

Abstract
We have identified two new lysozyme-like protein families by using a combination of sequence similarity searches, domain architecture analysis, and structural predictions. First, the P5 protein from bacteriophage φ8, which belongs to COG3926 and Pfam family DUF847, is predicted to have a new lysozyme-like domain. This assignment is consistent with the lytic function of P5 proteins observed in several related double-stranded RNA bacteriophages. Domain architecture analysis reveals two lysozyme-associated transmembrane modules (LATM1 and LATM2) in a few COG3926/DUF847 members. LATM2 is also present in two proteins containing a peptidoglycan binding domain (PGB) and an N-terminal region that corresponds to COG5526 with uncharacterized function. Second, structure prediction and sequence analysis suggest that COG5526 represents another new lysozyme-like family. Our analysis offers fold and active-site assignments for COG3926/DUF847 and COG5526. The predicted enzymatic activity is consistent with an experimental study on the zliS gene product from Zymomonas mobilis, suggesting that bacterial COG3926/DUF847 members might be activators of macromolecular secretion.
Keywords: lysozyme, structure prediction, bacteriophage φ8, lysozyme-associated transmembrane modules, macromolecular secretion
A major component of the bacterial cell wall is peptidoglycan, which is made from two alternating sugar monomers: N-acetylmuramic acid (MurNAc) and N-acetyl-glucosamine (GlcNAc) (Schleifer and Kandler 1972). Peptidoglycan layers are cross-linked by amino acids or amino acid derivatives. Lysozymes (Enzyme Committee [E.C.] number 3.2.1.17) degrade peptidoglycan by cleaving the β-1, 4 glycosidic bond between MurNAc and GlcNAc. Lysozymes are present in vertebrates or invertebrates to defend against bacteria, and in bacteriophages to infect bacteria. Bacterial lysozyme homologs help maintain cell wall structure during growth and division (Holtje 1998), and play an important role in many macromolecular transportation systems (Koraimann 2003). Several lysozymes, such as hen-egg white lysozyme (HEWL) and bacteriophage T4 lysozyme (T4L), have well-studied structures and catalytic mechanisms (Matthews et al. 1981). They also serve as model systems to study protein folding and stability (Imoto 1996; Matthews 1996; Merlini and Bellotti 2005).
Several known lysozyme families have shown remarkable divergence in their sequences. Some of them (e.g., chicken-type [C-type] lysozymes and T4 lysozymes) cannot be linked even with sensitive sequence similarity search tools such as PSI-BLAST (Altschul et al. 1997). However, the folds of all lysozyme families exhibit recognizable similarity. Their common features include a few secondary structural elements with similar orientation and contact patterns, and similar location of active site and catalytic residues (Robertus et al. 1998). In the Structural Classification of Proteins (SCOP) database (version 1.67) (Murzin et al. 1995), the “lysozyme-like” fold contains seven families with available three-dimensional structures: family 19 glycosidase, C-type lysozyme, phage T4 lysozyme, λ lysozyme, goose-type (G-type) lysozyme, bacterial muramidase catalytic domain, and chitosanase. All these enzymes cleave β-1, 4 glycosidic bonds, although some families show specificity for other polysaccharides than peptidoglycan. For example, family 19 glycosidase (E.C. 3.2.1.14) (Hart et al. 1995) and chitosanase (E.C. 3.2.1.132) (Marcotte et al. 1993) degrade chitin and chitosan, respectively. Many bacterial or phage lysozyme homologs are also lytic transglycosidases that, in addition to cleaving the glycosidic bonds between MurNAc and GlcNAc, form an intramolecular anhydrobond in the MurNAc moiety (Holtje et al. 1975). We refer to all seven SCOP families as “lysozyme-like” families.
Here, we report identification of two new lysozyme-like families (COG3926/DUF847 and COG5526) by using a combination of sensitive sequence similarity searches, domain architecture and gene structure analysis, and structure prediction techniques. These two families show high sequence divergence compared to known lysozyme-like families with structures, yet they are confidently predicted to adopt the same fold and have the same active site location and catalytic residues. Most members of these two families are not experimentally characterized, and many of them are classified as hypothetical proteins. Our predictions shed light on their function and catalytic mechanism.
Results and Discussion
COG3926 /DUF847 proteins have a new lysozyme-like domain
Double-stranded RNA (dsRNA) bacteriophage φ8 belongs to virus family Cystoviridae, which also includes bacteriophages φ6, φ12, and φ13 with complete genomes (Cuppels et al. 1980; Qiao et al. 2000, 2005; Gottlieb et al. 2002). All these evolutionarily related phages have three linear dsRNA segments. The P5 protein from the well-studied bacteriophage φ12 has been experimentally characterized as a lytic enzyme (Mindich and Lehman 1979; Bamford and Palva 1980) and classified as a peptidase with unknown mechanism (Caldentey and Bamford 1992; Barrett et al. 2004). Our previous sequence analysis indicates it is actually a distant homolog of lytic transglycosylases with a lysozyme-like fold (Pei and Grishin 2005). The P5 proteins from bacteriophage φ6 and φ13 also belong to lytic transglycosylases (Pei and Grishin 2005). The P5 protein from bacteriophage φ8 has the same gene location (at the end of the smallest RNA segment) as other P5 proteins. However, our previous sequence analysis has not revealed a homology relationship between the P5 protein from bacteriophage φ8 and other lysozyme-like families.
Transitive PSI-BLAST searches (e-value cutoff 0.001, other parameters are default; nonredundant database, April 2005, 2,430,773 sequences, 823,264,207 total letters) starting with the P5 protein from bacteriophage φ8 (gene identification [gi] number 17736969, 169 residues) converged to about 50 proteins, many of which are annotated as putative secretion activating protein or hypothetical protein. Conserved Domain Database (CDD) (Marchler-Bauer et al. 2002) searches suggest that these proteins form a family that is classified as COG3926 in the Clusters of Orthologous Groups (COGs) database (Tatusov et al. 2003) and as DUF847 (domain of unknown function) in the Pfam database (Bateman et al. 2004). No significant hits to known structures were found for them. We used the Meta Server (http://bioinfo.pl/meta) to predict the three-dimensional structure of this P5 protein. The top eight hits of 3D-JURY (Ginalski et al. 2003) are all T4 lysozyme structures with the best score of 43.75 (a score > 50 is considered to be significant). Fold recognition method Meta-Basic (Ginalski et al. 2004) also identified DUF847 as the best hit not annotated as a lyzozyme, using the phage P1 lysozyme structure (Xu et al. 2005) as a query (Protein Data Bank [PDB] ID 1xjt; Berman et al. 2002). Using one DUF847 sequence (gi|7379919) as a query, Meta-Basic identified putative peptidoglycan binding domain (Dideberg et al. 1982; Foster 1991; Krogh et al. 1998) as the closest hit, and the P1 lysozyme domain of structure 1xjt is among the top five hits. Interestingly, the putative peptidoglycan-binding motif is identified in an insertion to the structural core (Fig. 1 ▶, α-helix A4 and the loop after it, with two highly conserved sequence signatures, Leu–Gln and Asp–Gly, colored in pink). PSI-BLAST searches starting from T4 lysozyme did not yield significant hits to any member of the COG3926 family, nor did PSI-BLAST searches from other lysozyme-like families. These results indicate that COG3926/DUF847 family members could have a distant lysozyme-like domain with a putative peptidoglycan-binding motif insert.
Figure 1.
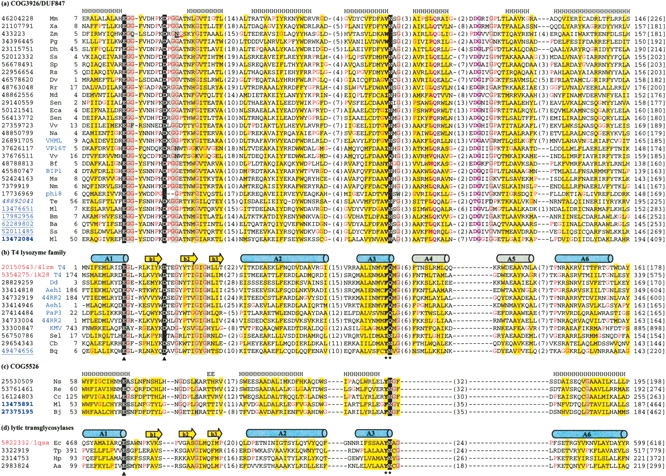
Multiple sequence alignments of representative sequences of COG3926/DUF847 (a), T4 lysozyme family (b), COG5526 (c), and lytic transglycosylases (d). The sequence identifiers on the left are NCBI gene identification (gi) numbers. The red gi numbers indicate sequences with known three-dimensional structures, with Protein Data Bank (PDB) IDs (Berman et al. 2002) following them. The blue gi number(s) in italic, underlines, and bold letters correspond to proteins with signal peptide, LATM1, and LATM2, respectively. The species name abbreviations are shown after the gi numbers, with bacteriophage species in blue letters. The first and the last residue numbers and the sequence length are shown. The numbers of residues between core blocks are shown in parentheses, with the exception of gi|433223, where there are seven residues inserted between the underlined “N” and “G.” The catalytic glutamate and aspartate, as well as the second residue in motif Pho-[Asn|Gln] are highlighted as white letters on black background. They are also marked under the alignments of T4 lysozyme family and lytic transglycosylase family with known structures (catalytic residues, triangles; Pho-[Asn|Gln] motif, stars). Conserved positions with mainly glycines are shaded in gray. Positions occupied by mainly hydrophobic residues are shaded in yellow. Two sequence signatures characteristic of peptidoglycan-binding motif are colored pink (Leu–Gln in α-helix A4 and Asp–Gly in the loop after it). Real secondary structural elements of T4 lysozyme (PDB ID 4lzm) and a lytic transglycosylase (PDB ID 1qsa) are shown as cartoon diagrams: cylinders as α-helices and arrows as β-strands. Blue α-helices are essential elements present in all lysozyme-like families and gray α-helices are not present in all of them. The predicted secondary structures for the COG3926/DUF847 family and COG5526 family are shown above their alignments. Species name abbreviations are 44RR2, Bacteriophage 44RR2.8t; Aa, Aquifex aeolicus; Aeh1, Bacteriophage Aeh1; Ba, Brucella abortus; Bf, Burkholderia fungorum; BIP1, Bordetella phage BIP1; Bj, Bradyrhizobium japonicum; Bm, Brucella melitensis; Bq, Bartonella quintana; Cb, Coxiella burnetii; Cc, Caulobacter crescentus; Dd, Dictyostelium discoideum; Dh, Desulfitobacterium hafniense; Dv, Desulfovibrio vulgaris; Ec, Escherichia coli; Eca, Erwinia carotovora; Hp, Helicobacter pylori; KMV, Bacteriophage φKMV; Md, Microbulbifer degradans; Ml, Mesorhizobium loti; Mm, Magnetospirillum magnetotacticum; Ms, Mannheimia succiniciproducens; Na, Novosphingobium aromaticivorans; Nm, Neisseria meningitidis; Ns, Nostoc sp.; PaP3, Pseudomonas aeruginosa phage PaP3; Pg, Porphyromonas gingivalis; φ8, Bacteriophage φ8; Re, Ralstonia eutropha; Rr, Rhodospirillum rubrum; Rs, Rhodobacter sphaeroides; Sen, Salmonella enterica; Sel, Synechococcus elongatus; Sp, Silicibacter pomeroyi; Ss, Silicibacter sp.; T4, Enterobacteria phage T4; Te, Trichodesmium erythraeum; Tp, Treponema pallidum; VHML, Vibrio harveyi bacteriophage VHML; VP16T, Vibrio parahaemolyticus phage VP16T; Vv, Vibrio vulnificus; Xa, Xanthomonas axonopodis; Zm, Zymomonas mobilis.
To further verify the possible homology relationship between COG3926/DUF847 family and T4 lysozyme family, we used PCMA program (Pei et al. 2003) to construct multiple sequence alignments from the COG3926/DUF847 family and T4 lysozyme family (Fig. 1a,b ▶) with manual inspection and adjustment. The alignments reveal that the two catalytic residues in T4 lysozyme (E11 and D20 in structure 4lzm) are also highly conserved in the COG3926/DUF847 family. Secondary structure elements of T4 lysozyme match the predicted secondary structures of COG3926/DUF847 family well in most of the α-helical regions. Hydrophobic patterns and a few positions occupied by mainly glycine residues are also consistently conserved between the two families.
The catalytic residue E11 of T4 lysozyme is situated at the end of the first α-helix (A1) (Fig. 1b ▶). It serves as a general acid that attacks the glycosidic bond. This glutamate is invariant in all lysozyme-like families as well as in COG3926/DUF847. There is a conserved glycine residue following E11 in both the COG3926/DUF847 and the T4 lysozyme family. This position is often occupied by a conserved serine in C-type lysozymes, G-type lysozymes, and lytic transglycosylases (Pei and Grishin 2005). The catalytic residue D20 of the T4 lysozyme is situated at the end of the first β-strand (b1). It functions to stabilize the intermediate using its negative charges, and is also highly conserved at the same position in the COG3926/DUF847 family. This aspartate is not required in some of the other lysozyme-related families, such as G-type lysozymes (Weaver et al. 1995) and lytic transglycosylases (Holtje 1998). The turn between the second and the third β-strands (Fig. 2b ▶, b2 and b3) of the T4 lysozyme family has a sequence signature of “Gly–Xaa–Gly–[His|Arg],” where “Xaa” is often a hydrophobic residue. However, the sequence signature of this turn in the COG3926/DUF847 family is more like the ones in other lysozyme-like families, which is often “Gly–Xaa–Xaa–Gln” (“Xaa”s are often hydrophobic residues) (Pei and Grishin 2005), although the “Gln” is not highly conserved in the COG3926/DUF847 family. Another highly conserved motif is the “Pho–[Asn|Gln]” sequence signature at the end of α-helix A3, where “Pho” is a hydroPhobic residue in the COG3926/DUF847 family and often an aromatic residue in T4 lysozyme family and other lysozyme-like families. The conserved asparagine or glutamine makes an important hydrogen-bond interaction with substrate. Both this residue and the catalytic aspartate are mutated in the P5 protein from bacteriophage φ8 (Fig. 1a ▶), indicating rapid evolution of this viral sequence and its potentially weakened catalytic activity.
Figure 2.

Domain architecture of COG3926/DUF847 and COG5526 proteins. The lengths of domains are approximately to scale. Horizontal braces together with the arrows under them suggest homology relationship between domains that can be linked by PSI-BLAST with significant e-values. The two lysozyme-associated transmembrane modules are shown with dotted boundary lines. Predicted transmembrane regions are marked rectangles, with one low confidence prediction shown with dotted boundary line. NCBI gene identification (gi) numbers and species names of representative sequences are shown. The number of proteins with a domain architecture is shown in parentheses after the domain diagram. (a) Single-domain proteins of COG3926/DUF847; (b) the COG3926/DUF847 member with a signal peptide; (c) COG3926/DUF847 members with LATM1; (d) T4 lysozyme family members with LATM1; (e) the COG3926/DUF847 member with LATM2; (f) COG5526 members with PGB and LATM2; (g) single-domain COG5526 members.
Domain architecture analysis of COG3926/DUF847 members reveals two lysozyme-associated transmembrane modules
Most of the COG3926/DUF847 members have a single lysozyme-like domain with sequence lengths < 200 residues (Fig. 2a ▶). A few members have additional modules such as predicted signal peptide or transmembrane regions. These proteins can be categorized into three groups as described below:
Group 1. The protein from Cyanobacteria Trichodesmium erythraeum (gi|48892041; Fig. 2b ▶) is the only COG3926/DUF847 member with a signal peptide in the N terminus, as predicted with high confidence by the SignalP 3.0 server (Bendtsen et al. 2004).
Group 2. A few COG3926/DUF847 members with sequence lengths between 250 and 300 residues contain a predicted transmembrane region at the C terminus, e.g., gi|17982956 from Brucella melitensis (Fig. 2c ▶). These transmembrane regions, together with a conserved segment at its N terminus, bear strong sequence similarity with each other. We name this putative domain “lysozyme-associated transmembrane module 1” (abbreviated as LATM1). A PSI-BLAST search using LATM1 from B. melitensis also detected a few proteins that do not belong to the COG3926/DUF847 family (Fig. 2d ▶). They are all from the newly sequenced human pathogens Bartonella quintana str. Toulouse or Bartonella henselae str. Houston-1 (Alsmark et al. 2004). Interestingly, these proteins are annotated as “phage-related lysozymes” or “phage-related proteins” and contain an N-terminal “endolysin_autolysin” domain suggested by CDD searches. The “endolysin_autolysin” domain (cd00737 in the CDD database) corresponds to the T4 lysozyme family. Therefore, LATM1 is a mobile module that co-occurs with two distinct lysozyme-like domains: COG3926/DUF847 and T4 lysozyme family (Fig. 2c,d ▶; the alignment of LATM1 is in the supplementary material).
Group 3. The COG3926/DUF847 member from Mesorhizobium loti (gi|13472084; Fig. 2e ▶) is the longest protein with a sequence length of 409 residues. The C-terminal 200 residues of this hypothetical protein are predicted to contain several transmembrane regions with no sequence similarity to LATM1. We term this putative domain “lysozyme-associated transmembrane module 2” (LATM2). A PSI-BLAST search using this LATM2 detected two other hypothetical proteins: gi|13475891 from Mesorhizobium loti, and gi|27375195 from Bradyrhizobium japonicum. These two proteins are longer (~460 residues) and also have the LATM2 domain situated at their C termini (the alignment of LATM2 is in the supplementary material). However, their N-terminal 260 residues do not bear significant sequence similarity to COG3926/DUF847 domain (Fig. 2f ▶). CDD searches revealed that their N termini have two modules: a domain corresponding to COG5526, and a peptidoglycan-binding domain (PGB, Pfam accession number PF01471) (Fig. 2f ▶). The PGB domain is a mobile domain co-occurring with many other domains that degrade the bacterial cell wall, such as muramoyl-pentapeptide carboxypeptidase, N-acetylmuramoyl-L-alanine amidase, autolytic lysozyme, and lytic transglycosylase (Dideberg et al. 1982; Foster 1991; Krogh et al. 1998).
Co-occurrence with other domains is not unique to lysozyme-like family COG3926/DUF847. For example, T4-related bacteriophages Aeh1 and 44R2, as well as phage T4, possess two proteins with a T4 lysozyme-like domain (Fig. 1b ▶). One of them (gene product gp5) is a multidomain protein that is an essential structural component of the tail baseplate (Vanderslice and Yegian 1974; Kikuchi and King 1975; Kanamaru et al. 2005). Crystal structure reveals that in addition to the middle lysozyme domain, gp5 has an N-terminal domain with an OB-fold (oligonucleotide/oligosaccharide-binding fold) (Murzin 1993), and a C-terminal domain with a triple-stranded β-helix fold (Kanamaru et al. 2002). The N-terminal OB-fold could function similarly to PGB domain and the C-terminal domain functions as a cell-puncturing device (Kanamaru et al. 2002). Some bacterial lytic transglycosylases are also multidomain proteins (van Asselt et al. 1999; Koraimann 2003).
COG5526 proteins have a new lysozyme-like domain
In the current COG database, COG5526 is annotated as “Uncharacterized conserved protein [function unknown]” (Tatusov et al. 2003). A PSI-BLAST search retrieved only five proteins belonging to COG5526. Two proteins are mentioned above, with a PGB domain and LATM2 (Fig. 2f ▶); the other three proteins are shorter and appear to be single-domain proteins (Fig. 2g ▶). Since the PGB domain frequently co-occurs with cell wall degrading domains, COG5526 could potentially also have such domains. We submitted COG5526 proteins to the protein structure prediction Meta Server (Ginalski et al. 2003). The top hits indicate that COG5526 has a new lysozyme-like domain with significant scores. For example, the N-terminal region (residues 1–180) of COG5526 member gi|13475891 from Mesorhizobium loti retrieved goose-type lysozymes as the top four predictions, with the best score above 80 (a score > 50 is considered to be significant) (Ginalski et al. 2003). Manual inspection of weak PSI-BLAST hits also indicates that COG5526 members are distantly related to the other lysozyme-like domains. For example, the second PSI-BLAST iteration with a COG5526 member from Ralstonia eutropha (gi| 53761461) identified a hypothetical protein from Bacillus clausii (gi|56963882, hit range residues 45–159) with an evalue of 0.068. This hypothetical protein has a “lytic transglycosylase (LT) and goose egg white lysozyme (GEWL) domain” (cd00254) according to CDD searches. The predicted catalytic glutamate and the Pho-[Asn|Gln] motif in gi|56963882 are aligned with those in gi|53761461 in the PSI-BLAST local alignment.
A multiple sequence alignment of COG5526 proteins (Fig. 1c ▶) indeed reveals an invariant glutamate residue at the end of a predicted α-helix as the predicted catalytic residue, as well as the conserved Pho-[Asn|Gln] motif. This alignment is merged with alignments of representative sequences of COG3924/DUF847 (Fig. 1a ▶), T4 lysozyme domains (Fig. 1b ▶), and lytic transglycosylases (Fig. 1d ▶) based on 3D-JURY alignments, secondary structure predictions, and conservation of motifs and hydrophobic patterns. Since only five members of COG5526 are available, the less conserved region corresponding to the three-stranded β-sheet is not reliably aligned to other families.
All COG5526 proteins have three additional predicted α-helices (not shown in Fig. 1 ▶) N-terminal to α-helix A1. Like those in the G-type lysozymes or some bacterial lytic transglycosylases, these additional N-terminal α-helices in COG5526 possibly wrap around α-helices A1 and A6 and result in their elevated hydrophobicity (Fig. 1c ▶). α-Helices A4 and A5 in COG3926/DUF847 and T4 lysozyme family seem to be missing in COG5526, according to the secondary structure prediction. These two α-helices (Fig. 1b ▶, shaded in gray) are not present in several lysozyme-like families such as C-type lysozyme and family 19 glycosidase (chitinase). On the other hand, α-helices A1, A2, A3, and A6 (Fig. 1b ▶, shaded in blue) are essential elements in all lysozyme-like families (except in C-type lysozymes, where A3 is almost deteriorated). A larger N-terminal region and a smaller C-terminal region suggest that COG5526 might be structurally and evolutionarily closer to the so-called “eukaryotic family” of lysozymes such as C-type lysozyme, chitinase, and G-type lysozyme, as proposed by Robertus et al. (1998). The 3D-Jury results and the weak PSI-BLAST hit also support the idea that COG5526 is more closely related to G-type lysozymes and lytic transglycosylases. Like the G-type lysozymes and lytic transglycosylases, COG5526 members do not have a conserved aspartate in the β-sheet region as the second catalytic residue (Fig. 1c ▶). On the other hand, the COG3926/DUF847 family is probably more similar to the “prokaryotic family” of lysozymes such as T4 lysozyme and chitosanase (Robertus et al. 1998), as it has no additional secondary structural elements in the N terminus, but has predicted α-helices A4 and A5 in the C terminus.
Phylogenetic distribution and putative cellular functions of COG3926/DUF847 and COG5526
Most of the COG3926/DUF847 members are found in proteobacteria, among which the α-proteobacteria is the most populated. There is only one sequence from bacteroidetesacteroides (gi|34396445, Porphyromonas gingivalis), cyanobacteria (gi|48892041, Trichodesmium, erythraeum), and firmicutes (gi|23115751, Desulfitobacterium hafniense), respectively. Four COG3926/DUF847 members are from bacteriophages. The hosts of all these phages belong to γ-proteobacteria. Gene structure analysis shows that a couple of proteins from bacteria are probably prophage proteins, such as the ones from Erwinia carotovora (gi|50121541) and Desulfovibrio vulgarish (gi|46578620). Such a phylogetic pattern suggests that COG3926/DUF847 could have originated within the proteobacteria and been horizontally transferred to other major branches of bacteria or bacteriophages. As for COG5526, four of its five members belong to proteobacteria and the other one is from cyanobacteria (Nostoc sp.).
The COG3926/DUF847 members from phages, such as the P5 protein from bacteriophage φ8, are likely to be the lytic enzymes for bacterial infection and/or cell lysis. Few experimental studies are available for bacterial COG3926/DUF847 and COG5526 members. However, the presence of signal peptide, transmembrane regions, or PGB domains in several members suggests that they are located in the vicinity of the cell wall and their general function is to cleave peptidoglycan, like other bacterial muramidases or lytic transglycosylases. The COG3926/DUF847 member from Zymomonas mobilis (gi|433223, the product of gene zliS) has been experimentally characterized as a protein that stimulates the secretion of extracellular levansucrase and invertase (Kondo et al. 1994). COG3926 is thus annotated as “zliS: putative secretion activating protein.” Since levansucrase and invertase are large proteins (> 400 residues), their extracellular secretion could be facilitated if a lysozyme breaks or rearranges the peptidoglycan layer during the secretion process. Therefore, the cellular function of gene zliS product is consistent with its predicted peptidoglycan-cleaving activity. In fact, many bacterial lysozymes or lytic transglycosylases have specialized functions in a variety of macromolercular transportation systems (Koraimann 2003). We submitted COG3926/DUF847 members to the STRING server (von Mering et al. 2003) for prediction of functional association with other genes. Although no strong associations were detected, a few members scored modestly (> 0.4, medium confidence) indicating functional association with putative membrane proteins or sugar transporters. Functional associations with other proteins cannot be predicted for any COG5526 member using the STRING server or gene structure analysis. The precise cellular roles of these two new lysozyme-like families await further experimental studies.
Materials and methods
The PSI-BLAST program (Altschul et al. 1997) was used to search for homologs of the P5 protein from bacteriophage φ8 against the NCBI nonredundant database (April 2005, 2,430, 773 sequences; 823,264,207 total letters). The e-value threshold was 0.001 for inclusion of sequences into a profile. Composition-based statistics (Schaffer et al. 2001) were applied and no filter for low complexity regions was applied. The other parameters were default. To ensure full coverage, found homologs were grouped by single-linkage clustering (1 bit per site threshold, ~50% sequence identity), and representative sequences from each group were used as queries for further PSI-BLAST iterations, as scripted by using the SEALS package (Walker and Koonin 1997). The same strategy was applied to other lysozyme domains and lysozyme-associated transmembrane modules.
Multiple sequence alignments were constructed by using the PCMA program (Pei et al. 2003) for representative sequences of COG3926/DUF847, T4 lyzozyme family, COG5526, and lytic transglycosylases. Manual adjustment of the multiple sequence alignment was made with guidance from available structures. Secondary structure predictions were made for representative sequences of COG3926/DUF847 and COG5 526 by using PSIPRED (Jones 1999). Domain architecture analysis was conducted using CDD (Marchler-Bauer et al. 2002), SMART (Letunic et al. 2004), and SignalP 3.0 (Bendtsen et al. 2004) servers. Transmembrane regions were predicted for individual proteins with programs TMHMM2.0 and TMPRED (Ikeda et al. 2002). Representative sequences were sent to the Meta Server coupled with 3D-JURY system (Ginalski et al. 2003) and Meta-Basic server (Ginalski et al. 2004) for structure prediction, and the STRING server (von Mering et al. 2003) for prediction of function association with other genes. The gene organization and co-occurrence analysis were also facilitated with the SEED database (http://theseed.uchicago.edu/FIG/index.cgi).
Electronic supplemental material
The multiple sequence alignments of LATM1 and LATM2 are available at ftp://iole.swmed.edu/pub/lysozymes/latm.doc.
Acknowledgments
We thank Drs. Lisa Kinch and James Wrabl, and an anonymous referee for critical reading of the manuscript and helpful comments and suggestions. This work was supported by NIH grant GM67165 to N.V.G.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051656805.
Supplemental material: see www.proteinscience.org
References
- Alsmark, C.M., Frank, A.C., Karlberg, E.O., Legault, B.A., Ardell, D.H., Canback, B., Eriksson, A.S., Naslund, A.K., Handley, S.A., Huvet, M., et al. 2004. The louse-borne human pathogen Bartonella quintana is a genomic derivative of the zoonotic agent Bartonella henselae. Proc. Natl. Acad. Sci. 101 9716–9721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford, D.H. and Palva, E.T. 1980. Structure of the lipid-containing bacteriophage φ 6. Disruption by Triton X-100 treatment. Biochim. Biophys. Acta 601 245–259. [DOI] [PubMed] [Google Scholar]
- Barrett, A.J., Rawlings, N.D., and Woessner, J.F. 2004. Handbook of proteolytic enzymes, 2nd ed. Elsevier Academic Press, New York.
- Bateman, A., Coin, L., Durbin, R., Finn, R.D., Hollich, V., Griffiths-Jones, S., Khanna, A., Marshall, M., Moxon, S., Sonnhammer, E.L., et al. 2004. The Pfam protein families database. Nucleic Acids Res. 32 D138–D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendtsen, J.D., Nielsen, H., von Heijne, G., and Brunak, S.2004. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340 783–795. [DOI] [PubMed] [Google Scholar]
- Berman, H.M., Battistuz, T., Bhat, T.N., Bluhm, W.F., Bourne, P.E., Burkhardt, K., Feng, Z., Gilliland, G.L., Iype, L., Jain, S., et al. 2002. The Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr. 58 899–907. [DOI] [PubMed] [Google Scholar]
- Caldentey, J. and Bamford, D.H. 1992. The lytic enzyme of the Pseudomonas phage φ 6. Purification and biochemical characterization. Biochim. Biophys. Acta 1159 44–50. [DOI] [PubMed] [Google Scholar]
- Cuppels, D.A., Van Etten, J.L., Burbank, D.E., Lane, L.C., and Vidaver, A.K. 1980. In vitro translation of the three bacteriophage φ 6 RNAs. J. Virol. 35 249–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dideberg, O., Charlier, P., Dive, G., Joris, B., Frere, J.M., and Ghuysen, J.M. 1982. Structure of a Zn2+-containing D-alanyl-D-alanine-cleaving carboxypeptidase at 2.5 Å resolution. Nature 299 469–470. [DOI] [PubMed] [Google Scholar]
- Foster, S.J. 1991. Cloning, expression, sequence analysis and biochemical characterization of an autolytic amidase of Bacillus subtilis 168 trpC2. J. Gen. Microbiol. 137 1987–1998. [DOI] [PubMed] [Google Scholar]
- Ginalski, K., Elofsson, A., Fischer, D., and Rychlewski, L. 2003. 3D-Jury: A simple approach to improve protein structure predictions. Bioinformatics 19 1015–1018. [DOI] [PubMed] [Google Scholar]
- Ginalski, K., von Grotthuss, M., Grishin, N.V., and Rychlewski, L. 2004. Detecting distant homology with Meta-BASIC. Nucleic Acids Res. 32 W576–W581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb, P., Potgieter, C., Wei, H., and Toporovsky, I. 2002. Characterization of φ12, a bacteriophage related to φ6: Nucleotide sequence of the large double-stranded RNA. Virology 295 266–271. [DOI] [PubMed] [Google Scholar]
- Hart, P.J., Pfluger, H.D., Monzingo, A.F., Hollis, T., and Robertus, J.D. 1995. The refined crystal structure of an endochitinase from Hordeum vulgare L. seeds at 1.8 Å resolution. J. Mol. Biol. 248 402–413. [PubMed] [Google Scholar]
- Holtje, J.V. 1998. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol. Mol. Biol. Rev. 62 181–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtje, J.V., Mirelman, D., Sharon, N., and Schwarz, U. 1975. Novel type of murein transglycosylase in Escherichia coli. J. Bacteriol. 124 1067–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda, M., Arai, M., Lao, D.M., and Shimizu, T. 2002. Transmembrane topology prediction methods: A re-assessment and improvement by a consensus method using a dataset of experimentally-characterized transmembrane topologies. In Silico Biol. 2 19–33. [PubMed] [Google Scholar]
- Imoto, T. 1996. Engineering of lysozyme. Exs 75 163–181. [DOI] [PubMed] [Google Scholar]
- Jones, D.T. 1999. Protein secondary structure prediction based on position- specific scoring matrices. J. Mol. Biol. 292 195–202. [DOI] [PubMed] [Google Scholar]
- Kanamaru, S., Leiman, P.G., Kostyuchenko, V.A., Chipman, P.R., Mesyanzhinov, V.V., Arisaka, F., and Rossmann, M.G. 2002. Structure of the cell-puncturing device of bacteriophage T4. Nature 415 553–557. [DOI] [PubMed] [Google Scholar]
- Kanamaru, S., Ishiwata, Y., Suzuki, T., Rossmann, M.G., and Arisaka, F. 2005. Control of bacteriophage T4 tail lysozyme activity during the infection process. J. Mol. Biol. 346 1013–1020. [DOI] [PubMed] [Google Scholar]
- Kikuchi, Y. and King, J. 1975. Genetic control of bacteriophage T4 base-plate morphogenesis. III. Formation of the central plug and overall assembly pathway. J. Mol. Biol. 99 695–716. [DOI] [PubMed] [Google Scholar]
- Kondo, Y., Toyoda, A., Fukushi, H., Yanase, H., Tonomura, K., Kawasaki, H., and Sakai, T. 1994. Cloning and characterization of a pair of genes that stimulate the production and secretion of Zymomonas mobilis extracellular levansucrase and invertase. Biosci. Biotechnol. Biochem. 58 526–530. [DOI] [PubMed] [Google Scholar]
- Koraimann, G. 2003. Lytic transglycosylases in macromolecular transport systems of Gram-negative bacteria. Cell Mol. Life Sci. 60 2371–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh, S., Jorgensen, S.T., and Devine, K.M. 1998. Lysis genes of the Bacillus subtilis defective prophage PBSX. J. Bacteriol. 180 2110–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic, I., Copley, R.R., Schmidt, S., Ciccarelli, F.D., Doerks, T., Schultz, J., Ponting, C.P., and Bork, P. 2004. SMART 4.0: Towards genomic data integration. Nucleic Acids Res. 32 D142–D144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer, A., Panchenko, A.R., Shoemaker, B.A., Thiessen, P.A., Geer, L.Y., and Bryant, S.H. 2002. CDD: A database of conserved domain alignments with links to domain three-dimensional structure. Nucleic Acids Res. 30 281–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte, E., Hart, P.J., Boucher, I., Brzezinski, R., and Robertus, J.D. 1993. Crystallization of a chitosanase from Streptomyces N174. J. Mol. Biol. 232 995–996. [DOI] [PubMed] [Google Scholar]
- Matthews, B.W. 1996. Structural and genetic analysis of the folding and function of T4 lysozyme. FASEB J. 10 35–41. [DOI] [PubMed] [Google Scholar]
- Matthews, B.W., Grutter, M.G., Anderson, W.F., and Remington, S.J. 1981. Common precursor of lysozymes of hen egg-white and bacteriophage T4. Nature 290 334–335. [DOI] [PubMed] [Google Scholar]
- Merlini, G. and Bellotti, V. 2005. Lysozyme: A paradigmatic molecule for the investigation of protein structure, function and misfolding. Clin. Chim. Acta 357 168–172. [DOI] [PubMed] [Google Scholar]
- Mindich, L. and Lehman, J. 1979. Cell wall lysin as a component of the bacteriophage φ 6 virion. J. Virol. 30 489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzin, A.G. 1993. OB(oligonucleotide/oligosaccharide binding)-fold: Common structural and functional solution for non-homologous sequences. EMBO J. 12 861–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzin, A.G., Brenner, S.E., Hubbard, T., and Chothia, C. 1995. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247 536–540. [DOI] [PubMed] [Google Scholar]
- Pei, J. and Grishin, N.V. 2005. The P5 protein from bacteriophage φ-6 is a distant homolog of lytic transglycosylases. Protein Sci. 14 1370–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei, J., Sadreyev, R., and Grishin, N.V. 2003. PCMA: Fast and accurate multiple sequence alignment based on profile consistency. Bioinformatics 19 427–428. [DOI] [PubMed] [Google Scholar]
- Qiao, X., Qiao, J., Onodera, S., and Mindich, L. 2000. Characterization of φ13, a bacteriophage related to φ 6 and containing three dsRNA genomic segments. Virology 275 218–224. [DOI] [PubMed] [Google Scholar]
- Qiao, J., Qiao, X., and Mindich, L. 2005. In vivo studies of genomic packaging in the dsRNA bacteriophage φ8. BMC Microbiol. 5 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertus, J.D., Monzingo, A.F., Marcotte, E.M., and Hart, P.J. 1998. Structural analysis shows five glycohydrolase families diverged from a common ancestor. J. Exp. Zool. 282 127–132. [PubMed] [Google Scholar]
- Schaffer, A.A., Aravind, L., Madden, T.L., Shavirin, S., Spouge, J.L., Wolf, Y.I., Koonin, E.V., and Altschul, S.F. 2001. Improving the accuracy of PSI-BLAST protein database searches with compositionbased statistics and other refinements. Nucleic Acids Res. 29 2994–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleifer, K.H. and Kandler, O. 1972. Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol. Rev. 36 407–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov, R.L., Fedorova, N.D., Jackson, J.D., Jacobs, A.R., Kiryutin, B., Koonin, E.V., Krylov, D.M., Mazumder, R., Mekhedov, S.L., Nikolskaya, A.N., et al. 2003. The COG database: An updated version includes eukaryotes. BMC Bioinformatics 4 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Asselt, E.J., Thunnissen, A.M., and Dijkstra, B.W. 1999. High resolution crystal structures of the Escherichia coli lytic transglycosylase Slt70 and its complex with a peptidoglycan fragment. J. Mol. Biol. 291 877– 898. [DOI] [PubMed] [Google Scholar]
- Vanderslice, R.W. and Yegian, C.D. 1974. The identification of late bacteriophage T4 proteins on sodium dodecyl sulfate polyacrylamide gels. Virology 60 265–275. [DOI] [PubMed] [Google Scholar]
- von Mering, C., Huynen, M., Jaeggi, D., Schmidt, S., Bork, P., and Snel, B. 2003. STRING: A database of predicted functional associations between proteins. Nucleic Acids Res. 31 258–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, D.R. and Koonin, E.V. 1997. SEALS: A system for easy analysis of lots of sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 5 333–339. [PubMed] [Google Scholar]
- Weaver, L.H., Grutter, M.G., and Matthews, B.W. 1995. The refined structures of goose lysozyme and its complex with a bound trisaccharide show that the “goose-type” lysozymes lack a catalytic aspartate residue. J. Mol. Biol. 245 54–68. [DOI] [PubMed] [Google Scholar]
- Xu, M., Arulandu, A., Struck, D.K., Swanson, S., Sacchettini, J.C., and Young, R. 2005. Disulfide isomerization after membrane release of its SAR domain activates P1 lysozyme. Science 307 113–117. [DOI] [PubMed] [Google Scholar]