

Abstract
The Src homology 3 (SH3) domain of the Src family kinase Lyn binds to the herpesviral tyrosine kinase interacting protein (Tip) more than one order of magnitude stronger than other closely related members of the Src family. In order to identify the molecular basis for high-affinity binding, the structure of free and Tip-bound Lyn-SH3 was determined by NMR spectroscopy. Tip forms additional contacts outside its classical proline-rich recognition motif and, in particular, a strictly conserved leucine (L186) of the C-terminally adjacent sequence stretch packs into a hydrophobic pocket on the Lyn surface. Although the existence of this pocket is no unique property of Lyn-SH3, Lyn is the only Src family kinase that contains an additional aromatic residue (H41) in the n-Src loop as part of this pocket. H41 covers L186 of Tip by forming tight hydrophobic contacts, and model calculations suggest that the increase in binding affinity compared with other SH3 domains can mainly be attributed to these additional interactions. These findings indicate that this pocket can mediate specificity even between otherwise closely related SH3 domains.
Keywords: SH3 domain, Lyn, extended binding motif, ligand affinity, NMR, structure, complex, NMR spectroscopy, fluorescence, docking proteins
The Src homology 3 (SH3) domain is one of the most commonly found modular protein domains in eukaryotic genomes. In the human genome, > 900 SH3 domains have been identified so far, attesting to its usefulness to the cell and its adaptability to a huge variety of specific interactions.
SH3 domains are relatively short (60–70 residues) non-catalytic protein modules (Morton and Campbell 1994) whose primary activity is to bind proline-rich ligands containing the consensus sequence xP–x–xP, in which “x” denotes any amino acid (Yu et al. 1994). The xP–x–xP motif forms a canonical type II polyproline helix with two possible binding orientations depending on the position of a flanking basic residue (predominantly arginine) at the N terminus (class I) or C terminus (class II) (Lim et al. 1994). For most SH3 domains, the ligand-binding site consists of two hydrophobic slots, each occupied by a xP dipeptide, and a third, negatively charged specificity pocket that accommodates the flanking basic residue of the ligand (Feng et al. 1994; Lim et al. 1994). Thus, upon binding, only five core ligand residues contact the SH3 domain, of which two are invariant prolines and one is the basic residue, leaving little space for specificity. It was recently shown that additional contacts between variable loops of the SH3 domain and ligand residues N-terminal or C-terminal to the core can greatly enhance specificity (Feng et al. 1995; Lee et al. 1996; Pisabarro and Serrano 1996; Pisabarro et al. 1998; Ghose et al. 2001; Fazi et al. 2002; Dutta et al. 2004).
The tyrosine kinase interacting protein (Tip) from Herpesvirus saimiri contains a proline-rich sequence that binds to the SH3 domains of several members of the Src family of kinases (Lck, Hck, Lyn, Src, Fyn) (Fig. 1 ▶) and exhibits the highest affinity for Lyn (Schweimer et al. 2002). NMR spectroscopic studies of the interaction of Tip with Lck showed that the sequence stretch C-terminally adjacent to the polyproline helix is involved in Lck recognition (Bauer et al. 2004). A detailed investigation of the nature of these interactions, however, failed due to the unfavorable dynamics of complex formation (fast-to-intermediate exchange on the NMR timescale) and the resulting paucity in intermolecular distance information (Bauer et al. 2004).
Figure 1.

(A) Sequence alignment of the SH3 domains from the tyrosine kinases Lyn, Lck, Hck, Src, Fyn, Csk, and Abl. The numbering scheme of the Lyn-SH3 domain used in the present study is given at the top. Arrows indicate those amino acids in Lyn-SH3 that show NOE contacts to the Tip ligand. The positions of the β-strands are highlighted by gray boxes. Amino acids involved in additional contacts between the SH3 domains and ligand residues outside the proline-rich recognition motif are shaded in black (Lck-SH3–Tip; [Bauer et al. 2004], Src-SH3–VSL12 [Feng et al. 1995], Csk-SH3–PEP-3BP1 [Ghose et al. 2001], Abl-SH3–p41 [Pisabarro et al. 1998]). (B) Sequence alignment of Tip isolates from different Herpesvirus saimiri subtype C strains and Tio from Herpesvirus ateles. Arrows indicate those amino acids in Tip that show NOE contacts to Lyn-SH3. The numbering scheme of Tip C488 is given at the top. Positions of identical, conserved, and semiconserved residues are indicated by *, :, and ., respectively.
In order to get insight into the details of molecular recognition between Tip and the SH3 domains of the Src family of kinases, we therefore determined the complex structure of the Lyn-SH3–Tip complex by NMR spectroscopy. In addition to obtaining general information about the role of the flanking residues in the recognition process, this study should give insight into the particular structural properties of Lyn-SH3 that result in an increased affinity for Tip compared with other Src family SH3 domains.
Results and Discussion
Structure determination of the free Lyn-SH3 domain
Using standard double and triple resonance NMR experiments, a complete resonance assignment for Lyn-SH3 was obtained. The calculation of the final structures was based on 638 interresidual distance restraints, 22 ϕ angle restraints, 17 hydrogen bond restraints, and 47 DN,HN and 40 DCα,Hα orientational restraints from residual dipolar couplings (RDCs) (Table 1). The flexible ends (residues 1–10 and 67–68) for which no medium or long-range distance information is available and which exhibit {1H}15N-NOE values of < 0.5 were considered to be highly flexible and were therefore excluded from the subsequent analysis. According to PROCHECK (Laskowski et al. 1996), analysis of the family of 20 structures, residues 11–66 (“core fold”) show energetically favorable backbone conformations: 80% of these residues are found in the most favored regions and 20% in the allowed regions of the Ramachandran plot. The structure of residues D11–L66 is well-defined (Fig. 2A ▶), showing average root mean square deviations (RMSDs) of 0.34 ± 0.04 Å and 0.87 ± 0.04 Å for backbone heavy atoms and all heavy atoms, respectively (Table 1).
Table 1.
Summary of structure calculation
| Lyn-SH3 | Lyn-SH3–Tip complex | |
| Experimental restraints for final structure calculation | ||
| Intermolecular NOEs | 124 | |
| Intramolecular NOEs (Lyn-SH3) | ||
| Sequential (|i-j| = 1) | 222 | 228 |
| Medium range (|i-j| ≤5) | 83 | 85 |
| Long range (|i-j| > 5) | 316 | 326 |
| Intraresidual NOEs | 17 | 19 |
| Intramolecular NOEs (Tip) | ||
| Sequential (|i-j| = 1) | 99 | |
| Medium range (|i-j| ≤5) | 33 | |
| Dihedral angle restraints | ||
| 3J(HN,Hα) | 22 | 21 |
| Hydrogen bonds | 17 | 17 |
| Salt bridges | 1 | |
| Restraints from dipolar couplings | ||
| DN,HN | 47 | 41 |
| DCα,Hα | 40 | 37 |
| Molecular dynamics statisticsa | ||
| Average energy (kcal/mol) | ||
| Etot | 43.9 ± 1.6 | 101.0 ± 3.1 |
| Ebond | 1.6 ± 0.3 | 3.7 ± 0.3 |
| Eangles | 21.8 ± 0.9 | 43.5 ± 3.2 |
| Eimproper | 5.0 ± 0.4 | 12.3 ± 1.1 |
| Evdw | 6.3 ± 1.0 | 20.8 ± 2.3 |
| ENOE | 5.0 ± 0.5 | 12.1 ± 1.5 |
| Ecdih | 0.001 ± 0.003 | 0.9 ± 0.4 |
| ERDC | 4.1 ± 0.4 | 7.9 ± 0.9 |
| RMSD from ideal distance (Å) | ||
| Bonds | 0.00125 ± 0.00011 | 0.00167 ± 0.00007 |
| NOE | 0.012 ± 0.001 | 0.016 ± 0.001 |
| RMSD from ideal angles (deg) | ||
| Bond angles | 0.282 ± 0.006 | 0.342 ± 0.012 |
| Improper angles | 0.347 ± 0.026 | 0.384 ± 0.014 |
| RMSD from dipolar couplings (Hz) | ||
| N–HN couplings | 0.144 ± 0.017 | 0.213 ± 0.027 |
| Cα–Hα couplings | 0.278 ± 0.018 | 0.367 ± 0.030 |
| Atomic RMS differences (Å) | ||
| SH3 coreb | ||
| Backbone | 0.34 ± 0.04 | 0.28 ± 0.06 |
| Heavy atoms | 0.87 ± 0.04 | 0.80 ± 0.06 |
| Tipc | ||
| Backbone | 0.38 ± 0.10 | |
| Heavy atoms | 0.57 ± 0.12 | |
| Complexb,c | ||
| Backbone | 0.42 ± 0.07 | |
| Heavy atoms | 0.88 ± 0.07 | |
a The final force constants used in the structure calculation were 1000 kcal · mol−1 · Å−2 for the bond length, 500 kcal · mol·1 · rad−2 for the bond angles and improper angles, 50 kcal · mol−1 · Å−2 for the NOE distance restraints, 55 kcal · mol−1 · rad−2 for the ϕ angle restraints, and 1.0 kcal · mol−1 · Hz−2 for the residual dipolar couplings.
b Calculated for the final set of 20 structures (residues 11–66).
c Calculated for the final set of 20 structures (residues 174–187).
Figure 2.

(A) Overlay of a set of 20 Lyn-SH3 structures (residues 9–68) in the unliganded state. (B) Average backbone structure of Lyn-SH3; residues V13, A15, L27, F29, M35, V37, A47, G56, I58, P59, and V63, forming the hydrophobic core of the protein, are shown in red.
Analysis of the fold reveals five anti-parallel β-strands (D11–A15, K34–E40, W44–S49, K54–P59, and V63–K65), which form two anti-parallel, triple-stranded β-sheets. These sheets are packed at almost right angles, thus constituting a compact β-barrel (Fig. 2B ▶). The five β-strands are connected by three loops (RT, n-Src, and distal loop) and by a helical turn connecting strands β4 and β5 (Fig. 2A,B ▶). The β-strands and the RT loop enclose a hydrophobic core that is formed by the non-polar amino acids V13, A15, L27, F29, M35, V37, A47, G56, I58, P59, and V63.
The structure of Lyn-SH3 determined here is highly similar to the previously solved solution structure of Lck-SH3 (Schweimer et al. 2002), as reflected in the backbone RMSD of 1.11 Å for the corresponding residues of the SH3 “core fold.” The RMSD of the regular secondary structure elements is even lower (0.74 Å), indicating that the differences are mainly located in the loops connecting the strands of the β-sheets.
Characterization of the Lyn-SH3–Tip interaction site
Information about those residues of Lyn-SH3 and Tip involved in binding was obtained from two sets of NMR titration experiments. First, unlabeled Lyn-SH3 was gradually added to 13C,15N-labeled Tip. Significant chemical shift changes were detected for the residues of the proline-rich sequence motif (T176–R182) and for the C-terminally adjacent stretch A184–G187. This is consistent with the {1H}15N-NOE of Tip, in which residues M174–G187 exhibit values > 0.4, while lower values were detected for all flanking residues (Supplementary Fig. 1 ▶).
In a second experiment, unlabeled Tip was gradually added to 13C,15N-labeled Lyn-SH3 (Fig. 3 ▶). The most prominent changes of the chemical shifts were observed for three stretches of the peptide chain (Y17–D26, E40–K46, and I58–Y62) corresponding to the RT loop, the n-Src loop, and the helical turn connecting strands β4 and β5, respectively (Fig. 4A ▶).
Figure 3.

NMR titration experiment showing the changes in the 1H,15N-HSQC spectrum of free Lyn-SH3 (red) upon gradual addition of Tip. Resonances belonging to the spectrum after the final step of titration (fivefold molar excess of Tip) are shown in green. Resonances are labeled with the corresponding sequence positions. Start and end point of resonances with a slow or intermediate exchange behavior on the NMR timescale are connected (solid lines). Side-chain NH2 resonances for glutamine and asparagine are connected (dashed lines).
Figure 4.
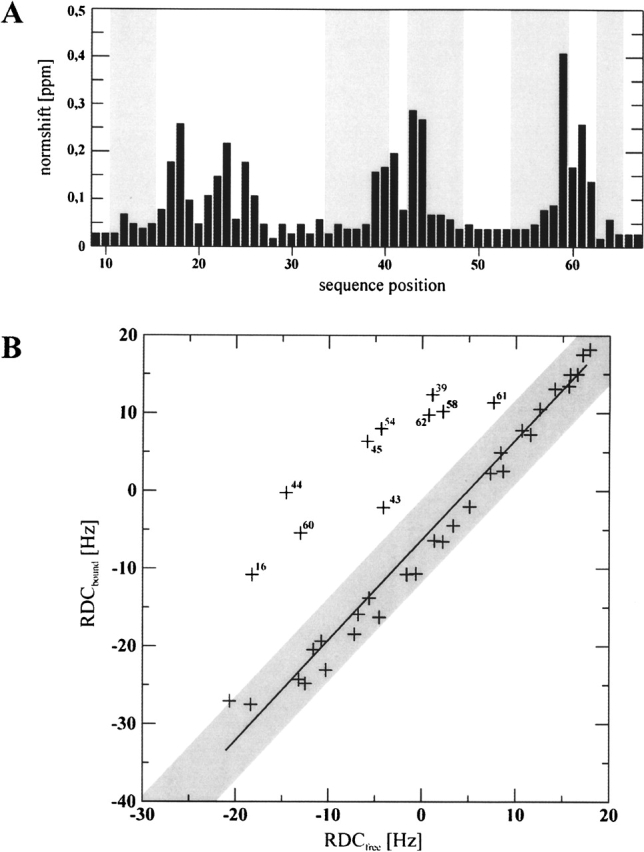
(A) Histogram of the normalized changes of the resonances of Lyn-SH3 upon Tip binding. Normshifts were calculated according to the equation given in Materials and Methods. The positions of the β-strands are highlighted by gray boxes. (B) Comparison of DN,HN residual dipolar couplings (RDCs) of the free and Tip-bound Lyn-SH3 domain. The solid line gives the linear regression fit with a correlation coefficient of 0.99 for the pairs of values in the gray area. Ten “outliers,” which are mainly part of the binding region, are labeled with the corresponding sequence positions and were excluded from the regression fit.
The binding regions in Tip and Lyn-SH3 are highly similar to those identified previously for the Lck-SH3–Tip interaction (Schweimer et al. 2002; Bauer et al. 2004). The two complexes of Tip with Lyn-SH3 and Lck-SH3, however, show different exchange behavior on the NMR timescale. Lck-SH3–Tip complex formation is in fast-to-intermediate chemical exchange (Schweimer et al. 2002), impeding the determination of a high-resolution Lck-SH3–Tip complex structure (Bauer et al. 2004). In contrast, for the Lyn-SH3–Tip complex, formation is in intermediate-to-slow chemical exchange (Fig. 3 ▶). The different exchange behavior on the NMR timescale detected for Lyn-SH3 and Lck-SH3 correlates well with their binding affinities determined by fluorescence spectroscopy (KD = 0.8 ± 0.1 μM and 16.8 ± 1.2 μM, respectively) (Fig. 5 ▶) and favored the determination of a high-resolution Lyn-SH3–Tip complex structure.
Figure 5.

Fluorescence binding studies of Tip to Lyn-SH3 (filled symbols) and Lck-SH3 (open symbols). Changes in the relative fluorescence are displayed by squares and circles for TipΔC and Tip, respectively. The fluorescence signal was detected at 340 nm after excitation at 280 nm. All intensities were normalized to a maximum value of 1. Curves were fitted according to the equation given in Materials and Methods.
The absolute values of the dissociation constants differ slightly from those determined previously for Tip binding to GST-fused SH3 domains from Lyn and Lck (Schweimer et al. 2002), which can most likely be attributed to the absence of blocked end groups in previous studies. In the light of the present study, which revealed that G187 is part of the binding interface, an unphysiological charge of the free C terminus at this position is expected to have significant effect on binding affinity and therefore was avoided here by using peptides with blocked end groups.
Structure determination of the Lyn-SH3–Tip complex
Using standard double and triple resonance NMR experiments, > 95% of the backbone and side-chain resonances could be assigned for Lyn-SH3 and for residues 169–191 of Tip. Due to severely reduced signal intensity in the NMR spectra caused by conformational dynamics on the intermediate chemical shift timescale or structural heterogeneity, no resonances for residues 140–168, which lie outside the SH3 binding region of Tip, were assigned (Supplementary Fig. 2 ▶).
Residual dipolar couplings of the free and the bound Lyn-SH3 correlate well, except for 10 “outliers” (L16, E39, E43–W45, K54, I58, S60–Y62), which are mainly part of or near by the ligand binding interface (Fig. 4B ▶). The correlation coefficient of 0.99 confirms that the free and the bound SH3 domains generally have similar structures, which undergo only minor changes upon ligand binding, indicating that the changes of the chemical shifts (Fig. 4A ▶) are due to very subtle structural rearrangements or result from direct interaction with the Tip peptide. The slope of the regression fit of 1.29 is due to variations in the concentrations of the orienting media used, giving rise to minor but uniform changes in the degree of orientation of the proteins.
For the Tip-bound Lyn-SH3, a total of 658 intramolecular distance restraints, 21 ϕ angle restraints, 17 hydrogen bond restraints, and 41 DN,HN and 37 DCα,Hα restraints from NMR experiments were derived. Using 13C/15N-labeled Tip in complex with unlabeled Lyn-SH3 and vice versa, a total of 132 intramolecular and 124 intermolecular distance restraints were obtained for the ligand (Table 1).
Intermolecular NOEs were observed for residues 175–187 of Tip (Fig. 1B ▶), consistent with {1H}15N-NOE measurements showing that residues 170–174 and 188–191 are very flexible on a picosecond-to-nanosecond timescale (Supplementary Fig. 1 ▶). The dynamics of these residues is also in accordance with fluorescence measurements showing that these residues do not contribute to binding affinity. The high number of distance restraints for residues 175–187 allowed structure calculations without any additional assumptions on the ligand binding site or the geometry of the polyproline helix.
Initial structure calculations revealed an electrostatic interaction between D26 of Lyn-SH3 and R182 of Tip. The set of calculated structures exhibits a distance of 3.3 ± 0.9 Å between the guanidino nitrogens of R182 and the side-chain oxygens of D26, which is slightly too large to infer unambiguously the presence of a salt bridge. This finding most probably results from the lack of an electrostatic potential term in the force field used for structure calculation. Therefore, a 1-nsec free molecular dynamics simulation of the complex that explicitly takes into account electrostatic interactions was run and confirmed the presence of a salt bridge, which was subsequently incorporated into the final stage of the structure calculation.
The final structure of the Lyn-SH3–Tip complex is well-defined, exhibiting RMSDs of 0.42 ± 0.07 Å and 0.88 ± 0.07 Å for the backbone and all heavy atoms, respectively (Table 1; Fig. 6A,B ▶). Comparison of the mean structures of free and bound Lyn-SH3 (Fig. 6C ▶) reveals an overall backbone RMSD of 1.11 Å. The most prominent differences are within the RT loop and n-Src loop, for which the backbone RMSD is 1.31 Å and 1.65 Å, respectively, whereas the backbone RMSD for all regular secondary structure elements and the distal loop is 0.91 Å and 1.14 Å, respectively. The backbone rearrangements of the RT loop are due to substantial changes of the hydrogen-bond network around the highly conserved D26, which forms a salt bridge upon ligand binding. Similar rearrangements have been described previously for the Fyn-SH3–Nef complex (Arold et al. 1997). The most prominent changes of the ϕ and ψ angle (44 °and 99 ° between the mean minimized structures, respectively) in Lyn-SH3 are observed for H41, located in the n-Src loop (Fig. 6C ▶). In the free conformation, H41 HN forms a hydrogen bond with the carbonyl oxygen of W44. After binding of Tip, this hydrogen bond is broken and results in a shorter β2 strand.
Figure 6.
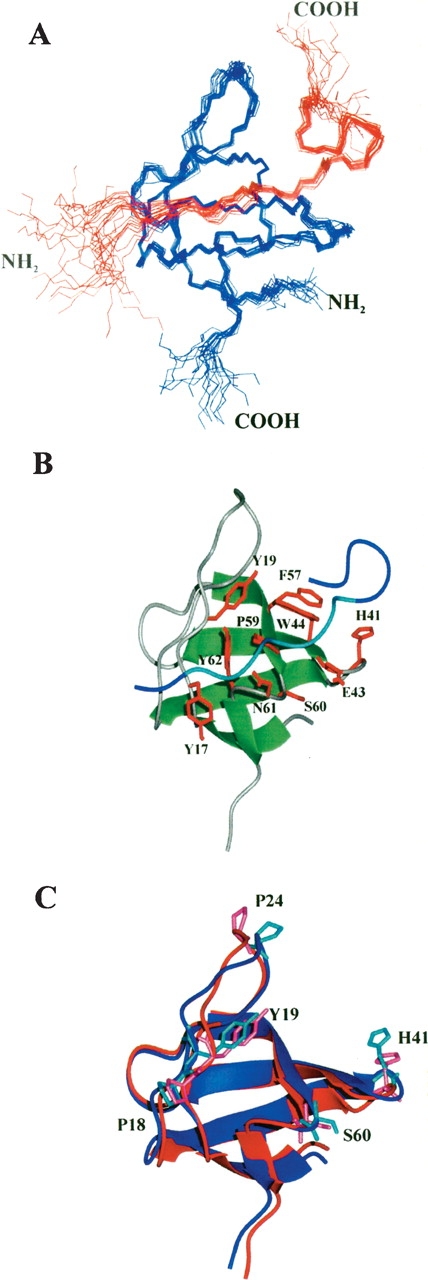
(A) Overlay of a set of 20 Lyn-SH3 structures (residues 9–68; blue) bound to Tip (residues 170–191; red). (B) Average backbone representation of the Lyn-SH3–Tip complex; the complex is shown in ribbon representation indicating the elements of secondary structure (Lyn-SH3, green and gray; Tip, blue). Residues of Lyn-SH3 that exhibit NOE contacts to the Tip ligand are shown as red sticks. The proline-rich region of Tip is shown in cyan. (C) Overlay of the average backbone representation of free (blue) and Tip-bound (red) Lyn-SH3. Amino acids differing in the values of ϕ and/or ψ angles by > 40 °are shown as sticks.
The 176TPPLPPR182 sequence stretch of Tip forms a PPII helix in which the two dipeptide moieties T176–P177 and L179–P180 of the ligand bind into two shallow hydrophobic slots on the Lyn-SH3 surface, which are formed by Y17 and Y62, and Y62, Y19, and W44, respectively (Fig. 7A ▶). R182 of Tip packs against the side chain of W44 and forms a salt bridge with D26 near the RT loop (Fig. 7B ▶), thus conforming all structural properties typically observed for proline-rich ligands binding in a class II orientation (Larson and Davidson 2000).
Figure 7.
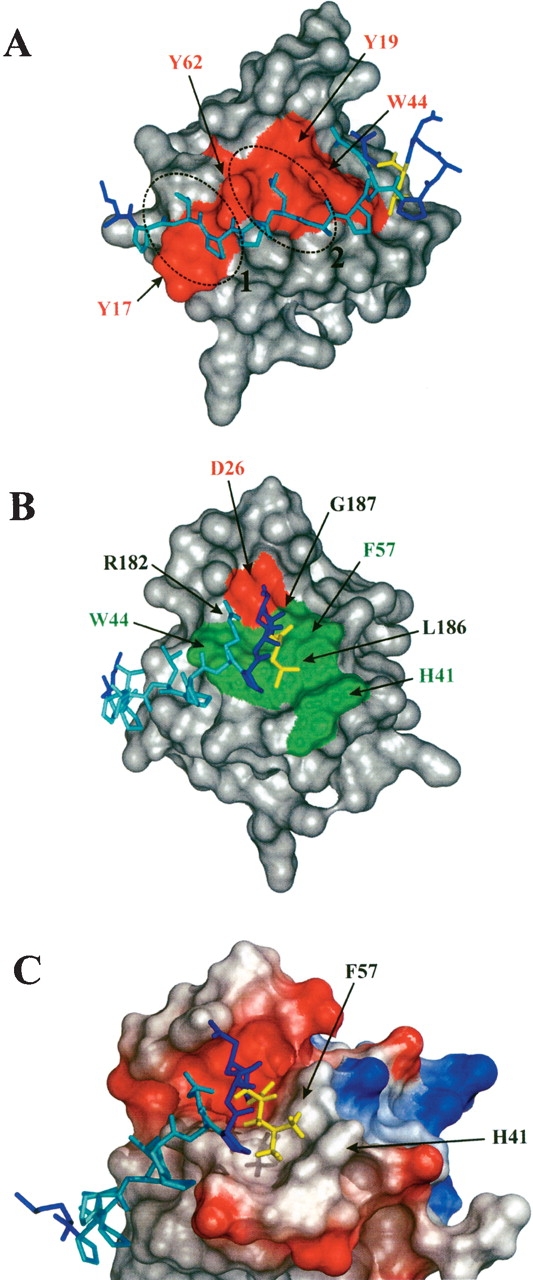
Contact surface of the Lyn-SH3 domain bound to Tip. (A) The hydrophobic surface patch of Lyn-SH3 formed by Y17, Y19, W44, and Y62 is shown in red, and ellipses mark the two slots that are occupied by residues T176, P177 and L179, and P180 of the ligand, respectively. The ligand is shown in stick presentation and the proline-rich sequence stretch is colored in cyan. (B) Additional contacts outside the proline-rich binding motif. R182 of Tip forms a salt bridge with D26 of Lyn-SH3 (red surface patch), and Tip L186 binds into a hydrophobic pocket formed by H41, W44, and F57 of Lyn-SH3 (green surface patches; figure rotated by 60 °along the Y-axis relative to A). (C) Detailed view of the interaction of Tip L186 with the hydrophobic pocket of Lyn-SH3; regions of positive and negative charges are shown in blue and in red, respectively. L186 (yellow) lies directly above the aromatic ring of F57, and the δ-methyl groups interact with H41 located in the n-Src loop of Lyn-SH3.
The flanking 183PANLG187 sequence is highly conserved among Tip isolates from different H. saimiri strains and the Tio protein of Herpesvirus ateles (Fig. 1B ▶). This five-residue stretch of Tip binds in a turn-like fashion into a valley between the n-Src and RT loops (Figs. 6B ▶, 7C ▶), as evidenced by NOEs between the β, γ, and δ protons of P183 and the α, β, and δ protons of L186. While the central residues of the turn, A184 and N185, exhibit no NOEs to the SH3 domain and point away from the binding interface, L186 and G187 occupy the center of the pocket formed by H41, W44, and F57 of Lyn-SH3 (Fig. 7B ▶). The extended side chain of L186 lies directly above the aromatic ring of F57 (Fig. 7C ▶), consistent with the strong upfield shifts of the side-chain protons (Hβ = 1.03 ppm and 1.07 ppm, Hγ = 0.67 ppm, and Hδ = 0.34 ppm and 0.13 ppm). Further NOEs indicate contacts between the δ-methyl groups of L186 and H41 located in the n-Src loop (Fig. 7C ▶).
Role of the C-terminal flanking region of Tip in Lyn-SH3 binding
The determination of the Lyn-SH3–Tip complex structure allowed a more detailed investigation of the role of particular residues for binding affinity and specificity. The role of L186 and G187 was assessed by determining the affinity of the C-terminally truncated TipΔC (amino acids A168–N185) for Lyn-SH3 and Lck-SH3. TipΔC binds to Lck-SH3 and Lyn-SH3 with KDs of 15.8 ± 1.6 μM and 15.0 ± 2.0 μM, respectively, showing that the proline-rich region in Tip alone exhibits almost identical affinities for both SH3 domains (Fig. 5 ▶). In contrast, the Tip peptide containing L186 and G187 binds significantly stronger to Lyn-SH3 than to Lck-SH3 (KDs of 0.8 ± 0.1 μM vs. 16.8 ± 1.2 μM). While the presence of L186 and G187 does not affect the affinity for Lck-SH3 within the error of the measurements, it leads to an increased affinity of more than one order of magnitude for Lyn-SH3. The affinity of Tip is among the highest ever reported for a natural proline-rich ligand of Src family SH3 domains and in most previous studies ligand design had to be employed to obtain similar (submicromolar) affinities (Feng et al. 1995; Pisabarro and Serrano 1996; Posern et al. 1998).
The results above demonstrate that residues L186/G187 form stabilizing contacts with Lyn-SH3 that cannot be formed in the complex with Lck-SH3. Structure analysis of the Lyn-SH3–Tip complex and sequence comparison to Lck-SH3 was performed in order to identify interactions that may account for this difference in binding affinity. One candidate is the hydrophobic interaction formed between L186 of Tip and H41 of Lyn-SH3 (Fig. 7C ▶). The latter residue is not conserved among the Src family of kinases, and in Lck-SH3 a serine is present at the respective sequence position (Fig. 1A ▶).
For a more detailed investigation of the role of H41 in Lyn-SH3 for tight binding, molecular mechanics/Poisson Boltzmann surface area (MM/PBSA) calculations were performed. This method has already applied to numerous systems, including SH3–ligand interactions (Wang et al. 2001), in the past and proved to be suitable to estimate differences in binding free energies (ΔΔGb) (Kollman et al. 2000). In order to test the reliability of the MM/PBSA method for the present system, a control simulation was initially performed to determine the differences in binding free energies (ΔΔGb) between the Lyn–Tip and Lyn–TipΔC complex. For this system an experimental ΔΔGb value is available from the differences of the fluorescence binding affinities (ΔΔGb = −1.7 kcal/mol) and can serve as a reference. The respective ΔΔGb value obtained from MM/PBSA (−0.8 kcal/mol; see Supplementary table) is of the same order of magnitude but slightly underestimates the effect of the truncation. Deviations of the same magnitude between experiment and simulation have been reported in a previous study that investigated SH3–ligand complexes using MM/PBSA (Wang et al. 2001), thus evidently representing the upper limit of accuracy that can currently be achieved by computational predictions for such systems.
The role of H41 for binding was investigated by a second simulation, in which H41 of Lyn was in silico replaced by serine to approximate the interface properties of the Lck-SH3–Tip complex. The MM/PBSA calculations give a difference in the binding free energies between the Lyn–Tip and the Lyn(H41S)–Tip complex of −1.0 kcal/mol. Comparison to the simulation of the Lyn–TipΔC complex shows that replacement of histi-dine by serine in Lyn causes an almost identical decrease of the binding affinity as a C-terminal truncation of Tip by two residues (ΔΔGb values of −1.0 kcal/mol and −0.8 kcal/mol, respectively), underlining the role of H41 for high-affinity binding.
This finding, together with the experimental data from fluorescence spectroscopy showing that the difference in binding affinity between the Lyn–Tip and Lck–Tip complexes is almost identical to that between the Lyn–Tip and Lyn–TipΔC complexes (ΔΔGb values of −1.8 kcal/mol and −1.7 kcal/mol, respectively), suggests that the tighter binding of Lyn compared with Lck can mainly be attributed to the sequence difference at position 41. Thus, the hydrophobic contacts between H41 of Lyn-SH3 and L186 of Tip outside the classical ligand-binding motif offer a plausible explanation for the tighter Tip binding of Lyn-SH3 compared with Lck-SH3. This tight binding of Tip to Lyn might allow an interaction between both proteins even when Lyn is present only at very low concentrations. This situation is present in H. saimiri C488 transformed T cells in which Lck is expressed at a significantly higher level compared with Lyn (Reiss et al. 2002).
General role of the residues outside the proline-rich region in SH3 binding
It is of interest to compare the contacts formed by the flanking residues in Tip to those found previously in other SH3–ligand complexes—in particular in the light of their role for modulating affinity and specificity of binding. L186 and G187 of Tip pack into a hydrophobic pocket on the Lyn surface. The candidate role of this pocket for increasing ligand affinity for Src family kinases has been shown previously for Src-SH3 by designing peptides that bind in a type I or type II orientation and exploit the hydrophobic pocket for additional contacts (Feng et al. 1995). For a peptide that binds in a type I orientation (VSL12), the residues N-terminally adjacent to the proline-rich motif were shown to bind into this pocket, and the structure of the respective complex was determined by NMR spectroscopy (Feng et al. 1995). For the APP12 peptide that binds like Tip in a type II orientation, however, no intermolecular NOEs were observed for the flanking residues, a fact attributed to the poor packing of the designed region (Feng et al. 1995).
Like Tip, both APP12 and VSL12 contain a leucine in the flanking region, which was shown to contact the hydrophobic pocket of Src-SH3 in complex with VSL12. The molecular details of the interaction, however, differ significantly between Tip and VSL12. The leucine (L3) of VSL12 is oriented toward the RT loop, while L186 of Tip interacts with the n-Src loop and the corresponding stretch of the peptide chain adopts a completely different backbone conformation. Another major difference is the surface accessibility of both leucines: While L3 in VSL12 has a solvent-accessible surface of 142 Å2 (53%), L186 of Tip has only 24 Å2 (9%).
Both in VSL12 and in APP12, the presence of the flanking residues that contact the hydrophobic pocket increases the affinity of binding by approximately one order of magnitude, which is similar to the effect caused by the presence of L186/G187 on the strength of the Lyn-SH3–Tip interaction. Similar to Tip, the increase of affinity caused by the flanking residues in VSL12 and APP12 is not equally large for different SH3 domains but was more pronounced for the original target Src-SH3 compared with the SH3 domain of PI3 kinase, which was used as a control (Feng et al. 1995).
This observation led to the conclusion that the valley between the RT and the n-Src loop, which exhibits different structural properties among various SH3 domains, represents a specificity pocket that might be used to specifically target one single SH3 domain or a subset of closely related SH3 domains (Fig. 1A ▶).
The role of the respective region for enhancing ligand specificity is further supported by studies from Pisabarro et al. (Pisabarro and Serrano 1996; Pisabarro et al. 1998) in which peptides (p40, p41) were designed that exploit the properties of this pocket to bind two to three orders of magnitude stronger to Abl-SH3 compared with Fyn-SH3. The structure of the Abl-SH3–p41 complex (Pisabarro et al. 1998) shows that a tyrosine of the flanking region plays a pivotal role in high-affinity binding. In addition to hydrophobic interactions, this residue can form two hydrogen bonds through its side-chain hydroxyl groups with the side chains of S12 and D14 in the RT loop.
A third example in which contacts formed to this part of the SH3 surface selectively increase binding affinity was reported for the C-terminal Src kinase (Csk) in complex with a peptide (PEP-3BP1) derived from the PEST domain of the natural SH3–ligand proline-enriched phosphatase (PEP) (Ghose et al. 2001). In the complex two hydrophobic residues of PEP-3BP1 (I21/V22), which are located C-terminal of the proline-rich recognition motif, specifically interact with A40, T42, and K43 of the Csk-SH3 n-Src loop. I21A and V22A, as well as I21V and V22L, mutations lead to a significant decrease of affinity (Ghose et al. 2001).
In summary, our results for the Lyn-SH3–Tip interaction are in line with these previous studies showing that the valley between the RT and the n-Src loop can be exploited by naturally occurring or by designed ligands to form additional contacts outside the proline-rich recognition motif, thus enhancing binding affinity. Since the respective surface patch exhibits considerable divergence in sequence among the family of SH3 domains (Fig. 1A ▶), ligand contacts in this region frequently increase not only affinity, but also specificity of binding. This is also evident from the present study, in which L186 of Tip forms tight contacts with H41, a residue that is not conserved even within the family of otherwise closely related Src kinases (Fig. 1A ▶). This information should be helpful in the future to design ligands that selectively target individual SH3 domains in order to interfere specifically with SH3-mediated signal transduction.
Materials and methods
Cloning, expression, and purification of Lyn-SH3 and Tip
Nucleotides comprising the SH3 domain of p59Lyn (amino acids 60–122, Lyn-SH3) were cloned via PCR from the IMAGE cDNA Clone IMAGp998F188710Q2 (RZPD, Deutsches Ressourcenzentrum für Genomforschung GmbH) into the BamHI and XhoI restriction sites of pGEX-6P-2 (Amersham Bioscience) using the oligonucleotides LynSH3_5′-(GGAGGAGGATCCCCAGAGG AACAAGGAGAC) and LynSH3_3′-(GGAGGACTCGAGT TAGGTGTTGAGTTTGGCCAC) (MWG-Biotech). The resulting vector pGEX-6P-LynSH3 provides an N-terminal glutathione S-transferase (GST) affinity tag cleavable with PreScission protease (Amersham Bioscience). PreScission cleavage of GST–Lyn-SH3 fusion protein leads to a Lyn-SH3 protein with an additional GPLGS sequence at the N terminus. The numbering scheme used throughout this paper will refer to the expressed protein.
Overexpression and purification of Tip (amino acids M140–A191) was performed as described in Bauer et al. (2004). Lyn-SH3 was overexpressed and purified as described for Lck-SH3, using PreScission protease cleavage instead of thrombine cleavage (Schweimer et al. 2002). For 13C and 15N labeling, M9 minimal medium was used with [U-99% 13C]-glucose and [U-98% 15N]-NH4Cl as the sole carbon and nitrogen sources, respectively. Fractions containing protein were identified by SDS-PAGE, dialyzed against 2 mM potassium phosphate (pH 6.4) with 1 mM NaCl, and concentrated by lyophilization.
Synthetic peptides
For fluorescence studies, two synthetic peptides comprising residues A168–G187 (“Tip”) or residues A168–N185 (“TipΔC”) of Tip were purchased from Coring. The length of these peptides was chosen based on the NMR spectroscopic data to either cover the complete Lyn-binding motif or only the proline-rich core binding motif. Because of their identical binding properties Tip(140–191) and Tip(168–187) will be consistently termed “Tip” throughout the manuscript. For undisturbed detection of SH3 fluorescence upon addition of ligand, W170 was replaced by leucine, which had been shown previously to have no effect on the binding affinity (Schweimer et al. 2002). In addition, the N and C termini of all peptides was acetylated and amidated, respectively, to adjust the propensities in the peptide to those of the intact protein (Chakrabartty et al. 1993).
Fluorescence spectroscopy and calculation of binding constants
All fluorescence spectra were measured in a F-4500 fluorescence spectrophotometer (Hitachi) at an excitation wavelength of 280 nm and an emission wavelength of 340 nm at 294 K. A semi-micro quartz fluorescence cell (light path 10 × 4 mm) with magnetic stirrer was used. Stock solutions of up to 5 mM of synthesized Tip or TipΔC were added in small increments to 700 μL of 0.5 μM SH3 domain in 50 mM Tris-HCl, 150 mM NaCl (pH 7.4), and subsequently stirred for 2 min. Afterward, the fluorescence was recorded for 30 sec and averaged. Since the concentration of the SH3 domain was low compared with the ligand, the experimental data were fitted to the standard equation (Posern et al. 1998)
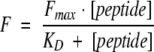 |
where [peptide] gives the final ligand concentration at each measurement point, F is the measured protein fluorescence intensity at the particular peptide concentration, and Fmax is the observed maximal fluorescence intensity of the protein when saturated with the peptide. Nonlinear regression curve fitting was carried out to fit the experimental data to the equation, with Fmax and KD as fitted parameters. The change in protein concentration that occurred as a result of peptide addition was properly corrected.
NMR spectroscopy
NMR experiments were recorded on Bruker 400-MHz, 600-MHz, 700-MHz, and 800-MHz spectrometers equipped with pulsed-field gradient capabilities at 298 K. NMR samples for the structure determination of the free Lyn-SH3 domain contained 1.6 mM 13C,15N-labeled protein in 100 mM potassium phosphate and 50 mM sodium chloride (pH 6.4), in H2O/D2O (9:1). For the structure determination of the Lyn-SH3–Tip complex, NMR samples containing either 1.6 mM 13C,15N-labeled Lyn-SH3 and 3.4 mM unlabeled Tip, or 1.6 mM 13C,15N-labeled Tip and 3.3 mM unlabeled Lyn-SH3 in 100 mM potassium phosphate and 50 mM sodium chloride (pH 6.4), in H2O/D2O (9:1) were used.
Standard double and triple resonance experiments were conducted in order to assign the resonances (Grzesiek and Bax 1993; Sattler et al. 1999). NOE distance restraints were derived from 3D 1H,13C-NOESY-HSQC (Cavanagh et al. 1996) and 1H,15N-NOESY-HSQC (Talluri and Wagner 1996) and from 4D 1H,13C-HMQC-NOESY-1H,13C-HSQC (Clore et al. 1991) and 1H,13C-HMQC-NOESY-1H,15N-HSQC (Kay et al. 1990) experiments with a mixing time of 120 msec.
Slowly exchanging amide protons were identified from a series of 1H,15N-HSQCs that were recorded after lyophilized protein was dissolved in D2O. The {1H}15N-NOE experiments were recorded using the pulse sequences of Dayie and Wagner (1994). The relaxation delay was 4 sec, and the proton saturation was performed by 120° high-power pulses with an inter-pulse delay of 5 msec for the final 3 sec of the relaxation delay of the saturation experiment.
For measuring DN,HN and DCα,Hα residual dipolar couplings, 10–18 mg/mL Pf1 phage suspension (Profos AG) was added and measurements were conducted at 600-MHz proton frequency by J-modulated HSQC experiments (Tjandra et al. 1997). The isotropic scalar couplings were measured using a sample without phages. For structure calculation, DCα,Hα residual dipolar couplings were weighted by a factor of −0.4784 relative to the DN,HN residual dipolar couplings (Bax et al. 2001).
The binding of Tip to Lyn-SH3 was followed by chemical shift disturbance measured by 1H,15N-HSQC experiments during titration of Tip to the 15N-labeled Lyn-SH3 domain (end concentrations of 0.2 mM for Lyn-SH3 and 0.9 mM Tip). The normalized shift was calculated according to
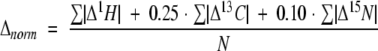 |
where Δ1H, Δ13C, and Δ15N represent the chemical shift in parts per million, and N, the number of shifts per amino acid.
Structure calculation and analysis
On the basis of 764 and 1031 experimental derived restraints (Table 1), the structures of the unbound Lyn-SH3 and the Lyn-SH3–Tip complex were calculated, respectively. NOE cross-peaks were manually classified as strong, medium, or weak according to their intensities and converted into distance restraints of < 2.7, 3.5, or 5.0 Å, respectively (Clore et al. 1987). Scalar 3JHN,Hα coupling constants of either < 6.0 Hz or > 8.0 Hz were restrained to adopt backbone torsion angles between −80° and −40° or between −160° and −80°, respectively (Karplus 1959; Pardi et al. 1984).
Slow exchanging hydrogens were identified from a series of HSQC spectra in D2O and appropriate acceptors were identified from initial structures calculated without hydrogen bond restraints. For each of the assigned 17 hydrogen bonds, the distance between the amide proton and the acceptor was restrained to < 2.3 Å and the distance between the amide nitrogen and the acceptor to < 3.1 Å. Salt-bridge restraints between D26 of Lyn-SH3 and R182 of Tip were incorporated as additional distance restraints after verification by a 1-nsec free molecular dynamics simulation of the complex in explicit water with Amber 7.0 using standard protocols (University of California, San Francisco).
This structural information served as an input for the calculation of 120 structures using restrained molecular dynamics with XPLOR-NIH-1.2.1 (Schwieters et al. 2003). A three-stage simulated annealing protocol (Nilges and O’Donoghue 1998) with floating assignment of prochiral groups (Folmer et al. 1997) was carried out using the following simulation procedure: For conformational space sampling, 60 psec with a time step of 3 fsec were simulated at a temperature of 2000 K, followed by 60 psec of slow cooling to 1000 K, and 30 psec of cooling to 100 K, both with a time step of 2 fsec. After simulated annealing, the structures were subjected to 400 steps of energy minimization. The 60 lowest energy structures were subject to refinement with RDCs (DN,HN and DCα,Hα) as described previously (Schweimer et al. 2002).
Of the 60 structures resulting from the final round of structure refinement, the 20 lowest energy structures having no NOE distance restraint violations > 0.1 Å and no ϕ angle restraint violations > 0.5 ° were selected for further characterization. The geometry of the structures, structural parameters, and elements of secondary structure were analyzed using the programs DSSP (Kabsch and Sander 1983), PRO-CHECK (Laskowski et al. 1996), PROMOTIF (Hutchinson and Thornton 1996), and LIGPLOT (Wallace et al. 1995). For the graphical presentation of the structures, MOLMOL (Koradi et al. 1996) was used.
The assignment and coordinates for the free Lyn-SH3 and the Lyn-SH3–Tip complex have been deposited in the BMRB (entries 6261 and 6456) and in the PDB (entries 1w1f and 1wa7).
Molecular dynamics simulations and binding energy calculation
Molecular dynamics (MD) simulations were done as described previously (Wartha et al. 2005). The binding free energy (ΔGb) is calculated following the standard molecular mechanics/Poisson Boltzmann surface area (MM/PBSA) approach (Srinivasan et al. 1998; Kollman et al. 2000; Wang et al. 2001) according to
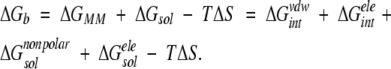 |
ΔGMM is calculated from the MM interaction energies between the ligand and the receptor, which is the sum of the respective van der Waals (ΔGintvdw) and electrostatic (ΔGintele) contributions. In analogy, the solvation energy ΔGsol is divided into two parts: ΔGsolnonpolar and ΔGsolele. ΔGb was obtained using the MM/PBSA module in the AMBER 7 program suite (Pearlman et al. 1995), which interfaces the program DelPhi 4 (Rocchia et al. 2001, 2002) for the calculation of the electrostatic contribution of the solvation energy (ΔGsolele). Entropy contributions (TΔS) to the binding were estimated by normal-mode analysis (Wang et al. 2001). In order to increase the accuracy of the results, all calculated energies were averaged over four independent MD simulations, which started from different structures of the NMR ensemble.
Electronic supplemental material
The supplementary material contains two figures showing the {1H}-15N heteronuclear NOE and the 1H,15N-HSQC spectrum of Lyn-SH3-bound Tip, respectively. The supplementary table contains the calculated free binding energies of LynSH3 for Tip and TipΔC.
Acknowledgments
This work was supported by grants from the Deutsche For-schungsgemeinschaft to H.S. and P.R. (SFB466, C11 and A5). We thank Prof. Franz X. Schmid for providing the opportunity to measure fluorescence spectra in his laboratory.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051563605.
Supplemental material: see www.proteinscience.org
References
- Arold, S., Franken, P., Strub, M.P., Hoh, F., Benichou, S., Benarous, R., and Dumas, C. 1997. The crystal structure of HIV-1 Nef protein bound to the Fyn kinase SH3 domain suggests a role for this complex in altered T cell receptor signaling. Structure 5 1361– 1372. [DOI] [PubMed] [Google Scholar]
- Bauer, F., Hofinger, E., Hoffmann, S., Ros sch, P., Schweimer, K., and Sticht, H. 2004. Characterization of Lck-binding elements in the herpesviral regulatory Tip protein. Biochemistry 43 14932–14939. [DOI] [PubMed] [Google Scholar]
- Bax, A., Kontaxis, G., and Tjandra, N. 2001. Dipolar couplings in macromolecular structure determination. Methods Enzymol. 339 127–174. [DOI] [PubMed] [Google Scholar]
- Cavanagh, J., Fairbrother, W.J., Palmer III, A.G., and Skelton, N.J. 1996. Protein NMR spectroscopy. Academic Press, New York.
- Chakrabartty, A., Doig, A.J., and Baldwin, R.L. 1993. Helix capping propensities in peptides parallel those in proteins. Proc. Natl. Acad. Sci. 90 11332–11336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clore, G.M., Gronenborn, A.M., Nilges, M., and Ryan, C.A. 1987. Three-dimensional structure of potato carboxypeptidase inhibitor in solution. A study using nuclear magnetic resonance, distance geometry, and restrained molecular dynamics. Biochemistry 26 8012–8023. [DOI] [PubMed] [Google Scholar]
- Clore, G.M., Kay, L.E., Bax, A., and Gronenborn, A.M. 1991. Four-dimensional 13C/13C-edited nuclear Overhauser enhancement spectroscopy of a protein in solution: Application to interleukin 1 β. Biochemistry 30 12–18. [DOI] [PubMed] [Google Scholar]
- Dayie, K.T. and Wagner, G. 1994. Relaxation-rate measurements for 15N-1H groups with pulsed-field gradients and preservation of coherence pathways. J. Magn. Reson. 111 121–126. [Google Scholar]
- Dutta, K., Shi, H., Cruz-Chu, E.R., Kami, K., and Ghose, R. 2004. Dynamic influences on a high-affinity, high-specificity interaction involving the C-terminal SH3 domain of p67phox. Biochemistry 43 8094–8106. [DOI] [PubMed] [Google Scholar]
- Fazi, B., Cope, M.J., Douangamath, A., Ferracuti, S., Schirwitz, K., Zucconi, A., Drubin, D.G., Wilmanns, M., Cesareni, G., and Castagnoli, L. 2002. Unusual binding properties of the SH3 domain of the yeast actin-binding protein Abp1: Structural and functional analysis. J. Biol. Chem. 277 5290–5298. [DOI] [PubMed] [Google Scholar]
- Feng, S., Chen, J.K., Yu, H., Simon, J.A., and Schreiber, S.L. 1994. Two binding orientations for peptides to the Src SH3 domain: Development of a general model for SH3-ligand interactions. Science 266 1241–1247. [DOI] [PubMed] [Google Scholar]
- Feng, S., Kasahara, C., Rickles, R.J., and Schreiber, S.L. 1995. Specific interactions outside the proline-rich core of two classes of Src homology 3 ligands. Proc. Natl. Acad. Sci. 92 12408–12415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folmer, R.H., Hilbers, C.W., Konings, R.N., and Nilges, M. 1997. Floating stereospecific assignment revisited: Application to an 18 kDa protein and comparison with J-coupling data. J. Biomol. NMR 9 245–258. [DOI] [PubMed] [Google Scholar]
- Ghose, R., Shekhtman, A., Goger, M.J., Ji, H., and Cowburn, D. 2001. A novel, specific interaction involving the Csk SH3 domain and its natural ligand. Nat. Struct. Biol. 8 998–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzesiek, S. and Bax, A. 1993. The importance of not saturating H2O in protein NMR. Application to sensitivity enhancement and NOE measurements. J. Am. Chem. Soc. 115 12593–12594. [Google Scholar]
- Hutchinson, E.G. and Thornton, J.M. 1996. PROMOTIF—A program to identify and analyze structural motifs in proteins. Protein Sci. 5 212–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch, W. and Sander, C. 1983. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22 2577–2637. [DOI] [PubMed] [Google Scholar]
- Karplus, M. 1959. Contact electron-spin coupling of nuclear magnetic moments. J. Chem. Phys. 30 11–15. [Google Scholar]
- Kay, L.E., Clore, G.M., Bax, A., and Gronenborn, A.M. 1990. Four-dimensional heteronuclear triple-resonance NMR spectroscopy of interleukin- 1 β in solution. Science 249 411–414. [DOI] [PubMed] [Google Scholar]
- Kollman, P.A., Massova, I., Reyes, C., Kuhn, B., Huo, S., Chong, L., Lee, M., Lee, T., Duan, Y., Wang, W., et al. 2000. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 33 889– 897. [DOI] [PubMed] [Google Scholar]
- Koradi, R., Billeter, M., and Wuthrich, K. 1996. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 14 51–55, 29–32. [DOI] [PubMed] [Google Scholar]
- Larson, S.M. and Davidson, A.R. 2000. The identification of conserved interactions within the SH3 domain by alignment of sequences and structures. Protein Sci. 9 2170–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski, R.A., Rullmann, J.A., MacArthur, M.W., Kaptein, R., and Thornton, J.M. 1996. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8 477–486. [DOI] [PubMed] [Google Scholar]
- Lee, C.H., Saksela, K., Mirza, U.A., Chait, B.T., and Kuriyan, J. 1996. Crystal structure of the conserved core of HIV-1 Nef complexed with a Src family SH3 domain. Cell 85 931–942. [DOI] [PubMed] [Google Scholar]
- Lim, W.A., Richards, F.M., and Fox, R.O. 1994. Structural determinants of peptide-binding orientation and of sequence specificity in SH3 domains. Nature 372 375–379. [DOI] [PubMed] [Google Scholar]
- Morton, C.J. and Campbell, I.D. 1994. SH3 domains. Molecular “Velcro.” Curr. Biol. 4 615–617. [DOI] [PubMed] [Google Scholar]
- Nilges, M. and O’Donoghue, S.I. 1998. Ambiguous NOEs and automated NOE assignment. Prog. Nucl. Mag. Res. Sp. 32 107–139. [Google Scholar]
- Pardi, A., Billeter, M., and Wuthrich, K. 1984. Calibration of the angular dependence of the amide proton-C α proton coupling constants, 3JHN α, in a globular protein. Use of 3JHN α for identification of helical secondary structure. J. Mol. Biol. 180 741–751. [DOI] [PubMed] [Google Scholar]
- Pearlman, D.A., Case, D.A., Caldwell, J.W., Ross, W.S., Cheatham 3rd, T.E., DeBolt, S., Ferguson, D., Seibel, G., and Kollman, P. 1995. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 91 1–41. [Google Scholar]
- Pisabarro, M.T. and Serrano, L. 1996. Rational design of specific highaffinity peptide ligands for the Abl-SH3 domain. Biochemistry 35 10634–10640. [DOI] [PubMed] [Google Scholar]
- Pisabarro, M.T., Serrano, L., and Wilmanns, M. 1998. Crystal structure of the abl-SH3 domain complexed with a designed high-affinity peptide ligand: Implications for SH3–ligand interactions. J. Mol. Biol. 281 513–521. [DOI] [PubMed] [Google Scholar]
- Posern, G., Zheng, J., Knudsen, B.S., Kardinal, C., Muller, K.B., Voss, J., Shishido, T., Cowburn, D., Cheng, G., Wang, B., et al. 1998. Development of highly selective SH3 binding peptides for Crk and CRKL which disrupt Crk-complexes with DOCK180, SoS and C3G. Oncogene 16 1903–1912. [DOI] [PubMed] [Google Scholar]
- Reiss, C., Niedobitek, G., Hör, S., Lisner, R., Friedrich, U., Bodemer, W., and Biesinger, B. 2002. Peripheral T-cell lymphoma in herpesvirus saimiri-infected tamarins: Tumor cell lines reveal subgroup-specific differences. Virology 294 31–46. [DOI] [PubMed] [Google Scholar]
- Rocchia, W., Alexov, E., and Honig, B. 2001. Extending the applicability of the nonlinear Poisson-Boltzmann equation: Multiple dielectric constants and multivalent ions. J. Phys. Chem. 105 6507–6514. [Google Scholar]
- Rocchia, W., Sridharan, S., Nicholls, A., Alexov, E., Chiabrera, A., and Honig, B. 2002. Rapid grid-based construction of the molecular surface and the use of induced surface charge to calculate reaction field energies: Applications to the molecular systems and geometric objects. J. Comput. Chem. 23 128–137. [DOI] [PubMed] [Google Scholar]
- Sattler, M., Schleucher, J., and Griesinger, C. 1999. Heteronuclear multi-dimensional NMR experiments for the structure determination of proteins in solution employing pulsed field grandients. Prog. Nucl. Mag. Res. Sp. 34 93–158. [Google Scholar]
- Schweimer, K., Hoffmann, S., Bauer, F., Friedrich, U., Kardinal, C., Feller, S.M., Biesinger, B., and Sticht, H. 2002. Structural investigation of the binding of a herpesviral protein to the SH3 domain of tyrosine kinase Lck. Biochemistry 41 5120–5130. [DOI] [PubMed] [Google Scholar]
- Schwieters, C.D., Kuszewski, J.J., Tjandra, N., and Clore, G.M. 2003. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 160 65–73. [DOI] [PubMed] [Google Scholar]
- Srinivasan, J., Cheatham III, T.E., Cieplak, P., Kollman, P.A., and Case, D.A. 1998. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate-DNA helices. J. Am. Chem. Soc. 120 9401– 9409. [Google Scholar]
- Talluri, S. and Wagner, G. 1996. An optimized 3D NOESY-HSQC. J. Magn. Reson. 112 200–205. [DOI] [PubMed] [Google Scholar]
- Tjandra, N., Omichinski, J.G., Gronenborn, A.M., Clore, G.M., and Bax, A. 1997. Use of dipolar 1H-15N and 1H-13C couplings in the structure determination of magnetically oriented macromolecules in solution. Nat. Struct. Biol. 4 732–738. [DOI] [PubMed] [Google Scholar]
- Wallace, A.C., Laskowski, R.A., and Thornton, J.M. 1995. LIGPLOT: A program to generate schematic diagrams of protein–ligand interactions. Protein Eng. 8 127–134. [DOI] [PubMed] [Google Scholar]
- Wang, W., Lim, W.A., Jakalian, A., Wang, J., Wang, J., Luo, R., Bayly, C.I., and Kollman, P.A. 2001. An analysis of the interactions between the Sem-5 SH3 domain and its ligands using molecular dynamics, free energy calculations, and sequence analysis. J. Am. Chem. Soc. 123 3986–3994. [DOI] [PubMed] [Google Scholar]
- Wartha, F., Horn, A.H.C., Meiselbach, H., and Sticht H. 2005. Molecular dynamics simulations of HIV-1 protease suggest different mechanisms contributing to drug resistance. J. Chem. Theory Comput. 1 315–324. [DOI] [PubMed] [Google Scholar]
- Yu, H., Chen, J.K., Feng, S., Dalgarno, D.C., Brauer, A.W., and Schreiber, S.L. 1994. Structural basis for the binding of proline-rich peptides to SH3 domains. Cell 76 933–945. [DOI] [PubMed] [Google Scholar]