

Abstract
We present here the structure of Yer010c protein of unknown function, solved by Multiple Anomalous Diffraction and revealing a common fold and oligomerization state with proteins of the regulator of ribonuclease activity A (RraA) family. In Escherichia coli, RraA has been shown to regulate the activity of ribonuclease E by direct interaction. The absence of ribonuclease E in yeast suggests a different function for this family member in this organism. Yer010cp has a few supplementary secondary structure elements and a deep pseudo-knot at the heart of the protein core. A tunnel at the interface between two monomers, lined with conserved charged residues, has unassigned residual electron density and may constitute an active site for a yet unknown activity.
Keywords: structural genomics, crystal structure, pseudo-knot
The aim of structural genomics (SG) projects worldwide is the high-throughput determination of protein structures with the main emphasis on discovering new folds (Stevens et al. 2001; Burley and Bonanno 2002; Quevillon-Cheruel et al. 2004). The rationale behind this choice was to sufficiently explore the protein structure universe to facilitate modeling of the proteins identified in genomes sequencing projects, thereby bridging the gap between the wealth of protein sequences available and the comparably scarce structural data available for these proteins.
In our Yeast Structural Genomics project (http://genomics.eu.org/) (Quevillon-Cheruel et al. 2004) proteins encoded by the Saccharomyces cerevisiae genome without any close structural homologs were chosen (sequence identity with proteins of known structure inferior to 30%), frequently targeting proteins of unknown biochemical or cellular function. In many cases, the newly solved structure belongs to a large structural superfamily, whose members harbor a wide range of functions, or more rarely contain entirely new folds (Leulliot et al. 2004; Quevillon-Cheruel et al. 2005). In this case, de novo function identification is greatly aided by bioinformatical methods, for example, to aid identification of active sites (Stark et al. 2004) and by information from high-throughput functional genomics studies (localization, systematic deletions, interacting partners, etc.).
The Yer010c gene, one of our YSG targets, codes for a 25-kDa protein of unknown function whose fold could not be predicted at the start of the project. In the NCBI Conserved Domain Database and PFAM, Yer010cp was found to belong to the demethylmenaquinone methyltransferase superfamily. Members of this family are enzymes that convert dimethylmenaquinone to menaquinone (or vitamin K2) in the final step of menaquinone biosynthesis. While finishing the structure of Yer010cp, two groups independently published the structure from homologs of Yer010cp from Escherichia coli, Thermus thermophilus (Monzingo et al. 2003; Rehse et al. 2004), and Mycobacterium tuberculosis (Johnston et al. 2003). These structures showed that these proteins had no known methyltransferase fold and clarified a wrongful annotation of the E. coli ortholog. Moreover, the E. coli homolog of Yer010cp was shown to be involved in the global modulation of RNA abundance in E. coli by inhibiting the activity of RNase E through direct interaction (Lee et al. 2003). The protein family was therefore renamed RraA (regulator of ribonuclease E activity A).
Here we present the crystal structure Yer010cp, the yeast member of the RraA-like family. The similarity to RraA is puzzling since no RNase E has been identified in yeasts, and raises the question of the function of this protein. Identification and analysis of similar domains in a variety of enzymes places proteins of the RraA family in a structural superfamily containing proteins with diverse enzymatic activities. The presence of residual electron density in a conserved pocket, suggests the location of a putative active site.
Results and Discussion
Overall structure
The structure of the Yer010cp protein was solved by multiple anomalous diffraction using selenium substituted protein (see Table 1 for data collection and refinement statistics). Six molecules are present in the asymmetric unit. Analysis of the packing and the quaternary arrangement indicates that Yer010cp crystallizes with two homotrimers in the asymmetric unit. Analytical size exclusion chromatography indicates that the protein migrates as a trimer in solution indicating that the crystal homotrimer represents the solution oligomeric state.
Table 1.
Data collection and refinement statistics
| Peak | Edge | Remote | Native | |
| Wavelength (Å) | 0.9793 | 0.9797 | 0.9763 | 0.934 |
| Unit-cell parameters a, b, c (Å) | 128.05, 255.63, 48.52 | 128.19, 255.07, 48.17 | ||
| Resolution (Å) | 2.56 | 1.70 | ||
| Total no. of refl. | 278,913 | 239,541 | 203,543 | 956,280 |
| Total no. of unique refl. | 47,594 | 47,380 | 48,116 | 174,329 |
| Multiplicity | 5.9 | 5.1 | 4.2 | 5.49 |
| Rmergea | 0.087 | 0.091 | 0.089 | 0.125 |
| I/σ(I) | 17.2 | 15.0 | 13.2 | 12.2 |
| Overall completeness (%) | 90.3 | 90.1 | 90.4 | 99.3 |
| FoM before/after density modification | 0.47/0.77 | |||
| Reflections (work/test) | 165,607/8721 | |||
| Rcryst/Rfreeb | 0.15/0.18 | |||
| Nonhydrogen atoms | 12,434 | |||
| Water molecules | 1670 | |||
| RMSD, bonds (Å) | 0.015 | |||
| Angles (°) | 1.53 | |||
| Mean B factor (Å2) | 13.4 |
aRmerge = ∑h∑i|Ihi − 〈Ih〉|/∑h∑i|hi, where Ihi is the ith observation of the reflection h, while 〈Ih〉 is the mean intensity of reflection h.
bRfactor = ∑||Fo| − |Fc||/|Fo|. Rfree was calculated with a set of randomly selected reflections (5%).
The Yer010cp monomer is composed of 10 β-strands and six α-helices, and is best described as an α–β–β–α sandwich (Fig. 1A ▶). The first β-layer is composed of a six-stranded β-sheet (S1) in the order β1β5β4β3β2β6, with the two exterior strands anti-parallel. The second β-layer is composed of two anti-parallel β-sheets, the first one (S2) containing the long bent β2 strand common to S1 and S2, β9, and β10, and the second one (S3) containing β7 and β8. The three β-sheets pack against each other by extensive hydrophobic interactions. The N-terminal helices α1 and α2 pack against S2 while α3 and α4, inserted between β3–β4 and β4–β5, respectively, pack against S1. The C terminus contains two long helices (α5 and α6) that are involved in oligomerization of the protein (see below).
Figure 1.
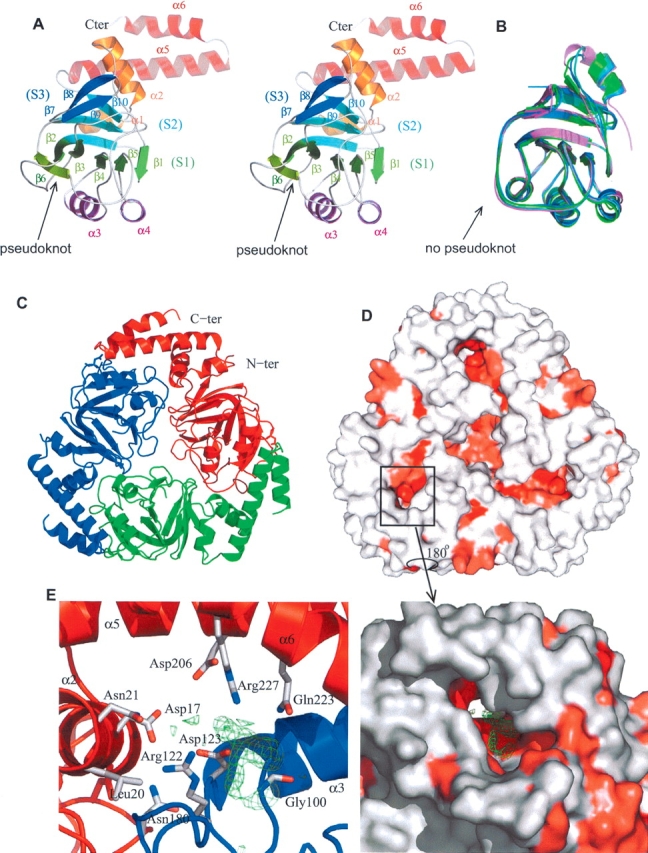
(A) Ribbon stereo representation of the structure of the Yer010cp monomer. The N- and C-terminal helices are colored in orange and in red, respectively. The second α-helical layer is colored purple. The (S1), (S2), and (S3) sheets are colored green, light blue, and dark blue, respectively. Protein figures are generated by Pymol (http://www.pymol.org). (B) Structural superposition of Yer010cp homologs in the same orientation as A: E. coli (dark blue), T. thermophilus (green), M. tuberculosis (light blue), and V. cholerae (purple). (C) Ribbon representation of the Yer010cp trimer. The three chains are in different colors. (D) Surface representation of the Yer010cp trimer. The surface is colored in increasing shades of red according to residue conservation (red most conserved) as determined by the consurf server (Glaser et al. 2003). (E) Close-up view of the conserved pocket between two monomers in surface (right) or cartoon (left) representation. The residual electron density (Fo−Fc map contoured at 3 σ) is shown in green. The residues lining the tunnel are shown in stick representation.
Quaternary structure
The Yer010cp monomer associates as a ring-shaped homotrimer of 65 Å outer and 9 Å inner diameter (Fig. 1C ▶). Monomer contacts involve packing of the α2 helix, the α1–β1 and β7–β8 loops with the β3–α3 and β5–β6 loops of the neighboring monomer. The C-terminal helices α5 and α6 pack against α3 on the exterior of the ring creating a propeller-like stacking arrangement of the three monomers. Oligomerization buries 2500 Å2 of surface-accessible area per monomer (20% of the total monomer surface), involving eight hydrogen bonds and one salt bridge (Asp17–Arg122).
Structural homologs
A search for structural homologs on the EBI ssm server (http://www.ebi.ac.uk/msd-srv/ssm/) retrieved proteins of the RraA-like (aka MenG) family: RraA from E. coli, P83846from T. thermophilus, Rv3853 from M. tuberculosis, and Q9KPK1 from Vibrio cholerae. On average, these proteins share 21% sequence identity with Yer010cp and superpose with an RMSD of 1.9 Å (for detailed statistics see Table 2). The bacterial and archaeal proteins share 44% sequence identity and superpose with an RMSD of 1.1 Å on average, making Yer010cp the most dissimilar member of this superfamily. All of these proteins seem to form trimers in the crystal, generated by either local or crystal symmetry axes. The trimers all adopt the same ring-like structure as Yer010cp with an average RMSD of 2 Å.
Table 2.
Structural neighbors of Yer010cp
| Protein | PDB code | RMSD (Å) | No. of residues aligned | Sequence identity |
| RraA-like family | ||||
| RraA E. coli | 1q5x | 2.08 Å | 145 | 18% |
| P83846 T. thermophilus | 1j3l | 1.70 Å | 137 | 23% |
| Rv3853 M. tuberculosis | lnxj | 1.95 Å | 138 | 21% |
| Q9KPK1 V. cholerae | 1vi4 | 1.95 Å | 138 | 23% |
| Other protein families | ||||
| Metal dependent metal hydrolase | 1r61 | 2.80 Å | 114 | 5% |
| 3-Isopropylmalate isomerase | 1v71 | 2.58 Å | 87 | 11% |
| Apical domain of GroEL | 1dkd | 3.21 Å | 86 | 5% |
| N-terminal domain of enzyme I | 3eze | 2.70 Å | 101 | 18% |
Although these proteins share the same fold, the yeast member of this family has some specific structural features. Most strikingly, the bacterial and archaeal proteins do not contain the N-terminal helix, α1, and the two C-terminal helices, α5 and α6. Therefore, one of the α helical layers is reduced to one α-helix in the other proteins, which were therefore described as β–β–α sandwiches. In Yer010cp, the two C-terminal helices are involved in intermolecular interactions. As these α-helices are absent in the other proteins of the RraA family, stabilization of the trimer does not seem to require the C-terminal extension, which in yeast could have a role in the formation of the protein’s active site (see below). From Blast sequence alignment the C-terminal extension as observed in Yer010cp seems also to be present in orthologs from other organisms (Fig. 2 ▶).
Figure 2.

Alignment of the Yer010cp sequence to eukaryotic (Candida albicans, Neurospora crassa, A. thaliana) to the E. coli and T. thermophilus orthologs whose structure has been solved and to the RraA-like domains in L. pneumophila and M. barkeri, which are fused to another protein domain (excluded from the alignment).
In the bacterial RraAs, the center of the ring is “hollow,” whereas in Yer010cp the longer loops between β3 and α3 from the three monomers interact at the center of the ring and effectively fill the hole (Fig. 1D ▶).
A knotable member of the RraA family
Another peculiarity of Yer010cp compared to other members of the family is the absence of an alpha helix between β2 and β3, although the loop has a helical turn (Fig. 1A,B ▶). This region of Yer010cp deviates from the bacterial proteins in its local structure. Indeed, in Yer010cp, the β6 strand is threaded inside the β2–β3 loop, whereas in the bacterial proteins the presence of the extra α-helix and a much shorter loop between this helix and β2 makes this impossible, and the chain simply stacks on top of the loop. In Yer010cp, this unique structural feature was initially identified by us as a deep knot located at residue 150, which would mean that at minimum 80 residues would have to be threaded in the loop for the protein to fold (Fig. 1A,B ▶).
Knots in proteins are known to be extremely rare; less than a dozen proteins with true knots have been identified (Taylor 2000). True topological knots in proteins are defined in analogy to strings by the presence of a “loop that tightens when pulled.” This definition has been incorporated by Taylor et al. in a computer algorithm to check for knots in protein structures. Application of this program on the Yer010cp structure revealed that the protein does not contain a topological knot: No knot is formed by pulling the protein chain from both ends. Closer inspection revealed that the knot in Yer010cp is in fact a pseudo-knot (Taylor and Lin 2003). A pseudo-knot is formed because proteins are different from pieces of string, as some regions can “stick” together, either through disulphide bridges (covalent knot), or by weaker hydrogen bonds (pseudo-knots).
The knot initially identified in the Set methylase domain was also shown to be a pseudo-knot formed by threading the 30 C-terminal acids through a loop fixed by hydrogen bonds to a short 3–10 helix and a β-strand (Taylor et al. 2003). The situation in Yer010cp is somewhat unique. The knot loop (residues 56–74), “rigidly” held by the seven hydrogen bonds of the two parallel β2 and β3 strands, wraps around the protein chain at residue 150, which leaves 80 or 150 residues on either side of the pseudo-knot. In contrast to the Set domain pseudo-knot, the chain on both sides of the Yer010cp pseudo-knot is extremely rich in secondary structure elements, precluding a mechanism where the chain is threaded through the loop, as the secondary structures would make it too bulky.
Location of a putative active site
During refinement of the structure, unassigned residual electron density was clearly visible in a tunnel located at the interface between two monomers (Fig. 1D ▶). This tunnel is formed by helix α3 and α4 from one monomer and helix α2, the β9–β10 loop and the α5 and α6 C-terminal helices from the other monomer (Fig. 1E ▶). This residual electron density is observed in the six copies of the tunnel present in the asymmetric unit. The residual density could not be modeled with any compounds present in the protein buffer or crystallization mother liquor and must arise from a solute in the E. coli broth bound by Yer010cp.
Although the residual electron density is quite heterogeneous in the different copies of Yer010cp in the asymmetric unit, several lines of evidence point to the biological relevance of the presence of a bound molecule at this location. Firstly, this pocket is lined with absolutely conserved residues in all the RraA family of proteins (Fig. 1D,E ▶). The solute is sandwiched between the charged residues, Asp17 from α2, Arg122 and Asp123 from the β4–α4 loop, Gly100, and the hydrophobic residues Leu102 and Met103 from α3, and Asp206 and Arg227 from the C-terminal helices α5 and α6 (Fig. 1E ▶). In contrast to Yer010cp, the bacterial and archaeal analogs do not contain the C-terminal helices that form a lid above this cavity, creating a groove rather than a tunnel. In the structure of Rv3853 from M. tuberculosis, residual density at the interface between two monomers revealed a bound tartrate ion from the crystallization buffer, in a location close to the S. cerevisiae-bound compound.
Comparison with other protein families
A search for structural relatives using the EBI ssm server retrieved a number of homologs with lower sequence and structural similarity that belong to different, well-characterized classes of enzymes. This places the RraA protein family in a bigger superfamily of proteins that have a similar fold but which have different oligomerization states or are fused to other domains. This protein family is classified in SCOP under the “swiveling” β/β/α domain fold. Proteins belonging to this diverse family include the phosphohistidine domain (present in pyruvate phosphate dikinase and enzyme I of the PEP:sugar phosphotransferase system), a domain of aconitase, a putative metal dependent hydrolase, and the GroEL apical domain, the carbamonyl phosphate synthetase (small subunit N-terminal domain), the swiveling domain of the glycerol dehydratase reactivase α subunit, and transferrin receptor ectodomain.
Among these, Yer010cp and the RraA-like proteins are most similar to the metal-dependent metal hydrolase (dimer), the 3-isopropylmalate isomerase from Pyrococcus horikoshii (tetramer) (Yasutake et al. 2004), the apical domain of GroEL (domain, 14mer) (Chen and Sigler 1999), and the N-terminal domain of enzyme I (Liao et al. 1996; see Table 2 for detailed similarity scores).
Function of Yer010cp
The present structure of Yer010cp raises interesting questions on the function of the members of the RraA family. E. coli RraA has been shown to bind to RNase E and to inhibit its activity (Lee et al. 2003). RNase E plays a key role in the degradation of mRNA, and overexpression of RraA in E. coli resulted in accumulation of RNase E targeted transcripts. The similarity of Yer010cp with RraA is therefore intriguing, since no RNase E has been detected in yeast or in other eukaryote and archaea genomes in which the RraA-like gene is found. This observation is backed by the fact that RNA degradation follows two distinct pathways in prokaryotes and eukaryotes.
Yer010cp has homologs in bacteria, archaea, fungi, and green plants that are either annotated as RraA like proteins or still misannotated as MenG methytransferases. In two cases, the RraA-like domain is fused to another domain (sequences aligned in Fig. 2 ▶). In one case, the protein fusion is found in three strains of Legionella pneumophila. The protein contains two domains: a C-terminal domain sharing 28% sequence identity with Yer010cp, and an N-terminal domain classified as an isocitrate/isopropylmalate dehydrogenase domain (COG473, PFAM00180), members of the β-decarboxylating dehydrogenase superfamily. This N-terminal domain is found as a fusion domain only in the proteins of these three Legionella strains. It is interesting to note that isopropylmalate isomerase, a member of the aconitase superfamily containing a domain structurally homologous to Yer010cp (Table 2), is the enzyme preceding isopropylmalate dehydrogenase in the leucine biosynthesis pathway. Enzymes of the aconitase and of the β-decarboxylating dehydrogenase superfamilies are involved in consecutive reactions in three distinct pathways: leucine biosynthesis, lysine biosynthesis, and the Krebs cycle (Yasutake et al. 2004). The size of the substrates involved in these reactions is compatible with the residual electron density seen in Yer010cp. Yer010cp may therefore be a metabolic enzyme from a yet unidentified pathway.
The second occurrence of domain fusion is found in six archaeal genomes: Methanosarcina barkeri, Methanococcoides burtonii, Methanococcus maripaludis, Methanosarcina mazei, Methanosarcina acetivorans, and Archaeoglobus fulgidus. The RraA-like domain at the C terminus is fused to a domain belonging to three enzyme families: 3-keto-l-gulonate 6-phosphate decarboxylase, 3- hexulose-6-phosphate synthase, and orotidine 5′ phosphate decarboxylase (COG0269 and related families). The proteins belonging to this suprafamily have been shown to adopt a (β/α)8-barrel structure, which is the fold associated with the most functions in the protein universe (Nagano et al. 2002). Providing clues to the function to the RraA-like proteins on the basis of the fusion of these domains is therefore challenging.
The localization of the well-conserved pocket in Yer010cp is very similar to that of the active site of the phosphohistidine domain containing pyruvate phosphate dikinase (Nagano et al. 2002) and enzyme I of the PEP:sugar phosphotransferase system (Liao et al. 1996). In these proteins, a conserved histidine located on the β4–α4 loop is used for phosphate transfer from ATP or phosphoenolpyruvate to a protein acceptor. The location of this absolutely conserved histidine coincides in Yer010cp with the loop harboring the conserved Arg122 and Asp123. In the E. coli phosphoenolpyruvate: sugar phosphotransferase system, substitution of the active center histidine for aspartate maintains phosphotransfer potential (Napper et al. 2001). It would be tempting to hypothesize that Asp123, conserved in most but not all RraA-like proteins, is involved in a phosphotransfer reaction on a small compound, but no clear residual density supports a covalent link between the compound and Asp123.
Conclusion
The structure of Yer010cp, a yeast member of the RraA family, raises several questions concerning the function of this protein in genomes where no ribonuclease E has been detected. This structure, the first for a eukaryotic RraA family member, contains an intriguing pseudo-knot. The presence of residual electron density in a charged well-conserved pocket and the fusion of some Yer010cp orthologs with other domains associated with some kind of enzymatic activity seem to point to a role in binding and perhaps catalysis of a small unidentified compound.
Materials and methods
Cloning, expression, purification, mutagenesis, and labeling
ORF Yer010c was amplified by PCR using genomic DNA of S. cerevisiae strain S288C as a template. An additional sequence coding for a six-histidine tag was introduced at the 3′ end of the gene during amplification. The PCR product was then cloned into a derivative of pET9 vector. Because only about 10% of the expressed protein was present in the soluble fraction of the Gold(DE3) cells growth at 37°C or 15°C, the coexpression of E. coli molecular chaperones was tried (Nishihara et al. 1998; Tresaugues et al. 2004). The expression of GroEL-GroES and DnaK-DnaJ-GrpE chaperones were induced by adding 2 mg of arabinose/mL of culture, 15 min before the addition of IPTG. This enabled the quasitotality of the protein to be recovered in the soluble state when the induction (0.3 mM IPTG) was done at 15°C overnight, in 2×YT medium (BIO101 Inc.) supplemented with kanamycin at 50 μg/mL. Cells were harvested by centrifugation, resuspended in 20 mM Tris-HCl (pH 7.5), 200 mM NaCl, 5 mMβ-mercaptoethanol, and stored overnight at −20°C. Cell lysis was completed by sonication. The his-tagged protein was purified on a Ni-NTA column (Qiagen Inc.), eluted with imidazole, and loaded on to a SuperdexTM200 column (Amersham Pharmacia Biotech) equilibrated in 20 mM Tris-HCl (pH 6.8), 150 mM NaCl, 5% glycerol, 10 mM β mercaptoethanol.
The homogeneity of the protein was checked by SDS-PAGE and mass spectrometry. An analytical size exclusion chromatography was performed on a calibrated SuperdexTM200 column and was consistent with a trimeric level of oligomerization state in solution.
As the native protein contained only two internal methionines for 234 residues in total, a mutant exhibiting three additional methionines in place of Phe31, Phe118, and Leu207 was constructed by directed mutagenesis for SeMet labeling. The labeling of the protein with Se-Met was performed according to standard protocols (Quevillon-Cheruel et al. 2005). The labeled mutant was purified according to the same experimental protocol as the wild-type protein, and checked by mass spectrometry to control the labeling level.
Crystallization and data collection
Both native and mutant proteins (10 mg/mL) crystallized at 293 K by the hanging drop vapor diffusion method from 1:1 μL drops of protein and precipitant containing 18% PEG 8K, 0.1 M sodium cacodylate (pH 6), 0.1 M magnesium sulphate. Crystals were transferred in the mother liquor containing 30% ethylene glycol prior to flash freezing in liquid nitrogen.
X-ray diffraction data from a crystal of the mutant SeMet substituted protein and from a native crystal were respectively collected on beamlines BM30A and ID14–2 at the ESRF. Data processing was carried out with MOSFLM and SCALA (Leslie 1992) and DENZO and SCALEPACK (Otwinowski and Minor 1997). The crystals belong to space group P21212 with six molecules per asymmetric unit, corresponding to 51% solvent content. The cell parameters and data collection statistics are reported in Table 1.
Structure solution and refinement
The structure was solved by multiple anomalous diffraction (MAD) using data to 2.56 Å resolution collected from a single selenated crystal at three different wavelengths and from a high-resolution 1.7 Å native data set from a nonselenated wild-type protein crystal. The program SHELXD (Schneider and Sheldrick 2002) was used to find an initial set of 10 Se sites. Refinement of the Se sites and phasing were carried out with the program SHARP (Bricogne et al. 2003). Additional sites were included in the refinement after inspection of residual maps. The final substructure model comprises 28 Se atoms. Phase refinement and extension were carried out via density modification with the program SOLOMON, and this resulted in a very high-quality map which allowed 90% of the residues to be built automatically using Arp/warp. The model was fully refined and completed from the native data set using REFMAC and O (Jones et al. 1991; CCP4 1994). Refinement statistics are shown in Table 1. The final model in the crystal contains residues 2–230. All the residues fall in favorable regions of the Ramachandran plot.
Acknowledgments
This work is supported by grants from the Ministère de la Recherche et de la Technologie (Programme Génopoles). We thank William R. Taylor (Division of Mathematical Biology, National Institute for Medical Research, London), who kindly provided us with his program to check for knots in proteins.
Abbreviations
SDS PAGE, sodium dodecyl sulphate polyacrylamide gel electrophoresis
ORF, open reading frame
SG, structural genomics
RMSD, root-mean-square deviation.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051684005.
References
- Bricogne, G., Vonrhein, C., Flensburg, C., Schiltz, M., and Paciorek, W. 2003. Generation, representation and flow of phase information in structure determination: Recent developments in and around SHARP 2.0. Acta Crystallogr. D Biol. Crystallogr. 59 2023–2030. [DOI] [PubMed] [Google Scholar]
- Burley, S.K. and Bonanno, J.B. 2002. Structuring the universe of proteins. Annu. Rev. Genomics Hum. Genet. 3 243–262. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project Number 4. 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50 760–763. [DOI] [PubMed] [Google Scholar]
- Chen, L. and Sigler, P.B. 1999. The crystal structure of a GroEL/peptide complex: Plasticity as a basis for substrate diversity. Cell 99 757–768. [DOI] [PubMed] [Google Scholar]
- Glaser, F., Pupko, T., Paz, I., Bell, R.E., Bechor-Shental, D., Martz, E., and Ben-Tal, N. 2003. ConSurf: Identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 19 163–164. [DOI] [PubMed] [Google Scholar]
- Johnston, J.M., Arcus, V.L., Morton, C.J., Parker, M.W., and Baker, E.N. 2003. Crystal structure of a putative methyltransferase from Mycobacterium tuberculosis: Misannotation of a genome clarified by protein structural analysis. J. Bacteriol. 185 4057–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.H., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron-density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Lee, K., Zhan, X., Gao, J., Qiu, J., Feng, Y., Meganathan, R., Cohen, S.N., and Georgiou, G. 2003. RraA.Aprotein inhibitor of RNase E activity that globally modulates RNA abundance in E. coli. Cell 114 623–634. [PubMed] [Google Scholar]
- Leslie, A. 1992. Joint CCP4 and EACMB newsletter protein crystallography. Daresbury Laboratory, Warrington, UK.
- Leulliot, N., Quevillon-Cheruel, S., Sorel, I., Graille, M., Meyer, P., Liger, D., Blondeau, K., Janin, J., and van Tilbeurgh, H. 2004. Crystal structure of yeast allantoicase reveals a repeated jelly roll motif. J. Biol. Chem. 279 23447–23452. [DOI] [PubMed] [Google Scholar]
- Liao, D.I., Silverton, E., Seok, Y.J., Lee, B.R., Peterkofsky, A., and Davies, D.R. 1996. The first step in sugar transport: Crystal structure of the amino terminal domain of enzyme I of the E. coli PEP:sugar phosphotransferase system and a model of the phosphotransfer complex with HPr. Structure 4 861–872. [DOI] [PubMed] [Google Scholar]
- Monzingo, A.F., Gao, J., Qiu, J., Georgiou, G., and Robertus, J.D. 2003. The X-ray structure of Escherichia coli RraA (MenG), a protein inhibitor of RNA processing. J. Mol. Biol. 332 1015–1024. [DOI] [PubMed] [Google Scholar]
- Nagano, N., Orengo, C.A., and Thornton, J.M. 2002. One fold with many functions: The evolutionary relationships between TIM barrel families based on their sequences, structures and functions. J. Mol. Biol. 321 741–765. [DOI] [PubMed] [Google Scholar]
- Napper, S., Brokx, S.J., Pally, E., Kindrachuk, J., Delbaere, L.T., and Waygood, E.B. 2001. Substitution of aspartate and glutamate for active center histidines in the Escherichia coli phosphoenolpyruvate:- sugar phosphotransferase system maintain phosphotransfer potential. J. Biol. Chem. 276 41588–41593. [DOI] [PubMed] [Google Scholar]
- Nishihara, K., Kanemori, M., Kitagawa, M., Yanagi, H., and Yura, T. 1998. Chaperone coexpression plasmids: Differential and synergistic roles of DnaK-DnaJ-GrpE and GroEL-GroES in assisting folding of an allergen of Japanese cedar pollen, Cryj2, in Escherichia coli. Appl. Environ. Microbiol. 64 1694–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collection in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Quevillon-Cheruel, S., Liger, D., Leulliot, N., Graille, M., Poupon, A., De la Sierra-Gallay, I.L., Zhou, C.Z., Collinet, B., Janin, J., and Van Tilbeurgh, H. 2004. The Paris-Sud yeast structural genomics pilot-project: From structure to function. Biochimie 86 617–623. [DOI] [PubMed] [Google Scholar]
- Quevillon-Cheruel, S., Leulliot, N., Graille, M., Hervouet, N., Coste, F., Benedetti, H., Zelwer, C., Janin, J., and Van Tilbeurgh, H. 2005. Crystal structure of yeast YHR049W/FSH1, a member of the serine hydrolase family. Protein Sci. 14 1350–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehse, P.H., Kuroishi, C., and Tahirov, T.H. 2004. Structure of the RNA-processing inhibitor RraA from Thermus thermophilis. Acta Crystallogr. D Biol. Crystallogr. 60 1997–2002. [DOI] [PubMed] [Google Scholar]
- Schneider, T.R. and Sheldrick, G.M. 2002. Substructure solution with SHELXD. Acta Crystallogr. D Biol. Crystallogr. 58 1772–1779. [DOI] [PubMed] [Google Scholar]
- Stark, A., Shkumatov, A., and Russell, R.B. 2004. Finding functional sites in structural genomics proteins. Structure (Camb.) 12 1405–1412. [DOI] [PubMed] [Google Scholar]
- Stevens, R.C., Yokoyama, S., and Wilson, I.A. 2001. Global efforts in structural genomics. Science 294 89–92. [DOI] [PubMed] [Google Scholar]
- Taylor, W.R. 2000. A deeply knotted protein structure and how it might fold. Nature 406 916–919. [DOI] [PubMed] [Google Scholar]
- Taylor, W.R. and Lin, K. 2003. Protein knots: A tangled problem. Nature 421 25. [DOI] [PubMed] [Google Scholar]
- Taylor, W.R., Xiao, B., Gamblin, S.J., and Lin, K. 2003. A knot or not a knot? SETting the record “straight” on proteins. Comput. Biol. Chem. 27 11–15. [DOI] [PubMed] [Google Scholar]
- Tresaugues, L., Collinet, B., Minard, P., Henckes, G., Aufrere, R., Blondeau, K., Liger, D., Zhou, C.Z., Janin, J., Van Tilbeurgh, H., et al. 2004. Refolding strategies from inclusion bodies in a structural genomics project. J. Struct. Funct. Genomics 5 195–204. [DOI] [PubMed] [Google Scholar]
- Yasutake, Y., Yao, M., Sakai, N., Kirita, T., and Tanaka, I. 2004. Crystal structure of the Pyrococcus horikoshii isopropylmalate isomerase small subunit provides insight into the dual substrate specificity of the enzyme. J. Mol. Biol. 344 325–333. [DOI] [PubMed] [Google Scholar]