, we show in this report that PMs are structurally clustered at the active sites of eight different TIM-barrel protein families.
Results and Discussion
Triosephosphate isomerase
The TIM-barrel fold is the most ubiquitous in nature (Wierenga 2001). All TIM-barrel active sites are defined by loop regions at the C-terminal end of the α/β barrel. Compared to the enzyme’s core, active-site loops are hypermutable without affecting the integrity of the fold. Therefore, evolutionary selected mutations within the active-site loops largely depend on the mechanistic requisites of each reaction catalyzed. This architecture is a classic example of a molecular scaffold upon which a wide variety of enzymes can be based (for an excellent review, see Nagano et al. 2002). In fact, TIM barrels are known to span five of the six enzyme commission (EC) classifications. Despite global conservation of the active site at the C-terminal end of the barrel, the exact position and identity of the catalytic residue(s) are variable. Most TIM barrels are multimeric with large, well defined protein-protein interfaces. Frequently, the canonical TIM-barrel fold is interrupted by inserted domains that expand the catalytic possibilities of the enzyme. The enolase superfamily is one such example. In this case, a globular α+β N-terminal domain provides several additional substrate binding interactions. TIM-barrel sequences are not as conserved as one might expect, based on their remarkably similar fold topologies (Nagano et al. 2002). In fact, the similarity between most interfamily TIM-barrel proteins is firmly within the “twilight zone” (Chung and Subbiah 1996). The observed sequence similarity, or dissimilarity for that matter, has led to a debate regarding TIM-barrel evolution. Whether TIM barrels have resulted from convergent or divergent evolution remains an open question; however, the general consensus (Reardon and Farber 1995; Copley and Bork 2000) is that TIM barrels are divergently evolved from some ancestral protein.
Triosephosphate isomerase (TIM) is the namesake of the TIM-barrel fold because it was the first example in which the fold was observed (Wierenga 2001). TIM is a ubiquitous glycolytic enzyme that interconverts dihydroxyacetone phosphate (DHAP) and glyceraldehyde-3-phosphate. Glu169 (using a common sequence alignment numbering scheme throughout) acts as a general base (Knowles 1991) that first abstracts a proton from the α-carbon of DHAP, and later abstracts a proton from the α-hydroxyl group of the enediol intermediate (Fig. 1 ▶). Polarization of the (α-, β-) carbonyl group, followed by stabilization of the oxyanion in the (forward, reverse) reaction by an oxyanion hole (Kursula et al. 2001) makes breaking the C-H bond energetically feasible. The residues responsible for polarization in the forward and reverse reaction are Lys11 and His97, and Asn9 and His97, respectively. Subtle rearrangements of the catalytic residues and substrate along the reaction pathway have been identified (Kursula et al. 2001). Further conformational changes occur within loop 6 of the protein (Joseph et al. 1990; Wierenga et al. 1992; Rozovsky and McDermott 2001; Rozovsky et al. 2001). On substrate binding, the flexible “lid” (loop 6) closes over the active site. Despite these conformational changes, the reaction catalyzed by TIM is very fast; in fact it approaches the diffusion limit (Stroppolo et al. 2001). Our previous report (La et al. 2005) demonstrates that all electrostatic (H-bond and salt bridge) interactions between TIM and substrate, as well as the flexible “lid,” are identified as PMs. Furthermore, the best-scoring PM covers the entire Prosite (Hulo et al. 2004) definition of the family. In this report we demonstrate that PMs also identify most of the conserved electrostatic interactions that maintain the catalytic pKa value of Glu169.
Figure 1.

The TIM reaction cycle (Kursula et al. 2001). Residue numbering reflects our multiple sequence alignment. Two different oxyanion holes stabilize the intermediate anionic oxygen. Along the reaction pathway, the catalytic Glu alternates between a general base and acid. The evolutionary importance of the catalytic Lys11, His97, and Glu169 is demonstrated as all correspond to phylogenetic motif residues. Asn9 is not predicted as a phylogenetic motif residue, but is immediately adjacent to one. Most of the residues that mediate the pKa value of Glu169 also correspond to phylogenetic motifs.
The TIM family is largely composed of three distinct subfamilies (Fig. 2A ▶). Sequence fragments with two or three positions (from a window width = 5) that approximate the complete tree are identified as PMs. The remaining positions within those fragments are generally well conserved, which leads to the observed similarity between traditional and phylogenetic motifs. Very few highly variable positions within high scoring windows are observed. Sequence logos (Crooks et al. 2004), shown in Figure 3 ▶, highlight this point. In addition to the catalytic Glu169, two of the three oxyanion hole residues correspond to PM residues; these three residues are 100% conserved within the multiple sequence alignment. The third oxyanion hole residue (Asn9), which is conserved better than 90% in the sequence alignment, is immediately adjacent to the first PM (the structure alignment is shown in Fig. 4A ▶). Because Asn9 is so well conserved, it fails to contribute any new phylogenetic information. This is why, in this case, the conserved position occurs just outside the identified PM. In other instances, conserved positions frequently occur within PMs because they are between tree-determinant positions.
Figure 2.

Unrooted phylogenetic trees of the (A) triosephosphate isomerase and (B) enolase families. As highlighted, the triosephosphate isomerase alignment can be divided into three different subfamilies, whereas the enolase alignment can be divided into two (plus two outlier sequences). Triosephosphate isomerase subfamily #1 is demonstrated to have several distinguishing electrostatic features compared to subfamilies #2 and #3. In spite of these subtle, yet significant differences, the catalytic mechanism of the family is expected to be conserved. Sequences with known structure are indicated by the arrows. The order of structures (from top to bottom) is the same as in Table 1 (triosephosphate isomerase) and Table 3 (enolase).
Figure 3.

Sequence logo (Crooks et al. 2004) representations of (A) the eight triosephosphate isomerase, (B) the 12 enolase, and (C) the seven transaldolase phylogenetic motifs. In all cases, the numbering is that of the multiple sequence alignment used during phylogenetic motif identification (not to be confused with the alignments shown in Fig. 4 ▶). Most of the conserved electrostatic interactions calculated correspond to invariant (or nearly so) positions within the alignment. In these instances, proximal alignment positions reproduce the overall tree. However, there are a few instances (e.g., Asp34, which is strongly interacting with the catalytic Lys138 of transaldolase) where significant familial variability within the electrostatic network is observed.
Figure 4.

(A) Sequence alignment of the 12 triosephosphate isomerase structures investigated. The identified phylogenetic motifs, indicated by the black line above the first sequence, frequently correspond to those residues that are generally responsible for defining the catalytic Glu (highlighted in bold) pKa values. The three oxyanion hole residues are also highlighted in bold. Stabilizing interactions, which lower the pKa value, are colored light gray, whereas destabilizing interactions are dark gray. The order (top to bottom) of the proteins in the alignment is the same as in Figure 2A ▶ and Table 1. (B) Sequence alignment of the four enolase structures investigated. Again, phylogenetic motifs generally correspond to those residues that are responsible for the extreme pKa values in Glu211, Lys357, and Lys408 (highlighted in bold). The pKa value of His164 (also bold), which has also been suggested as a catalytic residue, is not as shifted as the other three potential catalytic candidates. Residues involved in the electrostatic network stabilizing the charged forms of the above residues are colored light gray. Inclusion in the electrostatic network is defined by a single pairwise interaction with one of the four residues above that is greater than ±1.0 kcal/mol or more than two interactions greater than ±0.5 kcal/mol. The order (top to bottom) of the proteins in the alignment is the same as in Figure 2B ▶ and Table 3.
As implied in Figure 1 ▶, a dynamic pKa of Glu169 is necessary for catalysis to occur. At the beginning of the reaction cycle, Glu169 must be deprotonated (i.e., a low pKa value) in order for it to act as a general base. However, if the pKa is too low, then it is unlikely it will be able to accept a proton. Next, Glu169 must give up its proton to form the enediol intermediate. This acid/base cycle is repeated in the second half of the mechanism, finally resulting in G3P formation. Calculated pKa values of the 12 apo and seven substrate-bound structures are provided in Table 1. The Glu169 pKa values in the apo structures can be clustered into two groups (one from subfamily #1 and one from subfamilies #2 and #3). The pKa of the P. woesei catalytic residue is significantly higher (2.37) than that of the remaining structures (−1 to +1). Despite the quantitative difference in pKa values, differences in the percent deprotonated (calculated using the Henderson-Hasselbach equation at optimal growth pH) are marginal. For example, the P. woesei ortholog is calculated to be 99.98% deprotonated, whereas the S. cerevisiae ortholog is 100% deprotonated, meaning that a negatively charged Glu169 is ensured at the beginning of each reaction cycle.
Table 1.
Calculated pKa values of the TIM catalytic Glu residue
| Structure | Organism | pKa (apo)a | pKa (w/substrate)b |
| Subfamily #1 | |||
| 1hg3 | P. woesei | 2.37 | 9.07 |
| Subfamily #2 | |||
| 1b9b | T. maritima | −0.32 | n/a |
| 1btm | B. stearothermophilus | 0.51 | 7.12 |
| Subfamily #3 | |||
| 1lyx | P. falciparum | −0.94 | 7.23 |
| 1mo0 | C. elegans | −0.04 | n/a |
| 2ypi | S. cerevisiae | 0.77 | 8.00 |
| 1m6j | E. histolytica | 0.47 | n/a |
| 1n55 | L. mexicana | 0.09 | 8.05 |
| 1tcd | T. cruzi | 0.55 | n/a |
| 1kv5 | T. brucei | 0.38 | 7.45 |
| 1tre | E. coli | 0.72 | n/a |
| 1aw1 | V. marinus | 0.97 | 7.75 |
The model (aqueous) pKa value of Glu is 4.40.
a The homogeneity of pKa values from the true and computationally plucked apostructures leads us to conclude that significant induced fit conformational changes do not occur.
b All substrates are 2-phosphoacetic acid.
Eight PMs are identified within the TIM family (Table 2). Figure 4A ▶ provides the sequence alignment of the 12 TIM structures investigated; the identified PMs are highlighted. Titrating residues calculated to be strongly (more than ±0.5 kcal/mol) electrostatically interacting with the catalytic Glu are also indicated. The electrostatic calculations identify four conserved stabilizing and four conserved destabilizing interactions. Seven of the eight interactions correspond to PM residues; the remaining residue is weakly stabilizing. The average interaction energy of the three stabilizing PM interactions is −0.90, −1.95, and −0.70 kcal/mol, whereas the average of the non-PM interaction is −0.76 kcal/mol. Because the catalytic Glu is exposed on the surface of the protein (making desolvation effects irrelevant), the residues electrostatically interacting with it are uniquely responsible for keeping the pKa values so low. These interactions are generally conserved throughout all 12 structures. As expected, the most common and striking differences occur in the P. woesei ortholog. The observed differences between the P. woesei structure and the others are consistent with differences seen in the complete familial alignment. Figure 5A ▶ provides a structural representation of these results.
Table 2.
Sequence families and structures investigated in this work
| Protein family | COG # | # Seqa | PSZb | # PMsc | Structure(s) investigated |
| Dihydroorotase | 0044 | 69 | −1.8 | 7 | 1j79 |
| Enolase | 0148 | 73 | −1.8 | 12 | (see Table 3) |
| Triosephosphate isomerase | 0149 | 69 | −1.5 | 8 | (see Table 1) |
| Transaldolase | 0176 | 64 | −1.5 | 7 | 1onr |
| Fructose-bisP aldolase | 0191 | 71 | −2.0 | 6 | 1dos |
| Deoxyribose-P aldolase | 0274 | 44 | −2.0 | 6 | 1rvg |
| Thiamine-P synthetase | 0352 | 60 | −1.5 | 3 | 2tps |
| Methylenetetrahydrofolate reductase | 0685 | 51 | −2.0 | 6 | 1b5t |
a Number of sequences in the alignment.
b Phylogenetic similarity z-score threshold used in identification of the phylogenetic motifs.
c Number of phylogenetic motifs identified.
Figure 5.

Phylogenetic motifs (left) and electrostatic networks (right) of (A) triosephosphate isomerase, (B) enolase, and (C) transaldolase. (Left) Spheres represent phylogenetic motif α-carbons. In all cases, the identified phylogenetic motifs are structurally clustered at the C-terminal end of the barrel. (Right) All residues implicated in the functional electrostatic networks are indicated. Positions included here are based on a simple majority from the multiple sequence alignments in Figure 5 ▶. Catalytic residues are indicated by an asterisk; nonphylogenetic motif residues are underlined. For example, the catalytic residue of transaldolase (Lys133) is not part of any identified phylogenetic motif. Residue numbering is the same as in Figure 4 ▶.
Taken together, the PM and electrostatic results indicate subtle evolutionary variability within the catalytic residues of TIM. Regardless of the observed pKa value differences, it is unlikely that catalysis and/or reaction rates are substantially affected, because Glu169 is essentially 100% deprotonated in all cases. However, such a low pKa value conflicts with the later steps of the reaction. In the second and fourth steps of the reaction, the Glu169 must become protonated. The calculated pKa values are so small that their ability to become protonated is negligible. This quandary is resolved upon substrate binding, which shifts all 12 Glu169 pKa values similarly to the above seven, making protonation feasible. Less subfamily discrimination is observed in the calculated pKa values of the substrate-bound structures. Therefore, the low pKa value within the apo structures, in spite of the observed variability, ensures that the catalytic Glu is completely deprotonated, which is necessary for the reaction to begin. On substrate binding, the pKa is raised such that protonation becomes energetically feasible. In the case of S. cerevisiae, the catalytic Glu goes from 100% deprotonated to 96.94% protonated. Although not explicitly modeled here, the conformational rearrangements within the active site (Kursula et al. 2001) are expected to continually shift the pKa values, as needed, throughout the reaction pathway.
The electrostatic interactions highlighted in Figures 4A ▶ and 5A ▶ are from the apo structures. It should be pointed out that quantitatively similar pairwise values are calculated for the substrate-bound structures. In fact, the correlation coefficient between corresponding Glu169:X pairs, where X equals all other residues, in the apo and substrate-bound structures is greater than 0.9. Due to the technical manner in which the multiple-site titration procedure calculates pKa values, this initially surprising result should actually be expected. First, a so-called intrinsic pKa is calculated that accounts for solvent accessibility and neutral dipoles (note: substrate binding does not appreciably affect Glu169 accessibility). Next, the apparent pKa is calculated from the intrinsic value plus all pairwise electrostatic effects. Therefore, the electrostatic interaction, Φ, between Glu169 and X is not influenced by the substrate because it is assumed to be neutral when ΦGlu169:X is calculated, meaning that ΦGlu169:substrate is the only significant effect leading to the large pKa shift of Glu169.
All-to-all phylogenetic comparisons of TIM sequence windows reveal interesting results (Fig. 6 ▶). As expected, high-similarity regions correspond to PMs, only this time they are identified without recourse to complete familial tree comparisons. This result highlights intrafamily co-evolution within functional portions of the protein. Of course, a robust evolutionary description of any family should include both PM and non-PM regions. Nevertheless, the importance of conservation of function in protein family evolution is confirmed once more. Further, many conserved functionally important electrostatic interactions correspond to high-similarity regions. For example, the interactions mediating the pKa value of Glu169 are in the most phylogenetically similar regions. Many other conserved electrostatic interactions do not correspond to PMs. However, most of these interactions are structural, not catalytic. Note: In this study we define catalytic residues as the ones involved in the discussed electrostatic networks.
Figure 6.
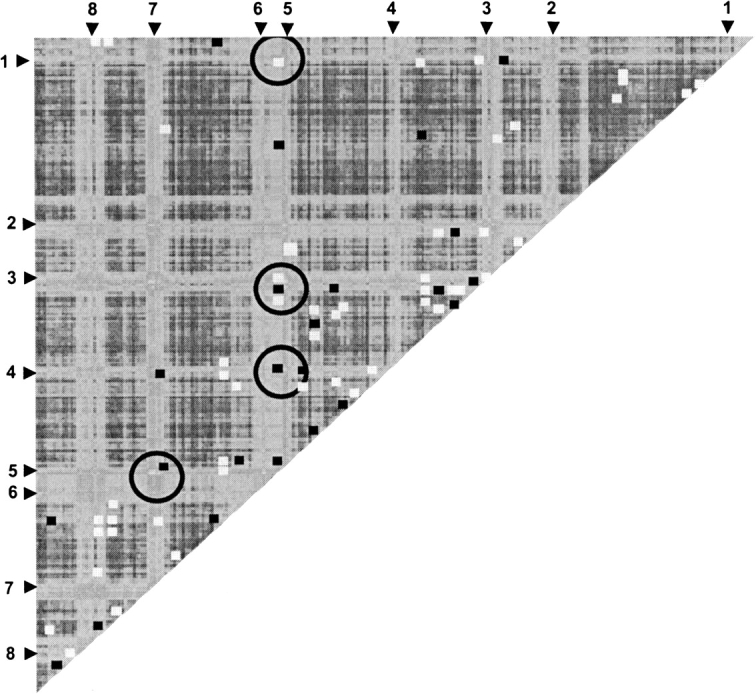
The all-to-all pairwise analysis of sequence window phylogenetic similarity is plotted. Lighter colors indicate higher similarity; darker colors indicate less. The identified phylogenetic motifs are numbered. Additionally, all strongly (greater than ±0.5 kcal/mol) interacting pairwise electrostatic interactions are laid on top of the phylogenetic comparisons. Stabilizing interactions are colored white; destabilizing interactions are gray. Many of the catalytic electrostatic network interactions are predicted by the phylogenetic comparisons. On the other hand, there are many electrostatic interactions, largely involved in stabilization of the structure, that are not predicted. Circles indicate the four phylogenetic motifs strongly interacting with Glu169 (see Fig. 5A ▶).
Enolase
Enolase, also a ubiquitous glycolytic enzyme, catalyzes the penultimate reaction of the pathway. Enolase catalyzes the reversible dehydration reaction converting 2-phosphoglycerate (2PG) to phosphenolpyruvate. As discussed above, enolase is a multidomain TIM-barrel protein. Both the TIM-barrel and N-terminal domains contribute active-site residues. Catalysis requires a general base to abstract the α-carbon proton from 2PG. In order for the reaction to proceed, several divalent metal ions (generally Mg2+ or Mn2+) are required at the active site (Wold and Ballou 1957), presumably to stabilize the carbanion intermediate. The catalytic residues of enolase have not been unequivocally determined; however, the conserved Lys357 (again using alignment numbering) is a likely candidate (Babbitt et al. 1996). In the forward reaction, Glu216 is thought to provide a proton to the leaving hydroxyl group (Cohn et al. 1970). Several other conserved acids are also present at the active site. Experimental profiles for Mg2+ activation led Vinarov and Nowak (1998) to refute the Lys357/Glu216 catalytic pair hypothesis. Their results suggest that Lys408 and His164 are the catalytic pair. Whether or not His164 is a catalytic residue, its functional importance is confirmed by the H164A mutation, which has 0.01% of wild-type activity (Vinarov and Nowak 1999).
Figure 2B ▶ indicates that the enolase family can be roughly divided into two subfamilies (plus two outlier sequences). Two structures per subfamily are currently available. Unlike TIM, where conserved subfamily differences in the pKa value of Glu169 are calculated, no clustering of the electrostatic properties is observed. Quantitative pKa values are highly protein-dependent and cannot be grouped based on subfamily. Rather, a large electrostatic network is conserved in all enolase structures, which results in qualitatively similar pKa values across the whole family. As before, many of the conserved electrostatic interactions that make up this functional network correspond to PMs.
Twelve enolase PMs are identified, which is roughly proportional to the number found in TIM after normalizing for alignment length. Based on our previous and ongoing studies (results not included), we have determined this to be a weakly consistent trend. Future large-scale analyses will attempt to quantify this qualitative observation. All four of the active-site residues discussed above are predicted as PM residues. The pKa values of Glu211, Lys357, and Lys408 are drastically shifted from their aqueous values (Table 3). The extent of the shifts highlights their functional importance (Elcock 2001). The extreme pKa values of these residues are stabilized by a conserved electrostatic network. Figures 4B ▶ and 5B ▶ highlight several conserved interactions within the network. A majority of the enolase electrostatic network residues are also identified as PMs, again confirming the evolutionary importance of catalytic electrostatic networks.
Table 3.
Calculated pKa values of the two potential enolase acid/base pairs
| Structure | Organism | His164/Lys408 | Glu211/Lys357 |
| Subfamily #1 | |||
| 1oep | T. brucei | 8.22/20.19 | −3.64/30.14 |
| 2one | S. cerevisiae | 3.63/28.66 | −8.29/21.37 |
| Subfamily #2 | |||
| 1iyx | E. hirae | 8.12/23.96 | −0.65/16.72 |
| 1e9i | E. coli | 4.71/27.11 | −7.48/21.84 |
The model (aqueous) pKa values of His, Lys, and Glu are 6.30, 10.40, and 4.40, respectively.
Transaldolase
In many cells, a constant supply of biosynthetic reducing power (in the form of NADPH) is provided by the pentose phosphate pathway (Wood 1986). Additionally, the pentose phosphate pathway can provide ribose-5-phosphate for nucleic acid biosynthesis and several glycolytic intermediates. NADPH is provided in the first (oxidative) half of the pathway, whereas transaldolase (TA) and transketolase provide a reversible link to glycolysis (Jia et al. 1996) in the nonoxidative portion of the pathway. TA catalyzes a three-carbon transfer from sedoheptulose-7-phosphate (S7P) to glyceraldehydes-3-phosphate. The products of this TA-catalyzed reaction are fructose-6-phosphate and erythrose-4-phosphate. The TA mechanism involves a nucleophilic attack of a deprotonated Lys (Lys128 in our alignment) on the carbonyl carbon of S7P, forming a Schiff base intermediate (Jia et al. 1997). The calculated pKa value of Lys128 (11.65) is approximately the model value. However, the calculated pKa value indicates that the deprotonated form of Lys128 is negligible at physiological pH. Presumably, substrate binding lowers the pKa such that the nucleophilic attack can occur.
Unlike TIM and enolase, the catalytic Lys of TA is not predicted as a PM. While not identified as a phylogenetic motif residue, the evolutionary importance of the stretch of residues surrounding Lys128 is confirmed. Lys128 is in the middle of a traditional motif; in fact, this stretch of residues around the catalytic residue is one of two Prosite (Hulo et al. 2004) definitions for the family. While the active-site motif does possess some variability, the variability is too random for a PM to be identified. The second TA Prosite definition is entirely covered by a PM. Conversely, most of the residues electrostatically interacting with Lys128 are identified as PMs. Seven PMs are identified in TA (Table 1). Five significant electrostatic interactions are calculated between Lys138 and the remaining residues (see Fig. 5C ▶). The calculated ΦLys138:X are listed in Table 4. Four of the five interactions correspond to PMs, including Asp34, which is one of the two most stabilizing interactions calculated. Unlike many PMs, the Asp34 PM is not also a traditional motif. The same is true for the less stabilizing, yet significant Asp16 interaction. From these and other (data not shown) traditional versus phylogenetic motif comparisons, we conclude that future efforts attempting to predict functional interactions from sequence alone (compared to our goal here of simply demonstrating correspondence) should employ various sequence feature identification strategies. These results are in line with the recent review by Jones and Thornton (2004) that classifies functional-site prediction strategies into two groups, one based on sequence conservation and the second based on feature identification.
Table 4.
Significant Lys128:X pairwise electrostatic interactions within transaldolase
| X = | Φ Lys128:Xa | Phylogenetic motif | Traditional motif b |
| Asp16 | −1.54 | ✓ | |
| Asp34 | −0.55 | ✓ | |
| Glu97 | −1.81 | ✓ | ✓ |
| Tyr129 | −0.56 | ✓ | |
| Lys128 | n/a | ✓ | |
| Arg177 | +0.72 | ✓ | ✓ |
Defined as greater than ±0.5 kcal/mol.
a Units in kcal/mol.
b As calculated by MINER using false positive expectations. See La et al. (2004) for a description of the approach.
Structural clustering of TIM-barrel phylogenetic motifs
Juxtaposed to our specific sequence/structure/function comparisons above, we also identified PMs in five other TIM-barrel protein families. In all cases, PMs are structurally clustered around the active-site region (Fig. 7 ▶). For the most part, PMs are solvent-exposed. However, in cases where substrates intercalate deeper into the core, PMs correspond to more buried regions as well. In all cases, defining PMs as functional is consistent with the structural information. Interestingly, many secondary structure elements (vs. random coil loops) are included within the PMs. Many (68%) PMs partially span the C-terminal end of the β-strands. A few even span the entire β-strand. PMs spanning α-helical regions are less common, yet still occur appreciably (36%). Only 13% of the identified PMs are segregated to only coil regions. Across the data set, PMs partially span all eight β-strands. PMs are most common in strands β1, β6, and β7, occurring four times. Conversely, PMs are least common in strand β3, where they occur only twice.
Figure 7.

Phylogenetic motifs of the five remaining TIM-barrel proteins investigated: (A) dihydroorotase, (B) methylenetetrahydrofolate reductase, (C) thiamine-phosphate synthetase, (D) deoxyribose-phosphate aldolase, and (E) fructose-bisphosphate aldolase. Spheres represent phylogenetic motif α-carbons. In all cases, the identified phylogenetic motifs are structurally clustered at the C-terminal end of the barrel. In some cases, extra domains, which may or may not be identified as phylogenetic motifs, interrupt the canonical fold.
Conclusions
The basic principle of protein evolution is conservation of function. Function is defined by the structural properties of a particular enzyme, which is encoded in sequence. We present the evolutionary variability within the sequence/ structure/function relationships of three important metabolic enzymes (triosephosphate isomerase, enolase, and transaldolase), all of which are TIM-barrel proteins. In the case of triosephosphate isomerase, quantitative differences in the pKa values of the catalytic Glu parallel subfamily differentiation. Further, phylogenetic motifs are shown to correspond to active-site electrostatic network residues within all three families. Finally, PMs are shown to structurally cluster around the active sites of eight different TIM-barrel families.
Materials and methods
Phylogenetic motifs
PMs are identified using a sliding sequence window algorithm to comprehensively evaluate the phylogenetic similarity between each window and the complete alignment. An input alignment is parsed into a series of windows (width = 5), which was previously demonstrated as ideal (La et al. 2005). All sequences (see Table 1) are taken from the most recent version of the clusters of orthologous groups (COG) database (Tatusov et al. 2003). A phylogenetic tree clusters each sequence fragment within the window. Similarity between the window and complete familial tree is quantified by the partition metric algorithm (Penny and Hendy 1985). The partition metric simply counts the number of topological differences between the two trees, meaning the smaller the partition metric score is, the greater the tree similarity. Multiple sequence alignments are calculated using CLUSTALW (Thompson et al. 1994). CLUSTALW is not always the best alignment method, especially in cases with appreciable sequence diversity. However, this is not a problem here due to the similarity of the sequences within each COG.
Phylogenetic trees are calculated using the distance-based algorithm within CLUSTALW. Due to the number of tree calculations required, distance-based trees are used to ensure computational efficiency. For example, in the case of TIM, a medium-sized protein, over 250 trees must be calculated. Additionally, as Kuhner and Felsenstein (1994) pointed out, distance-based approaches actually outperform maximum-likelihood approaches on short sequences. Phylogenetic similarity is quantified using z-scores calculated from the raw partition metric distribution. Plotting the phylogenetic similarity z-score (PSZ) against window number facilitates sequence analysis (Fig. 8 ▶). After all tree comparisons are made, the PSZ threshold can be adjusted to alter what constitutes a “hit.” The threshold can be raised or lowered to be more accommodating or stringent, respectively. Our previous report (La et al. 2005) suggested that PSZ thresholds between −1.5 and −2.0 are ideal. PSZ thresholds used here are given in Table 1. All overlapping windows scoring below the PSZ threshold are grouped as a single PM. MINER, our implementation of the PM identification algorithm, is freely available online at http://www.pmap.csupomona.edu/MINER/. Source code is available upon request.
Figure 8.

Plotting phylogenetic similarity z-scores (PSZ) vs. window number facilitates sequence comparison. Triosephosphate isomerase is shown as an example case. Phylogenetic motifs are defined as all overlapping sequence windows scoring past the PSZ threshold (dashed line), which in this case equals −1.5. Here, 22 (of 252) sequence windows score past the PSZ threshold. Grouping the overlapping windows identifies eight phylogenetic motifs.
Note that six triosephosphate isomerase PMs were identified in our previous study (La et al. 2005). Here, eight triosephosphate isomerase PMs were identified. The first difference arises from splitting a previously identified PM into two. In the second, a window that previously scored just shy of the PSZ threshold, now scores just past it. These differences occur because of two subtle differences in our PM identification strategy. First, and most important, highly (more than 50%) gapped alignment positions are purged from the alignment. Previously, we simply purged highly gapped windows. Second, a newer version of CLUSTALW is used. The second difference underscores the rather arbitrary nature of the PSZ threshold. We are currently using hierarchical clustering techniques to automate PSZ threshold determination (without any human subjectivity). This implementation should be completed in the next year and will be added to MINER when finished.
Continuum electrostatic calculations
Residue pKa values and intramolecular electrostatic interactions were calculated using the University of Houston Brownian Dynamics (UHBD) suite of programs (Madura et al. 1995). UHBD calculates pKa values using the single site titration procedure described by Gilson (1993) and Antosiewicz et al. (1994). Due to the number of Poisson-Boltzmann calculations required when calculating pKa values, the linear Poisson-Boltzmann equation is calculated using the Choleski preconditioned conjugate gradient method (Gibas and Subramaniam 1996; Livesay et al. 1999, 2003). The protein is centered on a 65 × 65 × 65 grid with each grid unit equaling 1.5 Å. Adaptive grids focus each grid unit to 1.2, 0.75, and 0.25 Å. A solvent dielectric constant of 80 and an interior protein dielectric of 20, which is best for reproducing experimental pKa values (Antosiewicz et al. 1996; Gibas and Subramaniam 1996), are used. Protein partial charges are taken from the CHARMM parameter set (Brooks et al. 1983) and radii from the optimized potentials for liquid systems (Jorgensen and Tirado-Rives 1988). In all cases, the temperature is 298 K and the ionic strength is 0.15 M.
Protein structures were prepared for the pKa calculation in a manner similar to that of previous reports (Livesay et al. 1999, 2003; Torrez et al. 2003). Currently, there are 15 triosephosphate isomerase orthologs within the PDB (Berman et al. 2000). Only 12 structures were investigated here, as the two mammalian and one chimeric structure are not representative of the COG database. Figure 2A ▶ indicates that the structural data set is representative of the sequences used in PM detection. Four microbial enolase and one microbial transaldolase structures were also investigated. A human transaldolase and a lobster enolase structure were excluded for the same reason as above. For computational efficiency, only monomers were investigated. Table 1 indicates all protein structures investigated in this work. Incomplete residues were corrected using the systematic rotamer search within MOE. [The Molecular Operating Environment (MOE) is a commercial implementation of many computational biology algorithms and tools. MOE is a trademark of Chemical Computing Group, Toronto, Canada.] Hydrogens were also added using MOE.
Seven of the 12 triosephosphate isomerase structures have a bound substrate analog. In all cases, the substrate is 2-phospho-acetic acid. Both apo and substrate-bound structures were investigated here. Apo structures of proteins with bound substrates were generated by simply removing the substrate coordinates from the PDB file. This approach for generating apo structures can potentially lead to incorrectly calculated pKa values. For example, drastic differences between the open and closed (both without substrate) forms of monoclonal antibody NC6.8, which is specific for a guanidiniumacetic acid derivative, were demonstrated previously (Livesay et al. 1999). However, no distinction within the calculated pKa values between the true and the deleted coordinate apo structures was observed (see Table 2). Due to the homogeneity of the results, we conclude that induced-fit conformational changes are not a concern. In the enolase and transaldolase cases, only apo structures were investigated.
Acknowledgments
We thank Dr. Shankar Subramaniam (University of California, San Diego) for several discussions relating to the phylogenetic and electrostatic methods employed here. We also thank Dr. Patrick Mobley and Dr. Sean Liu (both California State Polytechnic University, Pomona) for discussions concerning mechanistic issues of the enzyme catalyzed reactions. Patrick Mobley is also thanked for proofreading the manuscript. This work was partially supported by an American Chemical Society Petroleum Research Fund grant (36848-GB4), NSF MRI-grant (0321333), and a supercomputer allocation (MCB00018N) from the National Center for Supercomputing Applications to D.R.L.
Abbreviations
CuZnSOD, copper, zinc superoxide dismutase
PM, phylogenetic motif
COG, cluster of orthologous groups
TIM, triosephosphate isomerase
PSZ, phylogenetic similarity z-score
UHBD, University of Houston Brownian dynamics
PDB, Protein Data Bank
DHAP, dihydroxy-acetone phosphate
2PG, 2-phosphoglycerate
TA, transaldolase
S7P, sedoheptulose-7-phosphate
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041221105.
References
- Antosiewicz, J., McCammon, J.A., and Gilson, M.K. 1994. Prediction of pH-dependent properties of proteins. J. Mol. Biol. 238 415–436. [DOI] [PubMed] [Google Scholar]
- ———. 1996. The determinants of pKas in proteins. Biochemistry 35 7819–7833. [DOI] [PubMed] [Google Scholar]
- Babbitt, P.C., Mrachko, G.T., Hasson, M.S., Huisman, G.W., Kolter, R., Ringe, D., Petsko, G.A., Kenyon, G.L., and Gerlt, J.A. 1995. A functionally diverse enzyme superfamily that abstracts the α protons of carboxylic acids. Science 267 1159–1161. [DOI] [PubMed] [Google Scholar]
- Babbitt, P.C., Hasson, M.S., Wedekind, J.E., Palmer, D.R., Barrett, W.C., Reed, G.H., Rayment, I., Ringe, D., Kenyon, G.L., and Gerlt, J.A. 1996. The enolase superfamily: A general strategy for enzyme-catalyzed abstraction of the α-protons of carboxylic acids. Biochemistry 35 16489–16501. [DOI] [PubMed] [Google Scholar]
- Berman, H.M., Bhat, T.N., Bourne, P.E., Feng, Z., Gilliland, G., Weissig, H., and Westbrook, J. 2000. The Protein Data Bank and the challenge of structural genomics. Nat. Struct. Biol. (Suppl.) 7: 957–959. [DOI] [PubMed]
- Brooks, R.B., Bruccoleri, R.E., Olafson, B.D., States, D.J., Swaminathan, S., and Karplus, M. 1983. CHARMM, A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4 187–217. [Google Scholar]
- Chung, S.Y. and Subbiah, S. 1996. A structural explanation for the twilight zone of protein sequence homology. Structure 4 1123–1127. [DOI] [PubMed] [Google Scholar]
- Cohn, M., Pearson, J.E., O’Connell, E.L., and Rose, I.A. 1970. Nuclear magnetic resonance assignment of the vinyl hydrogens of phosphoenolpyruvate. Stereochemistry of the enolase reaction. J. Am. Chem. Soc. 92 4095–4098. [DOI] [PubMed] [Google Scholar]
- Copley, R.R. and Bork, P. 2000. Homology among (βα)(8) barrels: Implications for the evolution of metabolic pathways. J. Mol. Biol. 303 627–641. [DOI] [PubMed] [Google Scholar]
- Crooks, G.E., Hon, G., Chandonia, J.M., and Brenner, S.E. 2004. WebLogo: A sequence logo generator. Genome Res. 14 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elcock, A.H. 2001. Prediction of functionally important residues based solely on the computed energetics of protein structure. J. Mol. Biol. 312 885–896. [DOI] [PubMed] [Google Scholar]
- Gerlt, J.A. and Babbitt, P.C. 1998. Mechanistically diverse enzyme superfamilies: The importance of chemistry in the evolution of catalysis. Curr. Opin. Chem. Biol. 2 607–612. [DOI] [PubMed] [Google Scholar]
- Gibas, C.J. and Subramaniam, S. 1996. Explicit solvent models in protein pKa calculations. Biophys. J. 71 138–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilson, M.K. 1993. Multiple-site titration and molecular modeling: Two rapid methods for computing energies and forces for ionizable groups in proteins. Proteins 15 266–282. [DOI] [PubMed] [Google Scholar]
- Gu, X. 2003. Functional divergence in protein (family) sequence evolution. Genetica 118 133–141. [PubMed] [Google Scholar]
- Hulo, N., Sigrist, C.J., Le, S.V., Langendijk-Genevaux, P.S., Bordoli, L., Gattiker, A., De Castro, E., Bucher, P., and Bairoch, A. 2004. Recent improvements to the PROSITE database. Nucleic Acids Res. 32 (Database issue): D134–D137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia, J., Huang, W., Schorken, U., Sahm, H., Sprenger, G.A., Lindqvist, Y., and Schneider, G. 1996. Crystal structure of transaldolase B from Escherichia coli suggests a circular permutation of the α/β barrel within the class I aldolase family. Structure 4 715–724. [DOI] [PubMed] [Google Scholar]
- Jia, J., Schorken, U., Lindqvist, Y., Sprenger, G.A., and Schneider, G. 1997. Crystal structure of the reduced Schiff-base intermediate complex of transaldolase B from Escherichia coli: Mechanistic implications for class I aldolases. Protein Sci. 6 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, S. and Thornton, J.M. 2004. Searching for functional sites in protein structures. Curr. Opin. Chem. Biol. 8 3–7. [DOI] [PubMed] [Google Scholar]
- Jorgensen, W.L. and Tirado-Rives, J. 1988. The OPLS potential function for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Amer. Chem. Soc. 110 1657–1666. [DOI] [PubMed] [Google Scholar]
- Joseph, D., Petsko, G.A., and Karplus, M. 1990. Anatomy of a conformational change: Hinged “lid” motion of the triosephosphate isomerase loop. Science 249 1425–1428. [DOI] [PubMed] [Google Scholar]
- Knowles, J.R. 1991. Enzyme catalysis: Not different, just better. Nature 350 121–124. [DOI] [PubMed] [Google Scholar]
- Kuhner, M.K. and Felsenstein, J. 1994. A simulation comparison of phylogeny algorithms under equal and unequal evolutionary rates. Mol. Biol. Evol. 11 459–468. [DOI] [PubMed] [Google Scholar]
- Kursula, I., Partanen, S., Lambeir, A.M., Antonov, D.M., Augustyns, K., and Wierenga, R.K. 2001. Structural determinants for ligand binding and catalysis of triosephosphate isomerase. Eur. J. Biochem. 268 5189–5196. [DOI] [PubMed] [Google Scholar]
- La, D., Sutch, B., and Livesay, D.R. 2005. Predicting protein functional sites with phylogenetic motifs. Proteins 58 309–320. [DOI] [PubMed] [Google Scholar]
- Lichtarge, O., Bourne, H.R., and Cohen, F.E. 1996. An evolutionary trace method defines binding surfaces common to protein families. J. Mol. Biol. 257 342–358. [DOI] [PubMed] [Google Scholar]
- Lichtarge, O., Yamamoto, K.R., and Cohen, F.E. 1997. Identification of functional surfaces of the zinc binding domains of intracellular receptors. J. Mol. Biol. 274 325–337. [DOI] [PubMed] [Google Scholar]
- Lichtarge, O., Yao, H., Kristensen, D.M., Madabushi, S., and Mihalek, I. 2003. Accurate and scalable identification of functional sites by evolutionary tracing. J. Struct. Funct. Genomics 4 159–166. [DOI] [PubMed] [Google Scholar]
- Livesay, D.R., Linthicum, S., and Subramaniam, S. 1999. pH dependence of antibody-hapten association. Mol. Immunol. 36 397–410. [DOI] [PubMed] [Google Scholar]
- Livesay, D.R., Jambeck, P., Rojnuckarin, A., and Subramaniam, S. 2003. Conservation of electrostatic properties within enzyme families and superfamilies. Biochemistry 42 3464–3473. [DOI] [PubMed] [Google Scholar]
- Madura, J.D., Briggs, J.M., Wade, R.C., Davis, M.E., Lutty, B.A., Ilin, A., Antosiewicz, J., Gilson, M.K., Gagheri, B., Scott, L.R., et al. 1995. Electrostatics and diffusion of molecules in solution, simulations with the University of Houston Brownian dynamics program. Comput. Phys. Commun. 91 57–95. [Google Scholar]
- Nagano, N., Orengo, C.A., and Thornton, J.M. 2002. One fold with many functions: The evolutionary relationships between TIM barrel families based on their sequences, structures and functions. J. Mol. Biol. 321 741–765. [DOI] [PubMed] [Google Scholar]
- Penny, D. and Hendy, M. 1985. The use of tree comparison metrics. Syst. Zool. 34 75–82. [Google Scholar]
- Reardon, D. and Farber, G.K. 1995. The structure and evolution of α/β barrel proteins. FASEB J. 9 497–503. [DOI] [PubMed] [Google Scholar]
- Rozovsky, S. and McDermott, A.E. 2001. The time scale of the catalytic loop motion in triosephosphate isomerase. J. Mol. Biol. 310 259–270. [DOI] [PubMed] [Google Scholar]
- Rozovsky, S., Jogl, G., Tong, L., and McDermott, A.E. 2001. Solution-state NMR investigations of triosephosphate isomerase active site loop motion: Ligand release in relation to active site loop dynamics. J. Mol. Biol. 310 271–280. [DOI] [PubMed] [Google Scholar]
- Stroppolo, M.E., Falconi, M., Caccuri, A.M., and Desideri, A. 2001. Superefficient enzymes. Cell Mol. Life Sci. 58 1451–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov, R.L., Fedorova, N.D., Jackson, J.D., Jacobs, A.R., Kiryutin, B., Koonin, E.V., Krylov, D.M., Mazumder, R., Mekhedov, S.L., Nikolskaya, A.N., et al. 2003. The COG database: An updated version includes eukaryotes. BMC Bioinformatics 4 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrez, M., Schultehenrich, M., and Livesay, D.R. 2003. Conferring thermostability to mesophilic proteins through optimized electrostatic surfaces. Biophys J. 85 2845–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinarov, D.A. and Nowak, T. 1998. pH dependence of the reaction catalyzed by yeast Mg-enolase. Biochemistry 37 15238–15246. [DOI] [PubMed] [Google Scholar]
- ———. 1999. Role of His159 in yeast enolase catalysis. Biochemistry 38 12138–12149. [DOI] [PubMed] [Google Scholar]
- Wierenga, R.K. 2001. The TIM-barrel fold: A versatile framework for efficient enzymes. FEBS Lett. 492 193–198. [DOI] [PubMed] [Google Scholar]
- Wierenga, R.K., Borchert, T.V., and Noble, M.E. 1992. Crystallographic binding studies with triosephosphate isomerases: Conformational changes induced by substrate and substrate-analogues. FEBS Lett. 307 34–39. [DOI] [PubMed] [Google Scholar]
- Wold, F. and Ballou, C. 1957. Studies on the enzyme enolase. II. Kinetic studies. J. Biol. Chem. 227 313–328. [PubMed] [Google Scholar]
- Wood, T. 1986. Physiological functions of the pentose phosphate pathway. Cell Biochem. Funct. 4 241–247. [DOI] [PubMed] [Google Scholar]
- Yao, H., Kristensen, D.M., Mihalek, I., Sowa, M.E., Shaw, C., Kimmel, M., Kavraki, L., and Lichtarge, O. 2003. An accurate, sensitive, and scalable method to identify functional sites in protein structures. J. Mol. Biol. 326 255–261. [DOI] [PubMed] [Google Scholar]