

Abstract
For determination of multiple covalent intermediates bound to the ultra-large enzymes responsible for biosynthesis via nonribosomal peptide synthesis, mass spectrometry (MS) is a promising method to provide new mechanistic insight. Application of a quadrupole-Fourier-transform instrument (Q-FTMS) for direct analysis of aminoacyl intermediates is demonstrated for the first two modules (127 and 120 kDa) involved in the nonribosomal synthesis of gramicidin S. Cyanogen bromide digestions of recombinant proteins afforded detection of two active site peptides (both ~13 kDa) that provided direct evidence for modules copurifying with their preferred amino acid substrates. Given the ability to detect multiple covalent intermediates in tandem, a competition experiment among several nonnatural substrates in parallel was performed using the first module. This defined mixture of acyl-enzyme intermediates was used to probe the selectivity of the condensation step producing a diversity of noncognate dipeptides on the second module.
Keywords: Fourier-transform mass spectrometry, nonribosomal peptide synthesis, gramicidin S, covalent intermediates
Many of the longest genes known encode enzymes that harbor multiple covalently bound intermediates during the biogenesis of critical antibiotics and anti-tumor compounds. Such biosynthetic enzymes can be from 100 kDa to >1.5 MDa and fall into two classes: nonribosomal peptide synthases (NRPSs) and polyketide synthases (PKSs). For NRPS, a prototypical two-enzyme system produces the natural antibiotic gramicidin S, a cyclic decapeptide (Fig. 1A ▶). Gramicidin S synthetase I (GrsA) and gramicidin S synthetase II (GrsB) together harbor five modules that recognize, activate, and condense the five amino acid precursors to gramicidin S. GrsA (PheATE) contains three domains: adenylation (A), thiolation (T) or peptidyl carrier protein (PCP), and epimerization (E) (Stachelhaus and Marahiel 1995; von Dohren et al. 1997; Sieber and Marahiel 2005). The A domain binds phenylalanine (l-Phe), activates it by adenylation using ATP, and forms a Phe-S-intermediate covalently attached to the 4′-phosphopantetheine group that is post-translationally added to a highly conserved serine (Ser573) in the T domain. The E domain serves to epimerize l-Phe to d-Phe. GrsB (511 kDa) contains four modules and a thioesterase domain. ProCAT, the first module of GrsB, contains a condensation (C) domain that is responsible for forming a peptide bond between d-Phe and l-Pro. l-Val, l-Orn, and l-Leu are incorporated in a similar manner by the next three modules of GrsB. Following two iterations through the assembly line, the thioesterase domain (TE) cyclizes and releases the two peptides in a head-to-tail manner to give the final active product at an overall rate of ~10 molecules every minute per 1 molecule of enzyme (Vater et al. 1987).
Figure 1.
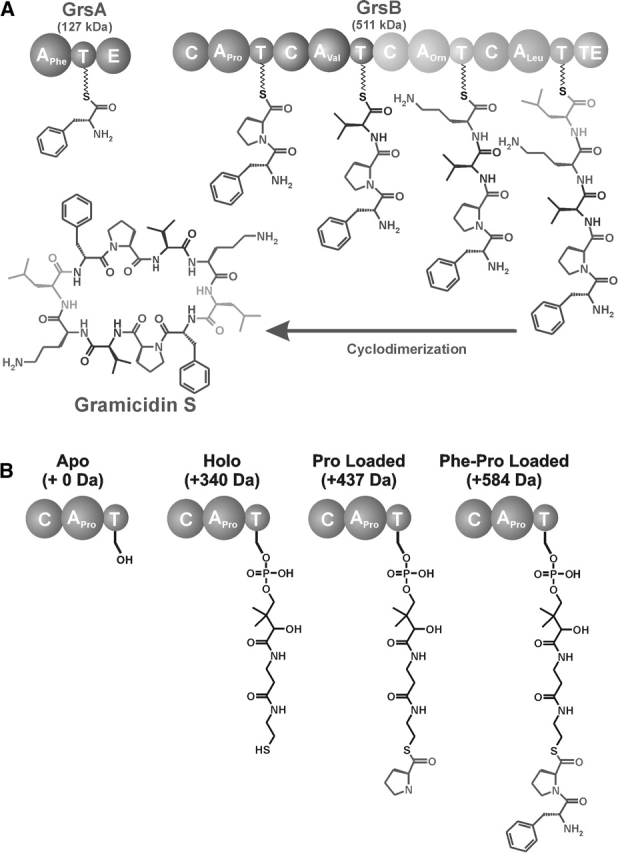
(A) Overview of the gramicidin S biosynthetic enzymes. GrsA (127 kDa), the initiation module, contains an adenylation (A), thiolation (T), and epimerization (E) domain and is responsible for incorporating d-Phe into gramicidin S. GrsB (511 kDa) contains four modules, each containing condensation (C), adenylation (A), and thiolation (T) domains. (B) The four possible covalent intermediates of the first module of GrsB, ProCAT. In the apo form, the active site Ser is unmodified; the holo form has an increase in mass of 340 Da due to the addition of a phosphopantetheine group from CoA. After the thiolation domain is converted to the holo form, the constituent amino acid, l-Pro, can be loaded, increasing the mass by 97 Da. If PheATE is present, d-Phe can be transferred onto the active site of ProCAT, therefore increasing the mass another 147 Da for a total increase of 584 Da from the apo form.
GrsA and ProCAT have been used as a model system for examining the loading and condensation events occurring in NRPS. This two-module system turns over by virtue of a d,l-diketopiperazine (d,l-DKP) that forms near neutral pH via a nonenzymatic release in the absence of a downstream TE domain (Stachelhaus et al. 1998). The kinetics of d-Phe transfer to Pro-S-ProCAT have been determined to be in the ~1 min−1 regime (Belshaw et al. 1999), with the epimerization shown to be ~3 sec−1 (Luo and Walsh 2001). Previous studies have also examined the specificity of the adenylation domain of PheATE. One such study using radiolabeled amino acids determined that l-Leu, l-Trp, and l-Tyr could be recognized and loaded onto the thiolation domain (Luo et al. 2001). This method, however, is limited in that only one intermediate is detected at a time, whereas mass spectrometry has the ability to detect multiple intermediates simultaneously (McLoughlin and Kelleher 2004).
The thiolation domains present in NRPSs exist in different covalent states throughout the process of nonribosomal peptide biosynthesis (Fig. 1B ▶). Since each covalent intermediate differs in mass, these systems are well suited for study by mass spectrometry. Quadrupole-Fourier transform mass spectrometry (Q-FTMS) is capable of analyzing complex proteolytic mixtures with a resolving power of >105 (McLafferty 1994), and, when coupled with chromatographic separation, provides high-resolution data allowing detailed characterization of acyl-enzyme intermediates in NRPS and PKS systems (Mazur et al. 2003; Hicks et al. 2004; McLoughlin and Kelleher 2004).
Here we interrogate a two-module system, PheATE and ProCAT (Stachelhaus et al. 1998), and quantify substrate analogs in their competition for enzyme occupancy and flux. Multiple nonnatural substrates were assayed in parallel, and their occupancy on the thiolation domain of PheATE was determined. Further, the ability of the noncognate amino acids to condense onto the T domain of the downstream module was assayed. For some enzymatic preparations, the cognate amino acids were shown to copurify with their A domains but could subsequently be removed by a type II thioesterase domain (TEII) (Schwarzer et al. 2002). Lastly, it was shown that even though the post-translational modifications that occur during nonribosomal peptide synthesis alter the overall composition of the proteins/peptides, the relative ratios of acyl-enzyme mixtures at a single carrier site can be determined to within 5% using electrospray ionization (ESI) mass spectrometry.
Results
Digestion of GrsA
Digestion of GrsA, a 127-kDa enzyme, by cyanogen bromide (CNBr) with subsequent separation by reverse-phase liquid chromatography (RPLC) yielded the data shown in Figure 2 ▶. The 29-min fraction yielded a spectrum in which 7 Mr values were observed. A 12,611.0–0 Da component (Fig. 2A ▶) was 26.6 Da lower than that predicted for a CNBr peptide generated from the published sequence (Hori et al. 1989). After DNA resequencing indicated a Ser541Asn error, the intact Mr value and the tandem MS (MS/MS) spectrum (not shown) confirmed the refined sequence for Arg533–Met641.
Figure 2.
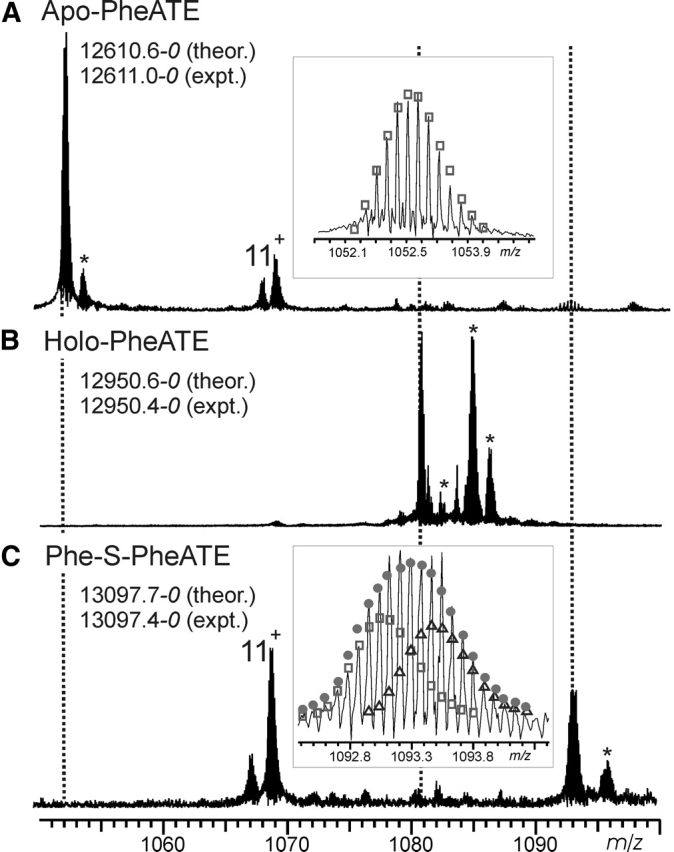
(A) Expansion of the region of the broadband spectrum of an RPLC fraction (29 min) containing the 12+ charge state of the CNBr digestion peptide of apo PheATE. Also present is the 11+ charge state of a peptide from PheATE that does not contain the active site (Leu140–Met243). (Inset) Expansion of isotopic distribution for the 12,611.0 Da component. (B) Spectrum containing the 12+ charge state of the holo form of the PheATE CNBr digestion peptide. (C) Region of the spectrum containing the 12+ charge state from material treated with a 1:1 mixture of d0- and d5-Phe before digestion. (Inset) Expansion of isotopic distribution for the 13,097.4 Da component. Open squares indicate the theoretical distribution for d0-Phe loaded peptide; open triangles, the d5-Phe loaded peptide. The actual ratio determined for the loading of d0- to d5-Phe was 53%:47%, for which the theoretical distribution is represented by the solid circles. An * indicates an adduct peak.
Treatment of PheATE with CoA and Sfp resulted in a shift of +339.8 Da from the predicted mass of the unmodified peptide, which is consistent with covalent attachment of the phosphopantetheinyl cofactor at Ser573 (Fig. 2B ▶). Treatment of this holo enzyme with a 50:50 mixture of d0:d5 phenylalanine followed by digestion and ESI/FTMS analysis yielded the Figure 2C ▶ spectrum. The broadened isotopic distribution (Fig. 2C ▶, inset) combined with the mass shift (combination of +147 and +152 Da) is in agreement with a phenylalanyl-S-PCP intermediate and does not require verification by MS/MS (Kelleher et al. 1997).
Digestion of ProCAT
ACNBr digest of ProCAT (120 kDa) was expected to yield 22 peptides ranging from 1 to 16 kDa in mass. The active site serine, Ser1007, is contained in a peptide of predicted mass 13,956.3–0 Da, which encompasses the C terminus, including the His tag. A peptide was observed at a mass of 13,497.2–0 Da, 459.1 Da lower than predicted (Fig. 3A ▶). MS/MS of these peptide ions yielded 17 b- and 19 y-type ions (data not shown). The y ions were all 459.1 Da lower than the predicted values, indicating that three to four residues were missing from the 11 C-terminal amino acids. The exact processing of this C-terminal region of ProCAT was not ascertained. The assignment was further confirmed by observing amass shift of +339.7 Da when Sfp and CoA were added to the reaction mixture before CNBr digestion (Fig. 3B ▶). Upon addition of proline and ATP, a new species was observed 97.4 Da higher than the holo form, indicating that the proline had been loaded onto the active site in near-quantitative yield (Fig. 3C ▶).
Figure 3.

(A) Expansion of the 16+ charge state of the active site containing the CNBr peptide of apo ProCAT. (B) Spectrum containing the 16+ charge state of the holo form of the peptide, as indicated by a mass shift of +339.7 Da due to loading of the phosphopantetheine arm. (C) The 16+ charge state of Pro-loaded form of ProCAT, which is evident by the +97.4 Da, corresponding to loading the constituent amino acid proline. An * indicates an adduct peak.
Substrate–A domain interaction
Attempted loading of PheATE with l-Leu, l-Tyr, or l-Trp resulted in minimal or no loading of the noncognate amino acid, but in each case a peak corresponding to the Phe-loaded CNBr digestion peptide was observed (data not shown). This suggested that the protein copurified with the cognate amino acid from the host organism throughout the purification. This was confirmed by simply incubating holo PheATE with ATP and observing a peak corresponding to Phe-loaded PheATE (Fig. 4B ▶). This demonstrates that the phenylalanine observed in the competition experiments (vide infra) was indeed protein-bound. Interestingly, a similar result was seen with ProCAT (Fig. 4 ▶, cf. C and D). Since ATP is required to observe aminoacylation, both PheATE and ProCAT are capable of copurifying with their cognate substrates through cell lysis and affinity chromatography to achieve ~40%–60% occupancy during in vitro ATP treatment. The occupancy of these aminoacyl-S-enzymes has been observed to be highly variable with different enzyme preparations. The observation that the substrates copurify with the apo enzyme is in agreement with the low Kd values for Phe and Pro, which are 6 and 0.6μM (Vater et al. 1985; Luo et al. 2001), respectively.
Figure 4.

Evidence for noncovalent association for amino acid substrates retained throughout protein purification. Shown is the region of the spectra containing 13+ ions (though all charge states were combined for quantitation) for the CNBr digestion peptides of PheATE after being treated with 0 (A) and 2 mM (B) ATP for 30 min. The region of the spectrum containing the 14+ charge states of the CNBr digest peptides of ProCAT following treatment with 0 (C) and 1 mM (D) ATP for 30 min; * indicates adduct peak.
Ionization efficiency of differentially modified ions
It has been shown that small proteins (~12 kDa) whose composition differs by a few amino acids can be relatively quantified by averaging the abundance over multiple charge states (Gordon et al. 1999). In the case of this study, the primary sequence of the peptides is identical, but the post-translational modifications change the overall composition of the peptide. Therefore, it was necessary to determine how the post-translational modifications affect the ionization efficiency of the peptides and the overall relative quantitation. A series of 50:50 mixtures of apo versus holo SrfB1 (a 13-kDa PCP domain fragment) was created and subjected to ESI/FTMS analysis (Fig. 5A ▶). After determining an apparent ratio for each charge state pair (Fig. 5B ▶), an overall ratio of 51% to 49% was determined using a weighted average for the intensity of each charge state. While ~25% fluctuations can be observed for low-abundance charge states (e.g., the 17+) and the apparent apo/holo ratio of individual charge states can alternate by 10%, an overall accuracy of <5% is possible. Further evidence was obtained by spraying defined apo/holo mixtures at ratios of 1:3, 3:1, 1:9, and 9:1. The values shown in Table 1 indicate that, even for large differences in the amount of apo versus holo (e.g., 1:9 and 9:1), the values are still within 5% of the anticipated result.
Figure 5.

(A) Broadband spectrum of apo SrfB1 and holo SrfB1 mixed in a 1:1 ratio. (B) The table at the bottom of the figure indicates the percentage of each form for each charge state. A weighted average, in which the most abundant charge state (in this case the 14+ charge state) contributes the greatest amount to the final average, was used to calculate the total percent apo vs. holo. For this spectrum the overall weighted average was apo (51%) and holo (49%).
Table 1.
Values obtained after mixing apo and holo SrfB1 in indicated ratios and measuring the percent occupancy over all charge states
| Experiment number | ||||||
| Mixing ratio | 1 | 2 | 3 | Average | Std. deviation | |
| 1:1 | Apo | 51% | 42% | 43% | 46% | 5% |
| Holo | 49% | 58% | 57% | 54% | ||
| 1:3 | Apo | 22% | 29% | 16% | 22% | 6% |
| Holo | 78% | 71% | 84% | 78% | ||
| 3:1 | Apo | 72% | 73% | 70% | 71% | 1% |
| Holo | 28% | 27% | 30% | 29% | ||
| 1:9 | Apo | 5% | 5% | 5% | 5% | <1% |
| Holo | 95% | 95% | 95% | 95% | ||
| 9:1 | Apo | 92% | 94% | 90% | 92% | 2% |
| Holo | 8% | 6% | 10% | 8% | ||
Since SrfB1 could be electrosprayed intact without prior digestion or chromatographic separation, it was not clear whether sample preparation steps used in digestion would artifactually alter observed ratios. Apo Phe-ATE and Phe-loaded Phe-ATE were mixed 1:1, digested with CNBr, and the resulting peptide mixture was fractionated by RPLC. The fractions were electrosprayed, and those containing either form of the CNBr digestion peptide were combined to obtain a final spectrum from which the ratios were determined (data not shown). The ratio across all charge states for the 1:1 sample was assessed as 57% apo and 43% Phe-loaded. A 1:3 mixture of apo to Phe-loaded yielded the ratio of 22% to 78%. The Δm for this PTM is 487 Da, with an addition of a phosphate, primary amine, amide, and phenyl functional groups and a 4% increase in mass. Despite these changes, our results show an accuracy of ~5% in the quantitation of relative ratios of closely related protein forms without the use of stable isotopes. This quantitation may be degraded substantially by using 1- to 3-kDa peptides versus the 10- to 30-kDa species often used in large molecule MS for prior NRPS/PKS studies (Mazur et al. 2003; Hicks et al. 2004; McLoughlin and Kelleher 2004) and detection of differently modified forms of histone proteins (—CH3, —PO4, —COCH3) (Medzihradszky et al. 2004; Pesavento et al. 2004; Zhang et al. 2004). Another advantage of using polypeptides >10 kDa for quantitative assays at high mass is their similar retention times of differentially modified forms during chromatographic separation by RPLC.
Condensation and DKP formation
In order to observe the dipeptide (Phe-Pro) on ProCAT, Phe-S-PheATE and Pro-S-ProCAT were mixed and the species trapped via acid quench after 45 sec and 5 min. The species dominant at >15 sec are both aminoacyl-substrates and holo forms (Fig. 6A,B ▶). Quadrupole enhancement of the region in which the dipeptide is expected to be observed did not reveal the dipeptide-loaded form. Cyclization and DKP formation occurs at a rate of 0.5 min−1 at pH 8 (Belshaw et al. 1999). It is possible that the DKP formation is faster than the rate of condensation, but it cannot be ruled out that the DKP is still forming during the 24-h CNBr digestion at pH 2.
Figure 6.

Dipeptide formation. (A) Quadrupole enhancement of the region in which the 16+ peptides of ProCAT and 15+ peptides of PheATE are observed, where a 1:1 ratio of ProCAT to PheATE was incubated for 45 sec. The lines indicate where each species would be for the 16+ charge states. (B) Shows the same region with a 2:1 ProCAT to PheATE ratio after a 5-min incubation. (C) Broadband spectrum with an expansion of the spectral region containing 16+ charge states for ProCAT and 15+ for PheATE in the holo, Ala, and Phe-Ala forms (as indicated) after 15 min of incubation.
Kinetics of condensation (Ala)
Alanine can be loaded onto ProCAT and used as a positive control for the condensation step between Phe-ATE and ProCAT, as the intermediate is not autocatalytically released from the enzyme. The Phe-Ala dipeptide was observed loaded on the T domain of Pro- CAT at 32% occupancy after a 15-min incubation at 37 °C, with 32% of ProCAT observed in the holo form and 36% in the Ala-loaded form (Fig. 6C ▶). Quenching the reaction at various time points allowed the percent occupancy of each form (holo-, Ala-, and Phe-Ala-S-ProCAT) to be determined by taking the average abundance over all charge states (Fig. 7 ▶). After 30 min the occupancy of Phe-Ala-S-ProCAT was 40%. After 5 h the percent occupancy remained the same, indicating that 100% occupancy could not be achieved. Using early time points (<60 sec) an initial rate of condensation was determined to be 1.8 min−1, which correlates well to the previously determined value of 1.4 min−1 (Belshaw et al. 1999).
Figure 7.

Kinetics of condensation, showing the increase over time in abundance of the Phe-Ala-S-ProCAT intermediate. The observed intermediates are labeled for the 16+ (ProCAT) and 15+ (PheATE) peptides after 0 sec (A), 10 sec (B), and 15 min (C ).
Competition experiment
After the realization that natural substrates can copurify with A domains, an external TEII domain was added to clear enzyme occupancy and then removed to allow full restoration of holo enzyme (data not shown). Treatment of this 100% holo-PheATE with an equimolar mixture of three alternative substrates (Trp, Tyr, and Leu) was followed by standard analysis of PheATE. After averaging the abundance of each species over all charge states it was observed that Trp was adenylated and loaded to the greatest extent (65%), followed by Leu (10%) and Tyr (5%), with 20% in the holo form (Fig. 8A ▶). No amount of Phe-loading was observed, indicating that the Phe had been completely removed from the enzyme active sites.
Figure 8.

(A) PheATE loading competition experiment between l-Leu, l-Tyr, and l-Trp. Each amino acid was added at a final concentration of 50 μM with 2 mM ATP and 1 nM SrfA-D. The lines indicate the peak corresponding to each amino acid (shown is the region containing the 14+ charge states of the CNBr digestion product). The percentage of each form for the entire spectrum is as follows: holo (20%), Leu (10%), Tyr (5%), and Trp (65%). An * indicates an adduct peak. (B) Dipeptide competition experiment. A quadrupole-enhanced region of the spectrum in which the 16+ charge states of ProCAT peptides and 15+ charge states for PheATE peptides are observed. d-Leu, d-Tyr, and d-Trp were added to PheATE in equimolar concentration and then allowed to condense onto Ala-S-ProCAT for 10 min.
A competition experiment was later performed with a preparation of PheATE that did not have Phe bound, therefore eliminating the need for the TEII. This experiment gave slightly different results in which Trp loaded to the greatest extent (77% ± 2), followed by Tyr (13% ± 3) and Leu (4% ± 1) (data not shown). When tested for activity in adenylation assays, these same amino acids gave Km values of 0.5, 1.1, and 0.3 mM, respectively, for l-Leu, l-Tyr, and l-Trp, compared with 0.03 mM for Phe (Luo et al. 2001). All kcat values were the same within experimental error. These values correlate qualitatively with the percent occupancies found in the FTMS-based competition experiments; though the Tyr:Leu ratio fluctuated, Trp always commanded the majority of enzyme occupancy.
The competition experiment was further extended to examine which noncognate amino acids would transfer to ProCAT. Using a mixture of the d-amino acids Trp, Leu, and Tyr loaded onto PheATE, a preferential loading of Trp (66% ± 5), followed by Tyr (25% ± 5) and then Leu (9% ± 1), was seen (data not shown). This mixture was then introduced to Ala-S-ProCAT, and after 10 min of loading both the Tyr-Ala and Trp-Ala dipeptides were observed (Fig. 8B ▶).
Discussion
The large size of PheATE and ProCAT (>100 kDa) prohibits their direct analysis by mass spectrometry with <1 Da mass accuracy. CNBr was employed to digest these enzymes into more manageable ~13-kDa peptides that harbored the carrier site Ser for both PheATE and ProCAT. Working with peptides of this size allows more reliable quantitation of differently modified forms of synthetase domains by virtue of their large size relative to typical tryptic peptides observed in an exhaustive digest (i.e., 1–2 kDa). In off-line ESI/MS analysis of RPLC-fractionated CNBr digests, the chromatographic shift in retention time that accompanies modification is typically <2 min, making the critical carrier peptides easier to find in complex peptide mixtures. Also, the multiple charge states observed in ESI/MS allows a weighted average of relative abundances to be calculated, balancing out differences in ionization efficiency and other unpredictable phenomena related to ESI.
Though many recent MS-based studies of closely related molecular forms of protein ions in the 5- to 60-kDa range have claimed “semiquantitative” analysis (i.e., ~10%–20% accuracy in the ratio determination of two or more species), the detailed characterization of the precision and accuracy in such experiments has not been studied in detail to date. By mixing various protein forms at known ratios, we determined that our quantitation is accurate to within 5%, even when the ratio was 1:9 (Table 1). This type of quantitation of high-mass species will likely find wider application in a proteomics context for 14N/15N-based determination of protein expression ratios.
A sequence error was detected in the thiolation domain of PheATE, and ProCAT was discovered to have undergone some post-translational processing as evident by a loss of 459.1 Da from the C terminus. The nature of this loss is still unknown, but likely involves the removal of at least three histidines from the C-terminal His tag. The assignment of each active-site peptide was confirmed by loading with the phosphopantetheine arm and observing a mass shift of +340 Da. An alternative method (Kelleher et al. 1997) was demonstrated using a mixture of d0:d5-ring labeled Phe producing a broadened isotopic distribution (Fig. 2C ▶, inset). Both methods provide ways to identify or verify an active-site peptide in NRPS or PKS assembly lines without requiring tandem MS of high-mass ions, an experiment most easily accomplished on expensive FT mass spectrometers.
When attempting to load PheATE with noncognate amino acids, the modified peptide corresponding to Phe-S-enzyme was always present, indicating that Phe was present not as the adenylate, but as the amino acid noncovalently bound to the adenylation domain. Only after ATP was added to the holo enzyme did the Phe-loaded peptide appear. This was observed for both Phe- ATE and ProCAT and was highly variable depending on enzyme preparation, with no binding of cognate amino acid in some cases. While it was not clear as to why this occurred, it was shown that the cognate amino acid could be readily removed and restored to the holo form by the use of TEII domain. Studies using radioactively labeled amino acid substrates would not detect a nonlabeled amino acid bound to the enzyme, which could potentially skew results. MS-based methods complement traditional methods in that multiple, nonlabeled intermediates can be observed in tandem.
PheATE and ProCAT have been used extensively to study the condensation event between two modules (Stachelhaus et al. 1998; Belshaw et al. 1999; Stachelhaus and Walsh 2000; Bergendahl et al. 2002). In this study, the Phe-Pro dipeptide was not observed as a covalently bound intermediate, but Ala could be used as a control, therefore allowing the determination of an initial rate of condensation for the Phe-Ala dipeptide of ~2 min−1, consistent with that determined by Belshaw and coworkers (Belshaw et al. 1999). FTMS provides a viable method for determining kinetic parameters, such as condensation rates, by looking at the relative change in percent occupancy of intermediates at defined time points.
Previous studies have probed the specificity of Phe- ATE, examining which amino acids could be adenylated and loaded onto the enzyme. In this study, we mimicked a likely situation confronting attempts to engineer thiotemplate assembly lines for combinatorial production of new natural products. PheATE was presented with an equimolar mixture of possible substrates, and we directly measured the preferred processing through multiple enzymatic steps in a competition experiment of nonnatural substrates. The results qualitatively track the far more careful studies of Luo and coworkers (Luo et al. 2001), in which each substrate was kinetically interrogated in isolation. The change in percent occupancy at the PCP domain is quite drastic for a small change in substrate Km. This has profound implications for combinatorial biosynthetic strategies, requiring more knowledge of substrate kinetic parameters than would be preferred for utilizing enzymes in vitro without much prior characterization by structure–activity relationships (SAR). This method will be extended to other systems to track the enzymatic flux of complex arrays of substrates being processed down a wild-type or a mutated assembly line.
Utilizing the high resolution and molecular specificity of FTMS has allowed simultaneous and highly quantitative identification of multiple intermediates in parallel. This, combined with our finding that A domains can bind their cognate amino acids from the cell throughout purification, truly complements nonMS-based methods in which multiple species cannot be observed, and will aid in the efforts to streamline production and troubleshooting of new engineered biocatalysts.
Materials and methods
Expression and purification of enzymes
PheATE and ProCATwere both expressed as previously described (Stachelhaus et al. 1998). The expressed GrsA gene product was found to contain a 26.6-Da mass discrepancy. Resequencing a 1100-bp region revealed a single base pair error changing Asn541 to Ser541 (Δm = 27.1 Da) (data not shown). Resequencing of ProCAT did not reveal any errors in the thiolation domain region. SrfB1 (Weinreb et al. 1998) and SrfA-D (Yeh et al. 2004) were purified as described elsewhere.
Conversion to holo enzyme
Enzymes were converted from apo to holo forms by incubation with CoA and Sfp (Lambalot et al. 1996). Reaction mixtures contained 5 μM protein, 25 μM CoA, and 1 nmol Sfp in assay buffer (10 mM MgCl2, 50 mM HEPES [pH 7.5], 1 mM TCEP) for a final volume of 200 μL and were incubated for 2 h at 37°C to ensure complete conversion.
Loading of amino acids
Aminoacylation of PheATE was initiated by the addition of 50 μM amino acid and 2 mM ATP. The reactions were then incubated for 20 min to 2 h at 37°C. For isotopic broadening, equal amounts of unlabeled phenylalanine and d5-ring labeled phenylalanine were added to the reaction mixture and incubated with ATP for 2 h at 37°C.
Holo-ProCAT was treated in the same manner. Fifty μM proline or alanine was added along with 2 mM ATP and incubated for 30 min at 37°C. For dipeptide experiments, holo-PheATE and holo-ProCAT were first loaded with their constituent amino acids for 30 min at 37°C, then the two enzymes were combined and acid quenched at various time points.
Digestions
Digestions were done with CNBr obtained from Aldrich. CNBr was dissolved in a 30% TFA/70% H2O solution that was then added to the reactions at a ratio of 2:1. The digestion proceeded in the dark for 24 h at 4°C and was terminated by freezing at −80°C (Zhang et al. 1996).
RPLC analysis
Digestions were lyophilized and resuspended in 500 μL (1 mL for dipeptide experiments) 6 M urea, 100 mM NH4OAc [pH 4.0], 5 mM TCEP, and incubated for 20 min at 37°C before injection onto an RPLC column. Off-line fractionation of the CNBr digests was done on a Jupiter C18 column 5 μm 300 Å 150 × 4.6 mm (Phenomenex) using a 10%–95% CH3CN/0.1% TFA gradient over 35 min. The peptide of interest eluted from 29 to 31 min for PheATE and 28 to 30 min for ProCAT. The fractions were lyophilized before MS analysis. For the dipeptide experiments, RPLC fractions containing active site peptides were dissolved in 30 μL H2O/0.1% TFA and combined. The combined sample was then desalted using a C18 ZipTip (Millipore).
Release of cognate amino acids
SrfA-D, a type II thioesterase, was added to the holo enzyme along with ATP and the noncognate amino acids. Fifty μM of each amino acid (l-Leu, l-Tyr, l-Trp) was added to holo-PheATE along with 2 mM ATP and 1 nM SrfA-D. The reaction was incubated for 4 h at 37°C. SrfA-D was then removed from the reaction using a Microcon centrifugal filter device (YM-100) and ATP. The amino acids were added again and the reaction was incubated for 15 min at 37°C before quenching for a CNBr digestion.
Competition experiments
After priming PheATE as above, 50 μM l-Leu, l-Tyr, and l-Trp (or d-amino acids, as indicated) were added with 2 mM ATP. After 30 min, the mixture was either quenched by doing a CNBr digestion (control experiments) or combined with Pro- CAT loaded with Ala (see above). Dipeptide reactions were quenched at various time points and treated as above.
Studies of ionization efficiency
Solutions with equimolar concentrations of apo and holo SrfB1 were generated from the same stock of protein; the only difference between apo and holo is that H2O was added in place of CoA in the apo reaction. The loading reactions were quenched by lowering the pH to 2 with formic acid. Apo and holo SrfB1 were added in the indicated ratios and then desalted on a C18 ZipTip (Millipore). Apo and Phe-loaded GrsA were generated in a similar manner as above. Each form was added into the CNBr digestion solution in the indicated ratios. RPLC was performed as previously described.
The total abundance of each form (including adduct peaks) was determined by adding the relative signal intensity for the top three isotopes. Percent occupancy of apo and holo for each charge state was obtained by dividing the total signal for each form by the combined signal for both. The overall ratio across all charge states was calculated using a weighted average, wherein the most abundant charge state contributed the greatest to the final ratio. Each experiment was done three times, and the values for each experiment were averaged.
ESI-Q-FTMS analysis
FTMS analysis was performed on a custom-built 8.5 T Quadrupole- FTMS, the basic design and operation of which is reported elsewhere (Patrie et al. 2004). Dried samples were resuspended in ESI spray solution (78% CH3CN, 20% H2O, 2% acetic acid, or 50% CH3CN, 48% H2O, 2% acetic acid). The ~50-μL samples were transferred at 300 nL/min through 50 μm i.d. silica tubing that terminated in a 50-μm tip (New Objective), held at ~2200 V. Alternatively, a NanoMate 100 (Advion BioSciences) was used to introduce the samples via automated nanospray into the instrument. Ions were then directed into the Q-FTMS by a series of ion optics (Patrie et al. 2004). Relative molecular weight (Mr) values are reported with an italicized number indicating the number of heavy isotopes in the peak referenced (Kelleher et al. 1999). Single-protein mode of the ProSight PTM software bundle was used to interpret MS/MS data. ProSight PTM is freely available at https://prosightptm.scs.uiuc.edu.
Acknowledgments
We thank Torsten Stachelhaus for the PheATE and ProCAT plasmids, and Ellen Yeh and Prof. Christopher Walsh for the SrfA-D and SrfB1 plasmids. We also thank Pieter Dorrestein and Leslie Hicks for their helpful discussions and assistance with this work. This work was supported by the NIH (GM 067725) and the Alfred P. Sloan Foundation.
Abbreviations
MS, mass spectrometry
Q-FTMS, quadrupole-Fourier-transform mass spectrometry
TEII, type II thioesterase
NRPS, nonribosomal peptide synthases
PKS, polyketide synthases
GrsA, gramicidin S synthetase I
GrsB, gramicidin S synthetase II
A, adenylation domain
T, thiolation domain
E, epimerization domain
C, condensation domain
TE, thioesterase domain
DKP, diketopiperazine
CNBr, cyanogen bromide
CoA, coenzyme A
TCEP, tris(2-carboxyethyl)phosphine hydrochloride
RPLC, reverse-phase liquid chromatography
ESI, electrospray ionization
Mr, relative molecular weight
MS/MS, tandem MS
Δm, change in mass
PTM, post-translational modification
PCP, peptidyl carrier protein.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051553705.
References
- Belshaw, P.J., Walsh, C.T., and Stachelhaus, T. 1999. Aminoacyl-CoAs as probes of condensation domain selectivity in nonribosomal peptide synthesis. Science 284 486–489. [DOI] [PubMed] [Google Scholar]
- Bergendahl, V., Linne, U., and Marahiel, M.A. 2002. Mutational analysis of the C-domain in nonribosomal peptide synthesis. Eur. J. Biochem. 269 620–629. [DOI] [PubMed] [Google Scholar]
- Gordon, E.F., Mansoori, B.A., Carroll, C.F., and Muddiman, D.C. 1999. Hydropathic influences on the quantification of equine heart cytochrome c using relative ion abundance measurements by electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. J. Mass Spectrom. 34 1055–1062. [DOI] [PubMed] [Google Scholar]
- Hicks, L.M., O’Connor, S.E., Mazur, M.T., Walsh, C.T., and Kelleher, N.L. 2004. Mass spectrometric interrogation of thioester-bound intermediates in the initial stages of epothilone biosynthesis. Chem. Biol. 11 327–335. [DOI] [PubMed] [Google Scholar]
- Hori, K., Yamamoto, Y., Minetoki, T., Kurotsu, T., Kanda, M., Miura, S., Okamura, K., Furuyama, J., and Saito, Y. 1989. Molecular cloning and nucleotide sequence of the Gramicidin S Synthetase 1 gene. J. Biochem. 106 639–645. [DOI] [PubMed] [Google Scholar]
- Kelleher, N.L., Nicewonger, R.B., Begley, T.P., and McLafferty, F.W. 1997. Identification of modification sites in large biomolecules by stable isotope labeling and tandem high resolution mass spectrometry. J. Biol. Chem. 272 32215–32220. [DOI] [PubMed] [Google Scholar]
- Kelleher, N.L., Lin, H.Y., Valaskovic, G.A., Aaserud, D.J., Fridriksson, E.K., and McLafferty, F.W. 1999. Top down versus bottom up protein characterization by tandem high-resolution mass spectrometry. J. Am. Chem. Soc. 121 806–812. [Google Scholar]
- Lambalot, R.H., Gehring, A.M., Flugel, R.S., Zuber, P., LaCelle, M., Marahiel, M.A., Reid, R., Khosla, C., and Walsh, C.T. 1996. A new enzyme superfamily—The phosphopantetheinyl transferases. Chem. Biol. 3 923–936. [DOI] [PubMed] [Google Scholar]
- Luo, L. and Walsh, C.T. 2001. Kinetic analysis of three activated phenylalanyl intermediates generated by the initiation module PheATE of gramicidin S synthetase. Biochemistry 40 5329–5337. [DOI] [PubMed] [Google Scholar]
- Luo, L., Burkart, M.D., Stachelhaus, T., and Walsh, C.T. 2001. Substrate recognition and selection by the initiation module Phe- ATE of gramicidin S synthetase. J. Am. Chem. Soc. 123 11208– 11218. [DOI] [PubMed] [Google Scholar]
- Mazur, M.T., Walsh, C.T., and Kelleher, N.L. 2003. Site-specific observation of acyl intermediate processing in thiotemplate biosynthesis by Fourier transform mass spectrometry: The polyketide module of yersiniabactin synthetase. Biochemistry 42 13393–13400. [DOI] [PubMed] [Google Scholar]
- McLafferty, F.W. 1994. High-resolution tandem FT mass spectrometry above 10 kDa. Acc. Chem. Res. 27 379–386. [Google Scholar]
- McLoughlin, S. and Kelleher, N.L. 2004. Kinetic and regiospecific interrogation of covalent intermediates in the nonribosomal peptide synthesis of yersiniabactin. J. Am. Chem. Soc. 126 13265–13275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzihradszky, K.F., Zhang, X., Chalkley, R.J., Guan, S., McFarland, M.A., Chalmers, M.J., Marshall, A.G., Diaz, R.L., Allis, C.D., and Burlingame, A.L. 2004. Characterization of Tetrahymena histone H2B variants and posttranslational populations by electron capture dissociation (ECD) Fourier transform ion cyclotron mass spectrometry (FT-ICR MS). Mol. Cell Proteomics 3 872–886. [DOI] [PubMed] [Google Scholar]
- Patrie, S.M., Charlebois, J.P., Whipple, D., Kelleher, N.L., Hendrickson, C.L., Quinn, J.P., Marshall, A.G., and Mukhopadhyay, B. 2004. Construction of a hybrid quadrupole/Fourier transform ion cyclotron resonance mass spectrometer for versatile MS/MS above 10 kDa. J. Am. Soc. Mass Spectrom. 15 1099–1108. [DOI] [PubMed] [Google Scholar]
- Pesavento, J.J., Kim, Y.B., Taylor, G.K., and Kelleher, N.L. 2004. Shotgun annotation of histone modifications: A new approach for streamlined characterization of proteins by top down mass spectrometry. J. Am. Chem. Soc. 126 3386–3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer, D., Mootz, H.D., Linne, U., and Marahiel, M.A. 2002. Regeneration of misprimed nonribosomal peptide synthetases by type II thioesterases. Proc. Natl. Acad. Sci. 99 14083–14088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieber, S.A. and Marahiel, M.A. 2005. Molecular mechanisms underlying nonribosomal peptide synthesis: Approaches to new antibiotics. Chem. Rev. 105 715–738. [DOI] [PubMed] [Google Scholar]
- Stachelhaus, T. and Marahiel, M.A. 1995. Modular structure of peptide synthetases revealed by dissection of the multifunctional enzyme GrsA. J. Biol. Chem. 270 6163–6169. [DOI] [PubMed] [Google Scholar]
- Stachelhaus, T. and Walsh, C.T. 2000. Mutational analysis of the epimerization domain in the initiation module PheATE of gramicidin S synthetase. Biochemistry 39 5775–5787. [DOI] [PubMed] [Google Scholar]
- Stachelhaus, T., Mootz, H.D., Bergendahl, V., and Marahiel, M.A. 1998. Peptide bond formation in nonribosomal peptide biosynthesis. J. Biol. Chem. 273 22773–22781. [DOI] [PubMed] [Google Scholar]
- Vater, J., Mallow, N., Gerhardt, S., Gadow, A., and Kleinkauf, H. 1985. Gramicidin S synthetase. Temperature dependence and thermodynamic parameters of substrate amino acid activation reactions. Biochemistry 24 2022–2027. [DOI] [PubMed] [Google Scholar]
- Vater, J., Schlumbohm, W., Palacz, Z., Salnikow, J., Gadow, A., and Kleinkauf, H. 1987. Formation of D-Phe-Pro-Val-Cyclo-Orn by gramicidin- S synthetase in the absence of L-leucine. Eur. J. Biochem. 163 297–302. [DOI] [PubMed] [Google Scholar]
- von Dohren, H., Keller, U., Vater, J., and Zocher, R. 1997. Multifunctional peptide synthetases. Chem. Rev. 97 2675–2706. [DOI] [PubMed] [Google Scholar]
- Weinreb, P.H., Quadri, L.E.N., Walsh, C.T., and Zuber, P. 1998. Stoichiometry and specificity of in vitro phosphopantetheinylation and aminoacylation of the valine-activating module of surfactin synthetase. Biochemistry 37 1575–1584. [DOI] [PubMed] [Google Scholar]
- Yeh, E., Kohli, R.M., Bruner, S.D., and Walsh, C.T. 2004. Type II thioesterase restores activity of a NRPS module stalled with and aminoacyl- S-enzyme that cannot be elongated. Chem. Bio. Chem. 5 1290– 1293. [DOI] [PubMed] [Google Scholar]
- Zhang, X., Dillen, L., Vanhoutte, K., Van Dongen, W., Esmans, E., and Claeys, M. 1996. Characterization of unstable intermediates and oxidized products formed during cyanogen bromide cleavage of peptides and proteins by electrospray mass spectrometry. Anal. Chem. 68 3422– 3430. [DOI] [PubMed] [Google Scholar]
- Zhang, L., Freitas, M.A., Wickham, J., Parthun, M.R., Klisovic, M.I., Marcucci, G., and Byrd, J.C. 2004. Differential expression of histone post-translational modifications in acute myeloid and chronic lymphocytic leukemia determined by high-pressure liquid chromatography and mass spectrometry. J. Am. Soc. Mass Spectrom. 15 77–86. [DOI] [PubMed] [Google Scholar]