

Abstract
Structures for protein domains have increased rapidly in recent years owing to advances in structural biology and structural genomics projects. New structures are often similar to those solved previously, and such similarities can give insights into function by linking poorly understood families to those that are better characterized. They also allow the possibility of combing information to find still more proteins adopting a similar structure and sometimes a similar function, and to reprioritize families in structural genomics pipelines. We explore this possibility here by preparing merged profiles for pairs of structurally similar, but not necessarily sequence-similar, domains within the SMART and Pfam database by way of the Structural Classification of Proteins (SCOP). We show that such profiles are often able to successfully identify further members of the same superfamily and thus can be used to increase the sensitivity of database searching methods like HMMer and PSI-BLAST. We perform detailed benchmarks using the SMART and Pfam databases with four complete genomes frequently used as annotation benchmarks. We quantify the associated increase in structural information in Swissprot and discuss examples illustrating the applicability of this approach to understand functional and evolutionary relationships between protein families.
Keywords: structural genomics, protein structure, domain family, evolution
Genome-sequencing projects have uncovered many proteins without known functions, and many experimental and computational approaches are now applied to better understand them. Detection of a structural similarity between proteins can often provide clues, since this is often accompanied by a similarity in function. This is often true even for very weakly similar sequences: Two proteins can have sequence identities below 10% and still perform similar functions (Murzin et al. 1995; Sowdhamini et al. 1998; Orengo et al. 2003). Many sequence comparison methods are able to find very remote homologs: Current domain databases like SMART (Letunic et al. 2002) or Pfam (Bateman et al. 2002) contain homologous proteins with very limited sequence similarity. However, new three-dimensional (3D) structures continue to show that similarities between structures are still not detected by sequence comparison (e.g., Alexander et al. 2002; Eswaramoorthy et al. 2003). Such similarities are usually captured in structural classification databases including SCOP (Lo Conte et al. 2002), CATH (Orengo et al. 2003), and FSSP (Holm and Sander 1996).
Structural genomics initiatives often aim to use 3D structure as a means to identify functions (Zhang and Kim 2003). New structures can reveal binding sites or features that give hints about function, but the best functional inferences come when a new structure reveals an overall similarity to another that was not apparent from the comparison of sequences (Zhang et al. 2000). Such similarities can often allow the two families to be merged and much functional information to be transferred between them (Fig. 1A ▶). This is a central goal of many initiatives, and this strategy has lead to many successful annotations of function (Heger and Holm 2001; Aloy et al. 2002).
Figure 1.
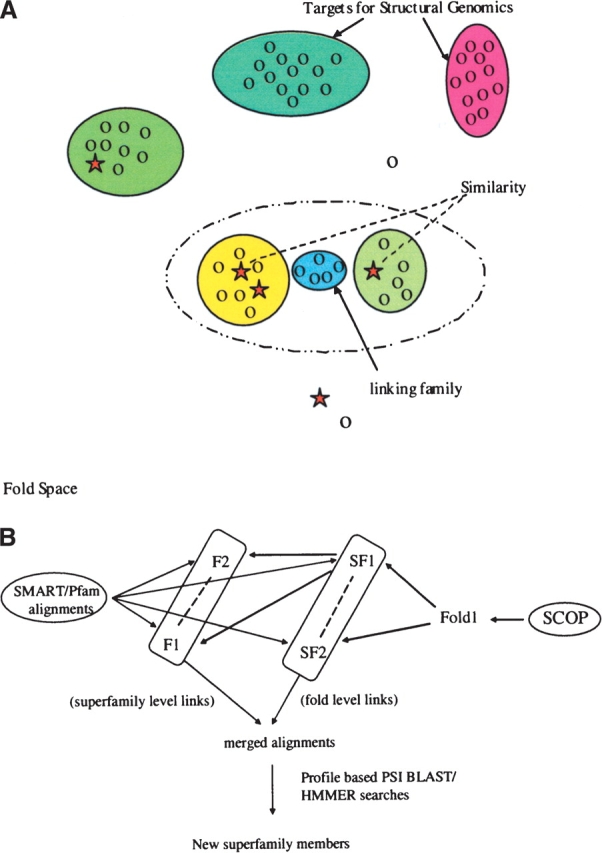
Merging sequences in folds space to find the bridging families. (A) Representation of fold space where related sequences are grouped into families (or higher-level groupings). Families related by structure (and assumed evolutionary relationship) can be merged to produce a powerful profile, which in turn can be utilized to find the sequences that occur at the “bridges.” Ovals of different colors represent different sequence families that populate fold space. Proteins of known structures are shown as stars; those without, as circles. (B) The strategy that we have used to identify bridging families. We use sequence information from SMART/Pfam and structural hierarchy of SCOP to merge different related families in to new superfamilies. We then use PSI-BLAST or HMMer profiles of the merged superfamily to search the sequence databases to identify the bridging families that may be part of the same superfamily.
A possible spin-off from such discoveries is the creation of new, more sensitive profiles (Griffiths-Jones and Bateman 2002; Panchenko and Bryant 2002), which can then be used to discover further relationships and transfer functional information. When a structural alignment can be used to merge to previously unassociated families, the combined sequence information can sometimes prove more sensitive than that for the separate families (Fig. 1A ▶). It is this concept that we explore in detail here. We use the SCOP database to identify pairs of domains within SMART or Pfam that are structurally similar. We then merge the separate alignments via a structure-based sequence alignment and use the new alignment to perform profile-based sequence database searches. We show that this procedure can identify relationships to other families, including many lacking a representative of known structure, and is thus able to suggest structures and often functional details for poorly understood sequence families.
Results
Overall strategy
We took structural mergers of Pfam and SMART alignments from a previous study (Aloy et al. 2002). In short, we aligned pairs of structures for representatives from Pfam/SMART domains using 3D-structure comparison (Russell and Barton 1992) and used these to merge the sequence alignments for the associated domains. These pairwise alignments were divided into two classes according to the nature of the structural similarity used to merge them as defined in the structural classification of proteins (SCOP) database (Fig. 1B ▶).
SCOP classifies proteins into a hierarchy of similarity. Within families protein domains are perhaps most similar in character to those contained in SMART or Pfam. Homology can be very remote, but typically the similarity is detectable by sensitive sequence comparison methods. Superfamilies are the next level up the hierarchy, where a common evolutionary origin has been inferred by comparison of 3D structures, typically owing to the presence of a common active or binding site, or unusual common features unlikely to arise by chance. At the next level are folds, where proteins adopt similar 3D structures without any compelling evidence for a common ancestor. At this level proteins might still be remote homologs, but there is insufficient evidence to argue the case convincingly. For this study we merged Pfam or SMART alignments using structures similar at the superfamily or fold level. Figure 1B ▶ shows the strategy described here. For each merged alignment we prepared profiles that we then used to search all sequences in the domain database. We then tested whether the searches retrieved additional members of the superfamily, fold, or new domain families lacking a representative of known structure.
Overall performance of structure-based profiles
Since many Pfam or SMART domains contain members of known 3D structures, we could easily define positive and negative matches (true or false) in database search results as those belonging to the same or different folds. In order to study the difference in the database search performance due to merged profiles, we chose to plot sensitivity and specificity versus E-value thresholds (Fig. 2 ▶, top and middle). Definitions of sensitivity and specificity are given in the Materials and Methods section. We have also plotted true positive rate versus false positive rate (ROC curves) as a standard evaluation of performance (Fig. 2 ▶, bottom). The number of true positives and false positives caused by the merged and unmerged profile searches are shown in Supplementary Table 1A,B for interested readers.
Figure 2.

Benchmarks with SMART and Pfam. Plots of Specificity (upper left) and Sensitivity (lower left) vs. BLAST/HMMer E-value (log scale). Labels indicate the type of profiles used: “Fold,” Pfam/SMART domains merged with structures sharing the same fold, but lying in different superfamilies; “Superfamily,” structures in the same superfamily; “Unmerged” separate Pfam/SMART domains. The vertical broken lines show the thresholds for HMMer (blue) and BLAST (red) chosen in the text to give the optimal results when searching. ROC curve (right) is also shown with the same labels. Curves for Unmerged profiles for Specificity are horizontal on top and sideways for ROC curves and may not be easily visible.
Table 1.
Number of profiles detecting additional sequences
| Profile type | Search method | Hierarchy | No. of profiles | No. of profiles detecting additional sequences | Percentage |
| Merged | HMMer | fold | 1185 | 167 | 14.09 |
| Merged | PSI-BLAST | fold | 1185 | 154 | 13.41 |
| Merged | HMMer | superfamily | 748 | 308 | 41.17 |
| Merged | PSI-BLAST | superfamily | 748 | 407 | 54.41 |
| Unmerged | HMMer | — | 903 | 56 | 6.20 |
| Unmerged | PSI-BLAST | — | 903 | 44 | 4.87 |
The plots in Figure 2 ▶ show that HMMer profiles are better overall than their BLAST equivalents, for they are both more sensitive and specific at comparable E-values. The plots also show, as expected, that profiles based on merged domains from the same superfamilies (superfamily level links) are more sensitive than those based on different superfamilies in the same fold (fold level links). Merging very diverse (or indeed non-homologous) alignments can lead to profiles that do not contain sufficient information to be used in searching.
Overall, Figure 2 ▶ also shows that structure-based merged profiles do not perform better than their unmerged counterparts: For BLAST they are marginally better, for HMMer marginally worse. However, inspection of the results (Table 1) shows that performance varies considerably: Some merged profiles are better than their unmerged equivalents. They detect more related sequences from the database. Database searches with 308 merged HMMer profiles and 407 of that with PSI-BLAST at the superfamily level find more sequences of the same fold (annotated with structures) or novels. Such searches with fold-level profiles with HMMer and PSI-BLAST obtain other members with 167 and 159 profiles, respectively, while only 4.87% of unmerged alignments find more related families with PSI-BLAST and 6.25% with HMMer. The combined strategy does find more members of diverse families, just not all of them. This is as expected, as some very distantly-related domains have key functional and structural residues conserved, while others show little beyond an overall similarity in structure. Thus, the best overall strategy is to extend searches with the merged profiles with the unmerged counterparts (Fig. 2 ▶, bottom).
Figure 3 ▶ shows examples of relationships found with merged profiles from particular folds. Typically more than one related family is identified by most profiles. For example, the merged profile of SMART domains HTH_ARSR and ETS finds families PAX, HTH_CRP, HTH_ICLR (with known structures), and HTH_DTXR (without representative structure). The merged profile of the Pfam, CheR, and RrnaAD domains finds Methyltransf_3, FtsJ, Fibrilarin, PCMT (with known structures), Methyltransf_2, Met_10, and Ubie_methyltransf (without representative structure). Families are often identified by more than one merged profile. For example, HTH_ICLR is found by both the HTH_ARSR/HSF and ETS/HTH_CRP profiles, Met_10 by both CheR/RrnaAD and PARP_reg/Methyltransf_3.
Figure 3.

Examples of similarities found using merged profiles. (A) A typical example from our profile searches using mergers of SMART domains of Helix-Turn-Helix fold. Domains with white boxes are from the starting sets. The solid black bars represent merged families. Domains in gray ovals are structurally uncharacterized and those in white ovals are structurally characterized (but not included in starting set). The gray dotted lines starting from the black bars represent the merged pair that brings out the “bridging” family. The relationships described here are a complex web where the same resultant families may be picked up by more than one different profile. (B) A similar figure for mergers of different Pfam families of the Methyltransferase fold.
In rare cases where there is a large difference in the size of families or huge evolutionary distances, merged profiles may reflect the characteristics of the major contributor, resulting in the presence of only one family during the database searches. Database searches with 38 merged HMMer profiles (5.08%) and 88 of that with PSI-BLAST (11.76%) at the superfamily level yield members of only one family out of two and no members of related families. The fold-level results for such searches for HMMer and PSI-BLAST are 7.08% (84 out of 1185) and 23.03% (273 out of 1185), respectively.
For further analysis (including benchmarking on genomes), we chose conservative E-value thresholds of 10−2 (BLAST) and 0.5 (HMMer) that do not introduce more false positives (specificity close to 1), but still uncover new similarities with the merged profiles (Fig. 2 ▶).
Annotation of structures in completed genomes
To quantify the gain in sensitivity, we searched the genome sequences of Mycoplasma genitalium (Fraser et al. 1995), Escherichia coli K12 (Blattner et al. 1997), Streptococcus pneumoniae R6 (Hoskins et al. 2001), and Saccharomyces cervisiae (Goffeau et al. 1996) with structure-based profiles. We chose these genomes as they are very well annotated. We compared our assignments to superfamily (HMM profiles of SCOP 1.59; Gough and Chothia 2002), AnDom (IMPALA profiles of SCOP 1.59; Schmidt et al. 2002), and BLAST assignments. The number of unique assignments by our profiles compared to other methods for each genome (assignment difference) is plotted in Figure 4 ▶ for HMMer (Fig. 4A ▶) and PSI-BLAST (Fig. 4B ▶) searches. Although the starting set is limited to alignments that can be merged via structural alignment, the respective profiles are much more powerful than BLAST and find more distant sequences than are present in either superfamily or AnDom assignments. On the other hand, we miss all the assignments provided by single member superfamilies in SCOP. Figure 4 ▶ shows a direct increase in sensitivity in identifying known structure families. It is also clear that HMMer searches are again better than the BLAST counterparts. For example, HMMer profiles assign approximately twice as many new domains as BLAST for the E. coli genome (i.e., over and above those assigned by BLAST with single sequences). As the genome of Mycoplasma genitalium has been frequently used as a benchmark, we sought to discover whether the combined profiles could improve structural annotation of this genome (Teichmann et al. 1999). We made an additional 56 (or 11%) assignments (comparison to assignments provided in the work of Teichmann and colleagues [Teichmann et al. 1999]) on the Mycoplasma genitalium genome. It should be noted that part of this improvement could come from increases in database size and diversity since the original study.
Figure 4.

Assignment and benchmarking on complete genomes. (A) Difference in number of fold assignments done using HMMer searches of merged profiles compared to the assignments of the same fold using methods like superfamily, BLAST and AnDom on full genomes of M. genitalium, E. coli K12, S. pneumoniae R6, and S. cerevisiae. (B) The assignment differences obtained with our profiles while searching genomes with PSI-BLAST.
New members of known superfamilies
A total of 56 domains (5 SMART and 51 Pfam) not currently assigned to a known structure were found to be similar to a known superfamily via the merged profiles (Table 2). As many superfamilies or folds contain more than two Pfam or SMART alignments, many domains were found by more than one profile. Only three of these domains were found using the same thresholds with any of the unmerged profiles. This is perhaps not surprising as significant similarities would typically lead to domain families becoming merged in SMART or Pfam.
Table 2.
Domains lacking known structures found with merged profile
| Superfamily/fold | New domain | Merged profile | HMMer | PSI-BLAST |
| Similarities found with SMART profiles | ||||
| 4-helical cytokines | DAGKa | CSF2/IL2 (SF) | — | 0.003 |
| knottins | ChtBD2a | PTI/EGF (F) | 1.5e-11 | — |
| P-loop ATPases | MCM | AAA/ART (SF) | 5.9e-11 | — |
| winged 3-α bundle | HTH_DEOR | HTH_ARSR/HSF (SF) | — | 0.003 |
| HTH_ICLR | ETS/HTH_CRP (SF) | 6.9e-07 | 2.0e-04 | |
| Similarities found with PFAM profiles | ||||
| Crotonase | DUF114 | CLP_protease/ECH (SF) | 0.27 | 1.0e-05 |
| DUF107 | CLP_protease/ECH (SF) | 0.047 | 6.0e-08 | |
| Cys-knot | DAN | TGF-β/Cys_knot (SF) | 0.029 | — |
| FAD/NAD binding | GIDA | pyr_redox/GDI (SF) | (0.023) | 1.0e-05 |
| Thi4 | GMC_oxred/FAD_binding_3 (SF) | 0.007 | 5.0e-05 | |
| FMO-like | pyr_redox/Monooxygenase (SF) | (0.074) | 9.0e-07 | |
| Phytoene_dh | pyr_redox/Monooxygenase (SF) | 0.011 | 5.0e-06 | |
| DAOa | GDI/FAD_binding_3 (SF) | (0.064) | 1.0e-04 | |
| Glu synth ATP binding | DUF201 | CPSase_L_chain/Dala_Dala_ligas (SF) | — | 2.0e-07 |
| Immunoglobulin-like | K1 | Neocarzinostat/ig (F) | 0.21 | — |
| λ-repressor | Sigma54_factors | lacl/pou (SF) | 0.001 | — |
| Transposase_12 | lacl/pou (SF) | 0.17 | — | |
| P-loop ATPases | bac_dnaA | arf/UvrD-helicase (SF) | — | 0.002 |
| FeoB | GTP_EFTU/arf (SF) | 0.17 | — | |
| CoaE | Adenylatekinase/SRP54 (SF) | — | 1.0e-03 | |
| MMR_HSR1 | recA/arf (SF) | (0.013) | 6.0e-06 | |
| CobA_CobO_BtuR | recA/UvrD-helicase (SF) | — | 1.0e-03 | |
| ATP-bind | PRK/SRP54 (SF) | — | 1.0e-04 | |
| PLP-dependent | GDC-P | Cys_Met_Meta_PP/aminotran_2 (SF) | — | 5.0e-06 |
| pyridoxal_deC | Cys_Met_Meta_PP/SHMT (SF) | — | 2.0e-05 | |
| Phosphatase | Phosphodiest | alk_phosphatase/Sulfatase (SF) | — | 1.0e-03 |
| Rossmann-like | NAD_Gly3P_dh | llvC/3HCDH (SF) | — | 2.0e-06 |
| Acetylald_DH | GFO_IDH_MocA/DapB (SF) | 0.17 | — | |
| DAOa | 3HCDH/adh_short_C2 (SF) | — | 0.002 | |
| Polysacc_synt_2 | Epimerase/adh_short_C2 (SF) | (5.5e-10) | 2.0e-22 | |
| Octopine_DH_N | GFO_IDH_MocA/3HCDH (SF) | 0.12 | — | |
| PDH | llvC/3HCDH (SF) | (0.078) | 1.0e-07 | |
| Isoflavone_redu | THF_DHG_CYH/3β_HSD (SF) | (0.013) | 4.0e-04 | |
| Homoserine_dh | GFO_IDH_MocA/3HCDH (SF) | 0.3 | — | |
| ODC_Mu_crystall | llvC/3β_HSD (SF) | — | 4.0e-04 | |
| DUF108 | GFO_IDH_MocA/DapB (SF) | (0.005) | 5.0e-05 | |
| P5CR | llvC/3HCDH (SF) | (0.160) | 0.003 | |
| Snake-toxins | PLA2_inh | Activin_recp/UPAR_LY6 (SF) | 0.099 | — |
| TIM-barrel | UPF0034 | oxidored_FMN/FMN_dh (SF) | — | 2.0e-07 |
| Glyco_hydro_70 | Glyco_hydro_10/α-amylase (SF) | 0.035 | — | |
| Glyco_hydro_39 | Glyco_hydro_1/cellulose (SF) | 0.085 | — | |
| NPD | IMPDH_C/IMPDH_N (F) | (0.058) | 2.0e-07 | |
| Thioredoxin-like | SCO1-SenC | AhpC-TSA/thiored (SF) | — | 0.003 |
| Zincins | Fragilysin | Reprolysin/Peptidase_M10 (SF) | — | 0.003 |
| α/β hydrolases | Thioesterase | Lipase_3/abhydrolase (SF) | 1.3e-04 | (1.0e-03) |
| Ndr | Peptidase_S9/abhydrolase (SF) | 0.023 | — | |
| Lipase_2 | Peptidase_S9/abhydrolase (SF) | (0.18) | 0.003 | |
| Methyltransferase | PCMT | Methyltransf_3/RrnaAD (SF) | — | 7.0e-09 |
| UPF0020 | DNA_methylase/RrnaAD (SF) | — | 4.0e-05 | |
| Nol1_Nop2_Sun | Methyltransf_3/RrnaAD (SF) | — | 1.0e-05 | |
| Met_10 | PARP_regulatory/Methyltransf_3 (SF) | — | 2.0e-05 | |
| Spermine_synth | Methyltransf_3/RrnaAD (SF) | — | 3.0e-04 | |
| Methyltransf_2 | CheR/Methyltransf_3 (SF) | — | 4.0e-04 | |
| Methyltransf_4 | Methyltransf_3/RrnaAD (SF) | — | 3.0e-05 | |
| Ubie_methyltran | Methyltransf_3/RrnaAD (SF) | (0.005) | 5.0e-05 | |
| Winged 3-α bundle | TFIIE_α | HTH_5/IRF (SF) | 0.27 | — |
a Proteins with similarities in structure lying in different SCOP folds (not included in the total).
For the remaining 53 domains (4 SMART, 49 Pfam), we compared our assignments to those PSI-BLAST (10 iterations, E-value threshold 10−4) and AnDom assignments using a random, phylogenetically diverse set of sequences from each family. PSI-BLAST and AnDom (Schmidt et al. 2002) assigned the same superfamily for 46 of 53. For the seven novel assignments, we also ran the fold-recognition methods 3D PSSM (Kelley et al. 2000), FUGUE (Shi et al. 2001) and SUPERFAMILY (Gough and Chothia 2002) and predicted secondary structures with Jpred2 (Cuff et al. 1998). These supported four of the novel assignments. The results are summarized in Table 3, and we discuss two examples below.
Table 3.
More detailed analyses of the seven novel predictions
| Family | E-value | Predicted fold | 3D PSSM | FUGUE | SUPERFAMILY | Sec Str (JPRED) |
| DagKaa | 0.003 | 4 helical cytokine | — | TAF(II)230 TBP-binding fragmentb | — | All α |
| Glyco_hydro_39 | 0.085 | TIM β/α barrel | TIM β/α barrel | TIM β/α barrel | TIM β/α barrel | α/β |
| K1 | 0.21 | Ig-like β-sandwich | Ig-like β-sandwichb | Ig-like β-sandwich | Ig-like β-sandwich | All β |
| Ndr | 0.023 | α/β hydrolase | α/β hydrolase | α/β hydrolaseb | α/β hydrolase | α/β |
| Phosphodiesta | 0.001 | Alkaline phosphatase- like | Alkaline phosphatase- like | Alkaline phosphatase- like | Alkaline phosphatase- like | α/β |
| Transposase 12 | 0.17 | λ-repressor like DNA binding | Sigma 70 from RNA polb | Methyltransferaseb | — | All α |
| Sigma54_factors | 0.0011 | λ-repressor like DNA binding | Sigma 70 from RNA polb | β graspb | — | All α |
a Families identified by PSI BLAST profiles, others are identified by HMM profiles.
b Hits with scores above significance threshold (as described in methodology).
Our method could be categorized with PSI-BLAST and HMMer as we carry out profile-sequence searches. However, we also compared our results to a profile–profile comparison method. Profile–profile alignment methods are relatively new and they are believed to be more powerful then the profile–sequence alignment procedure (Ohlson et al. 2004). We chose to compare our results with FFAS methods as they are available for Pfam families on the authors’ Web site (Jaroszewski et al. 2000; Rychlewski et al. 2000). FFAS03 detected 33 out of 49 Pfam families (67.34%) and missed 16 families in our results. In addition, FFAS03 detected 52 other Pfam families without a representative structure. We found 23 out of those 52 families also in our results that we had discarded (i.e., with E-values above the conservative thresholds used by us). Correct assignments of 23 families above the E-value threshold also suggest that our assignments are highly specific and our method is complementary to profile–profile methods. These additional assignments are available as Supplementary Table 2.
Similarities among proteins with different folds
We also found instances where profiles suggested relationships between different folds. For example, those for the FAD/NAD(P) binding domain fold find members of the NAD(P) binding Rossmann (Pfam pair GDI + FAD_binding_ 3; DAO family at HMMer E-value of 0.0067) and Nucleotide binding folds (3HCDH_N family at HMMer E-value of 0.67). These three folds have previously been proposed to share a common ancestor but have accumulated structural changes during the course of evolution (Grishin 2001).
Similarly the chitin-binding domain in SMART (ChtBD2) is detected by merged profiles from different families of knottins with very low E-values (e.g., EGF + EGF_Lam, HMMer E = 3.4 × 10−9). SCOP classifies representative structures of ChtBD2 domain as the only member of the Tachycitin fold and knottins as members of EGF/Laminin-like fold. Literature searches and superposition of representative structures show that chitin-binding proteins in invertebrates and plants comprise a common chitin-binding structural motif (Suetake et al. 2000).
Merged profiles of the zinc-finger motif also found small cysteine-rich repeats in many proteins. For example, merged profiles found two CXXC repeats in the TOPRIM domain of O29238, which are not identified by SMART or Pfam HMMs. The structure of the TOPRIM domain is known and contains an insertion zinc finger motif in some cases (Rodriguez and Stock 2002). These may reflect the power of profiles to identify a remarkable local similarity in proteins.
Unification of superfamilies in the same fold
There were also many examples where mergers linked different superfamilies within the same fold, suggesting that they might indeed share a common evolutionary origin. For example, profiles from the merged immunoglobulin (Ig) variable and fibronectin type III (FN3) domains detect the purple acid phosphoesterases family at an HMMer E-value of 0.043 (SMART pair IGv/FN3). The HMMer profile of the merger of the IGc1 and IGc2 domains also finds FN3 at an E-value of 5.1 × 10−6 (SMART pair IGc1 + IGc2). Within the helix-turn-helix (HTH) fold, merged profiles of the arsenic resistance operon repressor and homeodomains family find the lux regulon domain (SMART pair of HTH_ARSR + HOX at HMMer E = 0.13), which is classified as a C-terminal effector domain of bipartite response regulator superfamily under an HTH fold.
The Ndr family
The Ndr (N-myc oncogene Downstream Regulated) family is named after a representative member found in a developing mouse embryo (Shimono et al. 1999), which is also found in humans, C. elegans and Drosophila. The Ndr-containing MESK2 gene of Drosophila is implicated in the Ras oncogene signaling pathway (Huang and Rubin 2000) and thus cancer. However, little is known about the molecular function of these proteins. Merged alignments of the Peptidase_S9 and α/β hydrolase Pfam families (both of which belong to the SCOP α/β hydrolase superfamily) find members of the Ndr family with HMMer E-values of 0.023. The relationship is confirmed by 3D-PSSM and SUPERFAMILY and was also noticed as the highest scoring match in the noise of the α/β hydrolase family profile by the Pfam annotators (see http://Pfam.wustl.edu).
The predicted secondary structure of Ndr fits well with the predicted fold, and there are also hints of key residue conservation, e.g., two invariant glycines found at the “nucleophile elbow” next to β5 in all α/β hydrolases are also conserved in Ndr homologs. A triad comprising a nucleophile (usually serine or cysteine), an acid (glutamate or aspartate), and a histidine are key parts of the catalytic enzymes in α/β hydrolases, with residues forming the oxy-anion-hole usually located on β-strands 5 and 3. The reaction chemistry for these proteins is similar to that of serine protease and subtilisin families (Ollis et al. 1992; Heikinheimo et al. 1999). Although the catalytic triad of chloroperoxidase (the best match to a known structure), Ser, Asp, and His is not conserved in the Ndr family (replaced by Gly, Ser/Ala, and Gly/Asp, respectively), other conserved positions containing theses residues are close in space when modeled onto chloroperoxidase. Thus Ndrs might exhibit “active-site migration,” known to occur in α/β hydrolases (Ollis et al. 1992) and other enzyme families (e.g., Todd et al. 2001 e.g., Todd et al. 2002).
The accessory domain of diacylglyceryl kinase
The merged profile for the CSF2 and IL2 domains of SMART, which belong to the very diverse four helical cytokine superfamily, find members of the diacylglyceryl kinase accessory (DAGKa) domain (BLAST E = 0.003). This prediction was not confirmed by fold recognition methods, though the predicted secondary structure and certain hydrophobic residue conservation broadly support the assignment (Supplementary Fig. 1 ▶). This prediction is surprising since all four helical cytokines are extracellular effectors, whereas DAGKas are intracellular. A precedent for such phenomenon is known to exist: Several members of the fibroblast growth factors, which are also extracellular effectors, are known to be intracellular signaling molecules (Schoorlemmer and Goldfarb 2001). However, there is also a good chance that this prediction is an artifact, particularly as the BLAST E-value is comparatively high. Alignment of DAGKa family members with four helical cytokine fold members is available as Supplementary Figure 1 ▶.
Overall increase in structural knowledge
We also measured the increase in links to known structure as increasingly more sensitive sequence comparison methods are used for annotation (Fig. 5 ▶). Considering only the 99,023 Swissprot sequences containing SMART or Pfam domains (Fig. 5 ▶), simple identity or homology assigns links to 27,036 (PDB, 2090 or HSSP, 24,946). Many more links are added based on the presence of a domain with representatives of known structure (SMART + Pfam, 12,379). The 53 assignments in Table 1 add 1067 (“PB or Andom” in Fig. 5 ▶) additional links, of which 95 (“Unique”) come from the seven novels (Table 2). This brings the total number of links to 40,367, or 41% of the total, of which 1% comes from merged profiles.
Figure 5.

Current level of structural annotation of sequence databases. Progression (left to right) showing how the number of links to known structure in Swissprot increases as more sensitive methods are used. PDB shows those proteins of known structure; HSSP augments these with their close homologs; “SMART + Pfam” are links added by matches to domains themselves linked to structures; “PB or Andom” increases this further via PSI-BLAST or Andom assignments; “Unique” are those assigned by our seven new domains found with merged profiles (Table 2).
Discussion
We have shown that the unification of protein families, as typically happens when a structural similarity is uncovered, can be used to successfully identify other members of diverse protein families. These new relationships can suggest clues into the function of previously uncharacterized sequences, such as for the Ndr example above. They also increase the structural annotation of genomes, and as such can be used to reprioritize target selection during structural genomics initiatives that aim to cover structure space.
Structural information has been used to aid the detection of remote homologs. Structure-dependent gap penalties, residue secondary structure, accessibility, and interaction pair preferences have all been incorporated into methods of fold recognition (Williams et al. 2001; Dietmann et al. 2002; Schonbrun et al. 2002; McGuffin and Jones 2003). Most recently, these methods have added information about homologous sequences to improve sensitivity (Kelley et al. 2000; Koretke et al. 2001; Williams et al. 2001). Here we have assessed the added value of combining sequence information for families that can only be aligned using knowledge of their structures.
Our findings also largely agree with others (Kelley et al. 2000; Panchenko and Bryant 2002), which suggests that more diverse alignments are generally less able to find distant homologs than their separate constituents. However, we have seen that this is not systematic: Some mergers increase sensitivity, while others decrease it. This suggests that the best strategy is clearly a combined one, where both merged and separated searches complement each other to attain the best sensitivity. New structural similarities, when exploited using this strategy, will often uncover more new members of the superfamily than the unaligned families in isolation.
Structural genomics initiatives continue to find new relationships between diverse protein structures. It is important to make the best use of the discovery in order to find new members of diverse sequence families. Methods like that described here and elsewhere (Pandit et al. 2002) help to do this, and thus provide a more complete picture of the structure and function of proteins within genomes.
Materials and methods
Database of merged alignments and profiles
Pairs of SMART/Pfam (Bateman et al. 2002; Letunic et al. 2002) domains aligned via representatives of known structure within the same SCOP (Lo Conte et al. 2002) superfamily or fold were taken from a previous study (Aloy et al. 2002). There were 1089 aligned Pfam pairs at the fold level and 581 at the superfamily level; for SMART the equivalent numbers were 167 and 96. These were used to build PSI-BLAST (Altschul et al. 1997) and HMMer (Eddy 1998) profiles with default options.
Searching and evaluation of accuracy
BLAST and HMMer searches were made with a library of superfamily/fold alignment profiles using default search parameters against databases containing all protein sequences from Pfam and SMART. Profiles generated from Pfam alignments were used to search Pfam and those generated using SMART were used to search SMART. For evaluation purposes, we considered the subset of those sequences for which we knew SCOP superfamily and fold membership by homology to a known structure. From the results of the searches, we computed standard definitions of Specificity and Sensitivity (Ingelfinger et al. 1987; Russell et al. 1998):
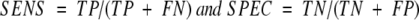 |
where TP, TN, FP, and FN denote true-positives, true-negatives, false-positives, and false-negatives, respectively. The SCOP definition of family, superfamily, and fold are used for the categorization of hits. Positives are those sequences with E-values at or better than a threshold value (described below) and are divided into true or false depending on whether or not they belong to the same superfamily or fold as the query profile. Negatives are those with E-values poorer than the threshold, divided into true or false in the opposite manner. As a control, we also did equivalent (HMMer and BLAST) searches for separated (i.e., unmerged) profiles.
Benchmarking on genomes
Protein sequences of Mycoplasma genitalium, Streptococcus pneumoniae R6, Escherichia coli K12 and Saccharomyces cervisiae genomes were downloaded from http://www.ncbi.nlm.nih.gov, searched using the above library of HMMer and PSI-BLAST profiles, and compared to assignments from SUPERFAMILY (Gough et al. 2001; Gough and Chothia 2002), AnDom (Schmidt et al. 2002), and BLAST (McGinnis and Madden 2004). We also compared our assignments for Mycoplasma genitalium to a standard benchmark by Teichmann et al. (1999), which combines assignments from many methods (Huynen et al. 1998; Wolf et al. 1999). AnDom assignments on genomes were done with the SCOP (1.59 release) profiles and an E-value filter value of 0.001 obtained from the authors (Schmidt et al. 2002). SUPERFAMILY assignments were filtered with an E-value of 0.01 as recommended by the SAM authors (Karplus et al. 1998). It should be noted here that we require at least two structures related at superfamily level to represent any given fold in our profiles. Hence, we miss all the assignments provided by single member superfamilies in SCOP. All data are available at http://www.bork.embl-heidelberg.de/~shah/pub/.
Fold prediction and sequence analysis
Fold predictions were done with the Web servers 3D PSSM (Kelley et al. 2000), SUPERFAMILY (Gough and Chothia 2002), and FUGUE (Shi et al. 2001) with several members for each family chosen from different branches of an evolutionary tree derived using the N-J tree option of CLUSTALX (Jeanmougin et al. 1998). Fold recognition thresholds were taken from the relevant Web sites (3D PSSM, E ≤ 0.05; SUPERFAMILY, E ≤ 0.01; FUGUE, Z ≥ 6.0). Secondary structures were predicted using Jpred2 (Cuff et al. 1998), Psipred (McGuffin et al. 2000), and PHD (Rost and Sander 1994). Structural alignment of representative structures of four helical cytokine families were prepared using STAMP (Russell and Barton 1992) and merged with the SMART alignment of the DagKa family. The Ndr family was merged with the structurally aligned thioesterase family members (STAMP).
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041187405.
Supplemental material: see www.proteinscience.org
References
- Alexander, J.M., Nelson, C.A., van Berkel, V., Lau, E.K., Studts, J.M., Brett, T.J., Speck, S.H., Handel, T.M., Virgin, H.W., and Fremont, D.H. 2002. Structural basis of chemokine sequestration by a herpesvirus decoy receptor. Cell 111 343–356. [DOI] [PubMed] [Google Scholar]
- Aloy, P., Oliva, B., Querol, E., Aviles, F.X., and Russell, R.B. 2002. Structural similarity to link sequence space: New potential superfamilies and implications for structural genomics. Protein Sci. 11 1101–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman, A., Birney, E., Cerruti, L., Durbin, R., Etwiller, L., Eddy, S.R., Griffiths-Jones, S., Howe, K.L., Marshall, M., and Sonnhammer, E.L. 2002. The Pfam protein families database. Nucleic Acids Res. 30 276–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattner, F.R., Plunkett 3rd, G., Bloch, C.A., Perna, N.T., Burland, V., Riley, M., Collado-Vides, J., Glasner, J.D., Rode, C.K., Mayhew, G.F., et al. 1997. The complete genome sequence of Escherichia coli K-12. Science 277 1453–1474. [DOI] [PubMed] [Google Scholar]
- Cuff, J.A., Clamp, M.E., Siddiqui, A.S., Finlay, M., and Barton, G.J. 1998. JPred: A consensus secondary structure prediction server. Bioinformatics 14 892–893. [DOI] [PubMed] [Google Scholar]
- Dietmann, S., Fernandez-Fuentes, N., and Holm, L. 2002. Automated detection of remote homology. Curr. Opin. Struct. Biol. 12 362–367. [DOI] [PubMed] [Google Scholar]
- Eddy, S.R. 1998. Profile hidden Markov models. Bioinformatics 14 755–763. [DOI] [PubMed] [Google Scholar]
- Eswaramoorthy, S., Gerchman, S., Graziano, V., Kycia, H., Studier, F.W., and Swaminathan, S. 2003. Structure of a yeast hypothetical protein selected by a structural genomics approach. Acta Crystallogr. D. Biol. Crystallogr. 59 127–135. [DOI] [PubMed] [Google Scholar]
- Fraser, C.M., Gocayne, J.D., White, O., Adams, M.D., Clayton, R.A., Fleischmann, R.D., Bult, C.J., Kerlavage, A.R., Sutton, G., Kelley, J.M., et al. 1995. The minimal gene complement of Mycoplasma genitalium. Science 270 397–403. [DOI] [PubMed] [Google Scholar]
- Goffeau, A., Barrell, B.G., Bussey, H., Davis, R.W., Dujon, B., Feldmann, H., Galibert, F., Hoheisel, J.D., Jacq, C., Johnston, M., et al. 1996. Life with 6000 genes. Science 274 546, 563–567. [DOI] [PubMed] [Google Scholar]
- Gough, J. and Chothia, C. 2002. SUPERFAMILY: HMMs representing all proteins of known structure. SCOP sequence searches, alignments and genome assignments. Nucleic Acids Res. 30 268–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough, J., Karplus, K., Hughey, R., and Chothia, C. 2001. Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J. Mol. Biol. 313 903–919. [DOI] [PubMed] [Google Scholar]
- Griffiths-Jones, S. and Bateman, A. 2002. The use of structure information to increase alignment accuracy does not aid homologue detection with profile HMMs. Bioinformatics 18 1243–1249. [DOI] [PubMed] [Google Scholar]
- Grishin, N.V. 2001. Fold change in evolution of protein structures. J. Struct. Biol. 134 167–185. [DOI] [PubMed] [Google Scholar]
- Heger, A., and Holm, L. 2001. Picasso: Generating a covering set of protein family profiles. Bioinformatics 17 272–279. [DOI] [PubMed] [Google Scholar]
- Heikinheimo, P., Goldman, A., Jeffries, C., and Ollis, D.L. 1999. Of barn owls and bankers: A lush variety of α/β hydrolases. Structure Fold. Des. 7 R141–R146. [DOI] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1996. The FSSP database: Fold classification based on structure–structure alignment of proteins. Nucleic Acids Res. 24 206–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins, J., Alborn Jr., W.E., Arnold, J., Blaszczak, L.C., Burgett, S., DeHoff, B.S., Estrem, S.T., Fritz, L., Fu, D.J., Fuller, W., et al. 2001. Genome of the bacterium Streptococcus pneumoniae strain R6. J. Bacteriol. 183 5709–5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, A.M. and Rubin, G.M. 2000. A misexpression screen identifies genes that can modulate RAS1 pathway signaling in Drosophila melanogaster. Genetics 156 1219–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynen, M., Doerks, T., Eisenhaber, F., Orengo, C., Sunyaev, S., Yuan, Y., and Bork, P. 1998. Homology-based fold predictions for Mycoplasma genitalium proteins. J. Mol. Biol. 280 323–326. [DOI] [PubMed] [Google Scholar]
- Ingelfinger, J.A., Mosteller, F., Thibodeau, L.A., and Ware, J.H. 1987. Biostatistics in clinical medicine. Macmillan, New York.
- Jaroszewski, L., Rychlewski, L., and Godzik, A. 2000. Improving the quality of twilight-zone alignments. Protein Sci. 9 1487–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanmougin, F., Thompson, J.D., Gouy, M., Higgins, D.G., and Gibson, T.J. 1998. Multiple sequence alignment with Clustal X. Trends Biochem. Sci. 23 403–405. [DOI] [PubMed] [Google Scholar]
- Karplus, K., Barrett, C., and Hughey, R. 1998. Hidden Markov models for detecting remote protein homologies. Bioinformatics 14 846–856. [DOI] [PubMed] [Google Scholar]
- Kelley, L.A., MacCallum, R.M., and Sternberg, M.J. 2000. Enhanced genome annotation using structural profiles in the program 3D-PSSM. J. Mol. Biol. 299 499–520. [DOI] [PubMed] [Google Scholar]
- Koretke, K.K., Russell, R.B., and Lupas, A.N. 2001. Fold recognition from sequence comparisons. Proteins Suppl. 5: 68–75. [DOI] [PubMed]
- Letunic, I., Goodstadt, L., Dickens, N.J., Doerks, T., Schultz, J., Mott, R., Ciccarelli, F., Copley, R.R., Ponting, C.P., and Bork, P. 2002. Recent improvements to the SMART domain-based sequence annotation resource. Nucleic Acids Res. 30 242–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Conte, L., Brenner, S.E., Hubbard, T.J., Chothia, C., and Murzin, A.G. 2002. SCOP database in 2002: Refinements accommodate structural genomics. Nucleic Acids Res. 30 264–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis, S. and Madden, T.L. 2004. BLAST: At the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 32 W20–W25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuffin, L.J. and Jones, D.T. 2003. Improvement of the GenTHREADER method for genomic fold recognition. Bioinformatics 19 874–881. [DOI] [PubMed] [Google Scholar]
- McGuffin, L.J., Bryson, K., and Jones, D.T. 2000. The PSIPRED protein structure prediction server. Bioinformatics 16 404–405. [DOI] [PubMed] [Google Scholar]
- Murzin, A.G., Brenner, S.E., Hubbard, T., and Chothia, C. 1995. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247 536–540. [DOI] [PubMed] [Google Scholar]
- Ohlson, T., Wallner, B., and Elofsson, A. 2004. Profile–profile methods provide improved fold-recognition: A study of different profile–profile alignment methods. Proteins 57 188–197. [DOI] [PubMed] [Google Scholar]
- Ollis, D.L., Cheah, E., Cygler, M., Dijkstra, B., Frolow, F., Franken, S.M., Harel, M., Remington, S.J., Silman, I., Schrag, J., et al. 1992. The α/β hydrolase fold. Protein Eng. 5 197–211. [DOI] [PubMed] [Google Scholar]
- Orengo, C.A., Pearl, F.M., and Thornton, J.M. 2003. The CATH domain structure database. Methods Biochem. Anal. 44 249–271. [DOI] [PubMed] [Google Scholar]
- Panchenko, A.R. and Bryant, S.H. 2002. A comparison of position-specific score matrices based on sequence and structure alignments. Protein Sci. 11 361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit, S.B., Gosar, D., Abhiman, S., Sujatha, S., Dixit, S.S., Mhatre, N.S., Sowdhamini, R., and Srinivasan, N. 2002. SUPFAM—A database of potential protein superfamily relationships derived by comparing sequence-based and structure-based families: Implications for structural genomics and function annotation in genomes. Nucleic Acids Res. 30 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez, A.C. and Stock, D. 2002. Crystal structure of reverse gyrase: Insights into the positive supercoiling of DNA. EMBO J. 21 418–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost, B. and Sander, C. 1994. Combining evolutionary information and neural networks to predict protein secondary structure. Proteins 19 55–72. [DOI] [PubMed] [Google Scholar]
- Russell, R.B. and Barton, G.J. 1992. Multiple protein sequence alignment from tertiary structure comparison: Assignment of global and residue confidence levels. Proteins 14 309–323. [DOI] [PubMed] [Google Scholar]
- Russell, R.B., Sasieni, P.D., and Sternberg, M.J. 1998. Supersites within super-folds. Binding site similarity in the absence of homology. J. Mol. Biol. 282 903–918. [DOI] [PubMed] [Google Scholar]
- Rychlewski, L., Jaroszewski, L., Li, W., and Godzik, A. 2000. Comparison of sequence profiles. Strategies for structural predictions using sequence information. Protein Sci. 9 232–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt, S., Bork, P., and Dandekar, T. 2002. A versatile structural domain analysis server using profile weight matrices. J. Chem. Inf. Comput. Sci. 42 405–407. [DOI] [PubMed] [Google Scholar]
- Schonbrun, J., Wedemeyer, W.J., and Baker, D. 2002. Protein structure prediction in 2002. Curr. Opin. Struct. Biol. 12 348–354. [DOI] [PubMed] [Google Scholar]
- Schoorlemmer, J. and Goldfarb, M. 2001. Fibroblast growth factor homologous factors are intracellular signaling proteins. Curr. Biol. 11 793–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, J., Blundell, T.L., and Mizuguchi, K. 2001. FUGUE: Sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J. Mol. Biol. 310 243–257. [DOI] [PubMed] [Google Scholar]
- Shimono, A., Okuda, T., and Kondoh, H. 1999. N-myc-dependent repression of ndr1, a gene identified by direct subtraction of whole mouse embryo cDNAs between wild type and N-myc mutant. Mech. Dev. 83 39–52. [DOI] [PubMed] [Google Scholar]
- Sowdhamini, R., Burke, D.F., Huang, J.F., Mizuguchi, K., Nagarajaram, H.A., Srinivasan, N., Steward, R.E., and Blundell, T.L. 1998. CAMPASS: A database of structurally aligned protein superfamilies. Structure 6 1087–1094. [DOI] [PubMed] [Google Scholar]
- Suetake, T., Tsuda, S., Kawabata, S., Miura, K., Iwanaga, S., Hikichi, K., Nitta, K., and Kawano, K. 2000. Chitin-binding proteins in invertebrates and plants comprise a common chitin-binding structural motif. J. Biol. Chem. 275 17929–17932. [DOI] [PubMed] [Google Scholar]
- Teichmann, S.A., Chothia, C., and Gerstein, M. 1999. Advances in structural genomics. Curr. Opin. Struct. Biol. 9 390–399. [DOI] [PubMed] [Google Scholar]
- Todd, A.E., Orengo, C.A., and Thornton, J.M. 2001. Evolution of function in protein superfamilies, from a structural perspective. J. Mol. Biol. 307 1113–1143. [DOI] [PubMed] [Google Scholar]
- ———. 2002. Plasticity of enzyme active sites. Trends Biochem. Sci. 27 419–426. [DOI] [PubMed] [Google Scholar]
- Williams, M.G., Shirai, H., Shi, J., Nagendra, H.G., Mueller, J., Mizuguchi, K., Miguel, R.N., Lovell, S.C., Innis, C.A., Deane, C.M., et al. 2001. Sequence-structure homology recognition by iterative alignment refinement and comparative modeling. Proteins Suppl. 5: 92–97. [DOI] [PubMed]
- Wolf, Y.I., Brenner, S.E., Bash, P.A., and Koonin, E.V. 1999. Distribution of protein folds in the three superkingdoms of life. Genome Res. 9 17–26. [PubMed] [Google Scholar]
- Zhang, C. and Kim, S.H. 2003. Overview of structural genomics: From structure to function. Curr. Opin. Chem. Biol. 7 28–32. [DOI] [PubMed] [Google Scholar]
- Zhang, H., Huang, K., Li, Z., Banerjei, L., Fisher, K.E., Grishin, N.V., Eisenstein, E., and Herzberg, O. 2000. Crystal structure of YbaK protein from Haemophilus influenzae (HI1434) at 1.8 Å resolution: Functional implications. Proteins 40 86–97. [DOI] [PubMed] [Google Scholar]