

Abstract
We introduce a novel approach to estimate differences in the β-aggregation potential of eukaryotic proteomes. The approach is based on a statistical analysis of the β-aggregation propensity of polypeptide segments, which is calculated by an equation derived from first principles using the physicochemical properties of the natural amino acids. Our analysis reveals a significant decreasing trend of the overall β-aggregation tendency with increasing organism complexity and longevity. A comparison with randomized proteomes shows that natural proteomes have a higher degree of polarization in both low and high β-aggregation prone sequences. The former originates from the requirement of intrinsically disordered proteins, whereas the latter originates from the necessity of proteins with a stable folded structure.
Keywords: aggregation, protein aggregation propensity, proteome, intrinsically disordered proteins
Even proteins not implicated in amyloid diseases have been shown to form fibrils in vitro under denaturing conditions, indicating that fibrillogenesis is a common feature of polypeptide chains, which can form intermolecular backbone–backbone hydrogen bonds (Chiti et al. 1999, 2003) and favorable side-chain interactions (Azriel and Gazit 2001; Gsponer et al. 2003; Makin et al. 2005). Although in lower eukaryotes amyloid fibrils could represent an inheritable phenotype related to specific cellular functions (Osherovich and Weissman 2002; Osherovich et al. 2004; Si et al. 2003b), the cytotoxicity of prefibrillar aggregates (Bucciantini et al. 2002) and their association with diseases such as Alzheimer’s, Parkinson’s, Huntington’s, prion disease, cystic fibrosis, and type II diabetes (Kelly 1998; Rochet and Lansbury 2000) suggest that amyloid aggregates are generally dangerous for higher eukaryotes (Dobson 1999; Stefani and Dobson 2003).
We have previously developed an equation to predict the propensity for ordered aggregation, which solely requires the polypeptide sequence as input (Tartaglia et al. 2004, 2005). Our model is based on the physicochemical properties of the residues and takes into account both amino acid composition and positional information. The aggregation propensity πil of an l-residue segment starting at position i in the sequence is evaluated as
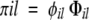 |
(1) |
The factor Φil contains exponential functions and is position-dependent
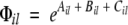 |
(2) |
where Ail, Bil, and Cil are functionals related to the aromaticity, β-propensity, and charge, respectively. The factor φil depends almost exclusively on the amino acid composition
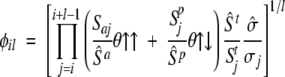 |
(3) |
where Sai, Spi, Sti, and σi—weighted by their average over the 20 standard amino acids (hatted values)—are the side-chain apolar, polar, total water-accessible surface area, and solubility, respectively. The functionals y↑↑ and y↑↓ include positional effects and reflect the parallel or anti-parallel tendency to aggregate if the majority of residues is apolar or polar, respectively. Details of the method are presented in the preceding paper (Tartaglia et al. 2005).
In the present work, we analyze complete proteomes of several eukaryotes to identify changes of β-aggregation propensity through organisms of different complexity. The 32,869 entries belonging to the human proteome database (Supplemental Material, Table 1) were decomposed in stretches of different sizes (5, 50, and 150 residues) to compute the β-aggregation propensity with Equation 1 and build the normalized histogram of β-aggregation propensity distribution, APD (Fig. 1A ▶). For each stretch size, the distribution is found to be nonsymmetric with respect to the average and skewed to the left, indicating that there are more stretches with low β-aggregation propensity (left tail of APD) than with high propensity (right tail). As pointed out in our previous study, short stretches are preferable to long stretches for the analysis of β-aggregation propensity because the latter contain folding features that deteriorate the signal-to-noise ratio (Tartaglia et al. 2005). Hence, a window size of five residues was used to analyze complete proteomes of Homo sapiens, Mus musculus, Drosophila melanogaster, Caenorhabditis elegans, Saccharomyces cerevisiae, and Paramecium tetraurelia (Supplemental Material, Table 1). Nonhuman eukaryotes show a larger amount of high-propensity stretches and a smaller amount of low-propensity stretches compared with H. sapiens (Fig. 1A ▶). Moreover, a clear trend is found with the increasing complexity of the organisms and their lifetime. To quantify this trend it is useful to introduce the APD deviation between two proteomes, x and y
Figure 1.

(A) (Inset) Distribution of the number of human polypeptide sequences as a function of β-aggregation propensity (APD) at three different window sizes. (Main plot) APD differences with respect to H. sapiens for complete proteomes of M. musculus, D. melanogaster, C. elegans, S. cerevisiae, and P. tetraurelia (window size of five residues). Life spans of organisms are reported in parentheses. (B) Unrooted tree diagram derived from the APD deviation (Equation 4). The deviation is computed from P. tetraurelia as a reference and magnified by a factor of 1000. The arrow indicates that lower eukaryotes have more high-propensity and fewer low-propensity stretches. This diagram is built using Phylodraw with the Fitch and Margoliash (1967) clustering algorithm. Data labeled with * belong to incomplete proteomes. (Phylodraw is available at http://pearl.cs.pusan.ac.kr/phylodraw/.) (C) Normalized histogram of the number of proteins as a function of the content of residues enriched in low-propensity and high-propensity stretches. Global contours are shown for all proteomes by solid lines. Isofrequency regions are shown for the human proteome, where red color indicates the most populated area, while blue fading color indicates the least-populated areas. (D) Same as C for shuffled proteomes.
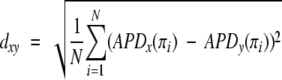 |
(4) |
where the β-aggregation propensity π is calculated by Equation 1 (Tartaglia et al. 2005) and i runs over the total number of bins N (N=100) in the APD histogram. With the addition of the proteomes of Danio rerio, Xenopus laevis, and Gallus gallus, the APD deviation was used to build the tree diagram of Figure 1B ▶. Except for the inversion between the amphibious X. laevis and the fish D. rerio (whose proteomes are not complete), the tree of Figure 1B ▶ is similar to the phylogenetic tree of cytochrome c (Dayhoff et al. 1972). Thus, the deviation calculated from P. tetraurelia, dxP, is an observable able to rank proteomes of organisms of increasing complexity. It is interesting to compare the amino acid frequencies in APD tails—defined for a subtended area of 0.05 in the histogram of Figure 1A ▶—with amino acid frequencies in entire proteomes (Table 1). This analysis reveals that for all proteomes stretches with low β-aggregation propensity are rich in A, G, H, K, P and R, whereas high-propensity stretches in C, F, I, L, N, Q, V, and Y. Figure 1C ▶ is a two-dimensional histogram that shows the number of proteins as a function of the content of residues enriched in low-propensity stretches and the content of residues predominant in high-propensity stretches. By increasing the organism complexity, the number of proteins with low-propensity residues increases, while the number of proteins with high-propensity residues decreases. A comparison with randomized proteomes is useful to further investigate the significance of such trends. Randomized proteomes were generated by shuffling amino acids within complete proteomes and keeping unchanged the global amino acid composition, number, and length of proteins. We stress that the β-aggregation propensity of five-residue stretches cannot differentiate natural and shuffled proteomes, because short segments describe mainly effects of the amino acid composition. Yet, differences between natural and shuffled proteomes are enhanced when residues belonging to low-/high-propensity stretches are used for the analysis of entire proteins. Comparing Figure 1, C and D ▶, it is evident that shuffled proteomes are less spread. In other words, natural proteomes reveal a sensible increase of sequences with residues predominant in low-propensity stretches as well as residues enriched in high-propensity stretches. While the amino acid global composition of proteomes is almost identical in higher eukaryotes, the content of low-propensity stretches increases significantly, indicating a clear change of protein features from proteome to proteome.
Table 1.
Amino acid frequencies in left or right APD tails of H. sapiens divided by their corresponding frequency in the whole proteome
| A | C | D | E | F | G | H | I | K | L | |
| Left/total | 1.1 | 0.2 | 0.4 | 0.5 | 0.4 | 1.3 | 1.6 | 0.5 | 2.1 | 0.5 |
| Right/total | 0.7 | 2.4 | 0.8 | 0.7 | 2.7 | 0.6 | 0.5 | 1.6 | 0.3 | 1.8 |
| M | N | P | Q | R | S | T | V | W | Y | |
| Left/total | 0.4 | 0.2 | 3.3 | 0.3 | 2.8 | 0.6 | 0.5 | 0.7 | 0.4 | 0.1 |
| Right/total | 0.8 | 1.5 | 0.2 | 1.2 | 0.2 | 0.7 | 0.8 | 1.2 | 0.8 | 2.7 |
Values exceeding 1.0 are shown in bold. Similar frequencies were found for all the proteomes.
It has recently been shown that natively unfolded proteins (or intrinsically disordered proteins, IDPs) are implicated in cellular regulation, signaling, and assembly/disassembly of macromolecular complexes (Dunker et al. 2002; Ward et al. 2004; Oldfield et al. 2005). The absence of a fixed structure suggests functional implications, which are required in complex organisms (Koonin et al. 2002). Interestingly, a larger diffusion of IDPs is found in higher eukaryotes than in lower eukaryotes and prokaryotes (Dunker et al. 2002; Liu et al. 2002; Linding et al. 2004). Using data from X-ray crystallography, nuclear magnetic resonance, and circular dichroism, Williams et al. (2001) found a high percentage of P, R, K, G, A, Q, S, and E in nonfolded segments of proteins, and F, Y, C, L, V, N, and W in folded segments. Except for Q, S, and E, Williams’ finding is in agreement with our tail composition analysis (Table 1), indicating that residues enriched in aggregating stretches promote both folding and β-aggregation, whereas residues predominant in stretches with low β-aggregation propensity are also enriched in IDPs.
To better understand the relationship between β-aggregation propensity and protein structure, we analyzed the APDs of polypeptide segments that assume a regular secondary structure, as well as IDPs (Supplemental Material, Table 1). As shown in Figure 2A ▶, strands have more β-aggregation potential than helices, and IDPs are the least prone to aggregate, in agreement with Linding’s analysis (Linding et al. 2004). Moreover, from lower to higher eukaryotes the APD deviation with respect to IDP decreases, while the APD deviation from strands increases (Fig. 2B,C ▶). The APD deviation of helices does not follow a monotonic trend and slowly increases from S. cerevisiae to H. sapiens. Compared with strands, helices display a lower amount of aggregation stretches, but it has to be mentioned that the transition helix-strand generates amyloidogenesis in some proteins (Selkoe 1996; Prusiner 1997).
Figure 2.

(A) APDs of five-residue stretches belonging to intrinsically disordered proteins (IDPs) or regular secondary structure elements within folded proteins and IDPs. (B) Deviation between the APD of entire proteomes and the APD of segments belonging to regular secondary structure or IDPs as a function of the organism complexity. The organism complexity is measured by the APD deviation from P. tetraurelia, dxP. Solid lines are drawn solely to guide the eye. (C) From lower to higher eukaryotes, the decrease of β-aggregation propensity is related to the increase of intrinsically disordered proteins.
To quantify interspecies shifts of amino acid compositions in the APD tails, we fitted the amino acid frequencies as a linear function of the APD deviation from P. tetraurelia, dxP (see Equation 4)
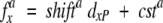 |
(5) |
where fax is the frequency of the amino acid a in the proteome x, shifta is the slope of the fit, and csta is the intercept. The sign “+” or “−” of the shifta was interpreted as a measure for the depletion or the enrichment of the amino acid a from P. tetraurelia to H. sapiens. Shifts obtained from high-confidence fits (Pearson’s correlation >0.80; Supplemental Material, Table 2) are
Right tails, i.e., high propensity: Decrease of Q, N, Y, and K and increase of L, V, A, W, R, H, G, and P.
Left tails, i.e., low propensity: Decrease of K, I, F, and N and increase of P, A, G, R, S, and E.
Interestingly, the decrease of Q, N, and Y in the right tails was already observed in higher eukaryote prion homologs of the yeast Sup35 prion protein (Balbirnie et al. 2001; Si et al. 2003a; Theis et al. 2003) and suggests that the trend does not affect only a specific family of proteins. In addition, we speculate that the increase of L, V, A, and W in the right tail is a consequence of the optimization of the “hydrophobic core” to stabilize the native state (Kellis et al. 1989; Richards and Lim 1993; Dill et al. 1995; Stefani and Dobson 2003).
The functional role of aggregation phenotypes in multicellular eukaryotes is still a matter of debate. Recently, it has been observed that the neuronal protein CPEB of Aplysia californica behaves like a prion switch that regulates long-term synaptic changes associated with memory storage (Si et al. 2003a,b). The switch mechanism involves the aggregation of the CPEB N terminus, rich in Q- and N- repeats that are missing in mammalian isoforms of CPEB (Theis et al. 2003). Motivated by these observations, we analyzed the data set of proteins expressed in neurons (Supplemental Material, Table 1). For a given proteome, the neuronal APD perfectly overlaps with the APD of the total proteome (data not shown), indicating that neuronal proteins are a descriptive subset of the total proteome and do not follow any specific trend. We thus cannot draw conclusions on particular links between memory mechanisms and aggregation phenotypes.
It has been shown that the frequency of N and Q repeats does not represent an observable able to describe amyloidogenic trends of proteomes (Michelitsch and Weissman 2000; Osherovich and Weissman 2002). Our findings indicate that to quantify aggregation trends, it is crucial to use an observable, such as the β-aggregation propensity, which accounts for the aggregation contribution of all amino acids including positional information.
In conclusion, we have introduced a novel approach to compare proteomes, which is based on the statistical analysis of ordered-aggregation propensity. From P. tetraurelia to H. sapiens, we have shown that proteomes of higher and more long-lived eukaryotes contain fewer sequences with high β-aggregation propensity and are accrued in proteins with low β-aggregation propensity. We also observed that, compared with random proteomes, natural proteomes are enriched in proteins with low β-aggregation potential, as well as proteins with high β-aggregation potential. Such polarization is a consequence of the dual evolutive requirement of IDPs with low β-aggregation propensity, as well as proteins with a stable fold, which comes at the cost of higher β-aggregation propensity. In the future, we plan to use gene ontology annotations of proteins with high predicted β-aggregation propensity to obtain insights into the specific role of some of the amyloidogenic proteins of unknown function.
Electronic supplemental material
This section contains two tables: Table 1 contains information for databases used in the article (origin of data sets, number of entries of the databases, and number of stretches used in our analysis); Table 2 contains fitting parameters for the amino acid shifts (see Equation 5).
Acknowledgments
We thank Dr. A.G. Abebe and M. Cecchini for very interesting discussions. This work was supported by the Swiss National Science Foundation and the NCCR “Neural Plasticity and Repair.”
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051473805.
Supplemental material: see www.proteinscience.org
References
- Azriel, R. and Gazit, E. 2001. Analysis of the minimal amyloid-forming fragment of the islet amyloid polypeptide. J. Biol. Chem. 276 34156–34161. [DOI] [PubMed] [Google Scholar]
- Balbirnie, M., Grothe, R., and Eisenberg, D. 2001. An amyloid-forming peptide from the yeast prion Sup35 reveals a dehydrated β-sheet structure for amyloid. Proc. Natl. Acad. Sci. 98 2375–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucciantini, M., Giannoni, E., Chiti, F., Baroni, F., Formigli, L., Zurdo, J., Taddei, N., Ramponi, G., Dobson, C.M., and Stefani, M. 2002. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416 507–511. [DOI] [PubMed] [Google Scholar]
- Chiti, F., Calamai, M., Taddei, N., Stefani, M., Ramponi, G., and Dobson, C.M. 1999. Studies of the aggregation of mutant proteins in vitro provide insights into the genetics of amyloid diseases. Proc. Natl. Acad. Sci. 99 16419–16426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti, F., Stefani, M., Taddei, N., Ramponi, G., and Dobson, C.M. 2003. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature 424 805–808. [DOI] [PubMed] [Google Scholar]
- Dayhoff, M.O., Park, C.M., and McLaughlin, P.J. 1972. Atlas of protein sequence and structure. National Biomedical Research Foundation, Silver Spring, MD.
- Dill, K.A., Yue, K., Fiebig, K.M., Yee, D.P., Thomas, P.D., and Chan, H.S. 1995. Principles of protein folding—A perspective from simple exact models. Protein Sci. 4 561–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson, C.M. 1999. Protein misfolding, evolution and disease. Trends Biochem. Sci. 24 329–332. [DOI] [PubMed] [Google Scholar]
- Dunker, A.K., Brown, C.J., Lawson, J.D., Iakoucheva, L.M., and Obradovic, Z. 2002. Intrinsic disorder and protein function. Biochemistry 41 6574–6582. [DOI] [PubMed] [Google Scholar]
- Fitch, W.M. and Margoliash, E. 1967. Construction of phylogenetic tree. Science 155 279–284. [DOI] [PubMed] [Google Scholar]
- Gsponer, J., Habertuer, U., and Caflisch, A. 2003. The role of side-chain interactions in the early steps of aggregation: Molecular dynamics simulations of an amyloid-forming peptide from the yeast prion Sup35. Proc. Natl. Acad. Sci. 100 5154–5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellis, J.T., Nyberg, K., and Fersht, A.R. 1989. Energetics of complementary side-chain packing in a protein hydrophobic core. Biochemistry 28 4914–4922. [DOI] [PubMed] [Google Scholar]
- Kelly, J.W. 1998. The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr. Opin. Struct. Biol. 8 101–106. [DOI] [PubMed] [Google Scholar]
- Koonin, E.V., Wolf, Y.I., and Karev, G.P. 2002. The structure of the protein universe and genome evolution. Nature 420 218–223. [DOI] [PubMed] [Google Scholar]
- Linding, R., Schymkowitz, J., Rousseau, J., Diella, F., and Serrano, L. 2004. A comparative study of the relationship between protein structure and β-aggregation in globular and intrinsically disordered proteins. J. Mol. Biol. 342 345–353. [DOI] [PubMed] [Google Scholar]
- Liu, J., Tau, H., and Rost, B. 2002. Loopy proteins appear conserved in evolution. J. Mol. Biol. 322 53–64. [DOI] [PubMed] [Google Scholar]
- Makin, O.S., Atkins, E., Sikorski, P., Johansson, J., and Serpell, L.C. 2005. Molecular basis for amyloid fibril formation and stability. Proc. Natl. Acad. Sci. 102 315–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelitsch, M.D. and Weissman, J.S. 2000. A census of glutamine/asparagine- rich regions: Implications for their conserved function and the prediction of novel prions. Proc. Natl. Acad. Sci. 97 11910–11915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldfield, C.L., Cheng, Y., Cortese, M.S., Brown, C.J., Uversky, V.N., and Dunker, A.K. 2005. Comparing and combining predictors of mostly disordered proteins. Biochemistry 44 1989–2000. [DOI] [PubMed] [Google Scholar]
- Osherovich, L.Z. and Weissman, J.S. 2002. The utility of prions. Dev. Cell 2 143–151. [DOI] [PubMed] [Google Scholar]
- Osherovich, L.Z., Cox, B.S., Tuite, M.F., and Weissman, J.S. 2004. Dissection and design of yeast proteins. PLoS Biol. 2 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner, S.B. 1997. Prion diseases and the BSE crisis. Science 278 245– 251. [DOI] [PubMed] [Google Scholar]
- Richards, F.M. and Lim, W. 1993. An analysis of packing in the protein folding problem. Q. Rev. Biophys. 26 423–498. [DOI] [PubMed] [Google Scholar]
- Rochet, J.C. and Lansbury Jr., P.T. 2000. Amyloid fibrillogenesis: Themes and variations. Curr. Opin. Struct. Biol. 10 60–68. [DOI] [PubMed] [Google Scholar]
- Selkoe, D.J. 1996. Amyloid β-protein and the genetics of Alzheimer’s disease. J. Biol. Chem. 271 18295–18298. [DOI] [PubMed] [Google Scholar]
- Si, K., Giustetto, M., Etkin, A., Hsu, R., Janisiewicz, A.M., Miniaci, M.C., Kim, J.H., Zhu, H., and Kandel, E.R. 2003a. A neuronal isoform of CPEB regulates local protein synthesis and stabilizes synapse-specific long-term facilitation in aplysia. Cell 115 893–904. [DOI] [PubMed] [Google Scholar]
- Si, K., Linquist, S., and Kandel, E.R. 2003b. A neuronal isoform of the aplysia CPEB has prion-like properties. Cell 115 879–891. [DOI] [PubMed] [Google Scholar]
- Stefani, M. and Dobson, C.M. 2003. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 81 678–699. [DOI] [PubMed] [Google Scholar]
- Tartaglia, G.G., Cavalli, A., Pellarin, R., and Caflisch, A. 2004. The role of aromaticity, exposed surface, and dipole moment in determining protein aggregation rates. Protein Sci. 13 1939–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2005. Prediction of aggregation rate and aggregation-prone segments in polypeptide sequences. Protein Sci. (this issue). [DOI] [PMC free article] [PubMed]
- Theis, M., Si, K., and Kandel, E.R. 2003. Two previously undescribed members of the mouse CPEB family of genes and their inducible expression in the principal cell layers of the hippocampus. Proc. Natl. Acad. Sci 100 9602–9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward, J.J., Sodhi, J.S., McGuffin, L.J., Buxton, B.F., and Jones, D.T. 2004. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 337 635–645. [DOI] [PubMed] [Google Scholar]
- Williams, R.M., Obradovic, Z., Mathura, V., Braun, W., Garner, E.C., Young, J., Takayama, S., Brown, C.J., and Dunker, A.K. 2001. The protein non-folding problem: Amino acid determinans of intrinsic order and disorder. Pac. Symp. Biocomput. 200 89–100. [DOI] [PubMed] [Google Scholar]