

Abstract
Amyloid-related diseases are often ascribed to protein “misfolding.” Yet in the absence of high-resolution structures for mature fibrils or intermediates, the connection between the mechanism of amyloid formation and protein folding remains tenuous. The simplistic view of amyloid fibrillogenesis as a homogeneous self-assembly process is being increasingly challenged by observations that amyloids interact with a variety of cofactors including metals, glycosaminoglycans, glycoproteins such as serum amyloid P and apolipo-protein E, and constituents of basement membranes such as perlecan, laminin, and agrin. These “pathological chaperones” have effects that range from mediating the rate of amyloid fibril formation to increasing the stability of amyloid deposits, and may contribute to amyloid toxicity. An increasing appreciation of the role of accessory molecules in amyloid etiology has paved the way to novel diagnostics and therapeutic strategies.
Keywords: Alzheimer Aβ peptide, protein folding, extracellular matrix proteins, heparan sulfate proteoglycans, basal lamina
According to the “Thermodynamic Hypothesis,” the functional native structure of a protein is determined by the full complement of interactions formed by the protein’s amino acid sequence in a given environment (Anfinsen 1973). Whereas in complementary duplex DNA one strand guides the structure of the other, all of the information for protein folding is contained within the protein sequence. In vivo, chaperones indirectly enhance the efficiency of protein folding by disentangling proteins trapped in aggregative side reactions but do not affect the structure or properties of the final native state (Dobson 2003). This has been demonstrated in among other experiments by the correct folding of proteins prepared by solid-phase methods in vitro (Anfinsen 1973; Dawson and Kent 2000). While the properties and structures of functional native proteins are encoded by their amino acid sequences (Anfinsen 1973), there is increasing evidence that the properties of amyloid deposits formed in vivo depend strongly on a variety of cofactors (Table 1). As such, the mechanism of amyloid formation may be more a consequence of improper heterologous interactions than improper protein folding.
Table 1.
Summary of principal amyloid cofactors and their effects
| Cofactor | Effecta | Reference |
| Zn2+ | Enhances Aβ aggregation | Bush et al. 1994 |
| Cu2+, Fe3+ | Increase Aβ toxicity via generation of free radicals | Bush 2003 |
| RNA | Converts PrPC prion to protease resistant form | Deleault et al. 2003 |
| Cholesterol | Metabolites may facilitate Aβ fibril nucleation | Zhang et al. 2004 |
| GAG | Promotes fibrillogenesis, stabilizes fibrils | Ancsin 2003 |
| Insulin | Inhibits IAPP fibril growth | Larson and Miranker 2004 |
| ApoE | Affects brain Aβ concentration, promotes fibrils | Bales et al. 2002 |
| ApoJ | Increases Aβ toxicity | DeMattos et al. 2002b |
| SAP | Stabilizes amyloid | Pepys 2001 |
| Agrin | Promotes Aβ fibrillogenesis, stabilizes fibrils | Cotman et al. 2000 |
| Perlecan | Promotes fibrillogenesis, stabilizes fibrils | Castillo et al. 1999 |
| Laminin | Inhibits Aβ fibrillogenesis | Castillo et al. 2000 |
a Where the specificities of cofactors for different types of amyloids are not given, the effects are thought to extend to all types of amyloid. For example, SAP appears to stabilize all known types of amyloid. In cases where specificities are given, the cofactors may play roles in additional processes. For example, Cu2+ may be involved in prion pathology and Parkinson’s disease in addition to its role in Alzheimer’s.
A large variety of proteins and peptides can self-assemble into aggregates that share tinctorial and morphological hallmarks of the amyloid fibrils found in situ (Rochet and Lansbury 2000). These include bacterial proteins (Alexandrescu and Rathgeb-Szabo 1999), proteins with an α-helical native structure such as myoglobin (Fandrich et al. 2001), and designed peptides as small as six amino acids (Lopez De La Paz et al. 2002). Inasmuch as polypeptides with unrelated sequences and functions can be driven to form fibrils in vitro (albeit under conditions removed from those for which the native state has been selected), it has been hypothesized that amyloid formation is a general property of proteins (Dobson 1999). Yet the amyloids associated with human disease are formed by only ~20 different polypeptide precursors (Coker et al. 2000). While much evidence suggests a correlation between protein stability and propensity for amyloid formation (Chiti et al. 2000), proteins implicated in amyloid pathology such as lysozyme, transthyretin, and β-microglobulin, do not seem remarkably unstable in light of estimates that as much of 40% to 60% of eukaryotic proteomes could be intrinsically disordered (Dunker and Obradovic 2001). A propensity to form partially folded conformations is also unlikely to be a hallmark of amyloidogenic proteins (Booth et al. 1997) given the proportion of large, multidomain proteins in the human proteome. Moreover, any protection afforded by intracellular chaperones against fibril deposition must precede the typical extracellular deposition of amyloids (Hirschfield and Hawkins 2003). With the exception of systemic amyloidosis where deposits occur in all tissues except the brain, amyloid deposits are focal; that is, amassed in specific locations (Sipe 1992; Bush 2002; Hirschfield and Hawkins 2003). Alzheimer’s β-amyloid (Aβ) is deposited in synapses and the basement membranes of brain blood vessels, in spite of the fact that Aβ is expressed in a variety of tissues (Cotman et al. 2000; Bush 2002). Deposits of hemodialysis-related β2-microglobulin amyloids are preferentially distributed in joints and connective tissues, and more rarely in visceral organs (Ohashi et al. 2002). Amyloids derived from islet amyloid polypeptide (IAPP) are deposited in the pancreatic islets of Langerhans. Deposition rarely occurs in persons without type II diabetes, although IAPP is expressed in normal individuals at levels manyfold higher than those required to form amyloid fibrils in vitro (Jaikaran et al. 2003; Potter-Perigo et al. 2003; Larson and Miranker 2004). Taken together, these observations challenge the view that amyloids result from protein misfolding, and suggest a prominent role for extrinsic cofactors in the etiology of amyloid-linked pathologies.
Although amyloid deposits are often described as inert, there is evidence that amyloid formation can be reversible, at least in some cases. Radioactively labeled serum amyloid P component (SAP) a cofactor present in all amyloid deposits formed in vivo, has been used to demonstrate dynamic turnover of fibrils and regression of deposits after the supply of amyloid precursor protein is removed (Pepys 2001; Pepys et al. 2002). In an elegant study, competition experiments involving 125I-labeled fibril precursors were used to demonstrate the reversibility of Aβ peptide deposition on plaques from Alzheimer’s disease (AD) brain tissue (Maggio et al. 1992). Reversible binding was attributed to a steady-state equilibrium between the Aβ peptide in amyloid deposits, and free in solution (Maggio et al. 1992). Fluorescence energy transfer between Aβ peptide analogs carrying either a tryptophan or AEDANS probe was used to demonstrate the reversibility of Aβ fibril formation, and to obtain kinetic rate constants for exchange between Aβ monomers and aggregates (Huang et al. 1997). Similarly, when model fibrils formed by the Escherichia coli protein CspA were incubated in D2O, all sites in the protein gave uniform hydrogen exchange rates; an observation most consistent with exchange occurring through dissociation of CspA from the fibrils (Alexandrescu 2001). Cofactors in amyloid diseases may shift the equilibrium between soluble and insoluble forms of the amyloidogenic proteins. Thus, depletion of metal ions by chelators facilitates aqueous dissolution of Aβ deposits isolated from postmortem AD brains (Cherny et al. 1999). Other types of cofactors appear to nucleate amyloids or have roles in amyloid assembly (Castillo et al. 1999; Cohlberg et al. 2002; Ancsin 2003; Deleault et al. 2003).
Accessory molecules in amyloid formation
Metals
The transition metals zinc, copper, and iron have been implicated in various aspects of Alzheimer’s disease (Bush and Tanzi 2002; 2003). Physiological concentrations of Zn2+ (μM) are sufficient to convert soluble Aβ peptide into protease-resistant amyloid-like aggregates (Bush et al. 1994; Bush 2003). Zinc-induced aggregation involves residue His13 of the Aβ peptide (Liu et al. 1999), which participates in His13 (Nɛ1)– Zn2+– His 13 (Nɛ1) cross-links between Aβ monomers (Miura et al. 2000). In contrast to the human peptide, rat Aβ, which has a His13 substituted by arginine, is not precipitated by physiological zinc concentrations (Bush et al. 1994). Extracellular zinc concentrations reach as high as ~0.3 mM during synaptic transmission but are subsequently reduced by an energy-dependent zinc-uptake transporter to levels below 0.5 μM (Bush and Tanzi 2002; Lee et al. 2002; 2003). Transport into synaptic vesicles requires the zinc transporter protein ZnT3. Mice bearing cerebral amyloid plaque pathology but lacking the ZnT3 gene show a marked reduction in Aβ deposits, and an increase in soluble Aβ (Lee et al. 2002).
While zinc is the only metal that can precipitate Aβ at physiological concentrations, copper and iron have more direct roles in Aβ toxicity (Bush 2003). Copper is found in synaptic vesicles at concentrations an order of magnitude lower than zinc, but Cu2+ binds Aβ (1–42) with 10−18 M affinity (Bush and Tanzi 2002). Because the affinity is so high, it is likely that the physiological form of Aβ is Cu2+-bound (Bush 2003). The Aβ peptide catalyzes hydrogen peroxide (H2O2) formation through the reduction of Cu2+ and Fe3+, using O2 and biological reducing agents as substrates (Bush 2003). Hydroxyl radicals are also produced in the reaction, and attack the susceptible Aβ peptide (Huang et al. 1999) leading to the formation of SDS-resistant Aβ oligomers with enhanced neurotoxicity (Walsh et al. 2002; Bush 2003). These oligomers may further seed zinc-induced amyloid deposition (Bush 2003). Copper and iron levels rise in senescence, and both metals are found in high concentrations in AD plaques (Bush 2003). The overexpression of amyloid precursor protein (APP), and increases in Aβ peptide levels, may themselves be responses to an increased concentrations of metal ions (Rogers et al. 2002). Although the precise biological function of APP remains unknown, the presence of Zn2+ and Cu2+ binding sites in the N-terminal and Aβ domains of the protein suggest a role in metal homeostasis (Maynard et al. 2002; Barnham et al. 2003).
In addition to AD, metal ions have been implicated in a number of other amyloid pathologies. Binding of Cu2+ to β-microglobulin is a contributing factor in hemodialysis-related amyloidosis (Morgan et al. 2001). Fibril formation by α-synuclein is accelerated by several metal ions (Al3+, Cu2+, Fe3+) implicated as risk factors for Parkinson’s disease (Uversky et al. 2001). The prion protein (PrP) binds copper and zinc and like APP may have a role in metal homeostasis (Watt and Hooper 2003). Removal of metal binding sites simultaneously affects the neurotoxicity and the ability of a 106–126 peptide fragment of human prion protein to form fibrils (Jobling et al. 2001). Binding of Cu2+ and Zn2+ have been suggested to enable interconversion between different subtypes of the infective PrPSc prion (Wadsworth et al. 1999).
Side-products of cholesterol metabolism
Derivatives of cholesterol containing aldehyde groups are generated as a result of antibody-catalyzed ozone production during the inflammation processes associated with atherosclerosis (Zhang et al. 2004). It was recently shown that some of the ozonolysis products of cholesterol markedly enhance the assembly of Aβpeptides into fibrils (Zhang et al. 2004). The aldehyde groups from the cholesterol metabolites can condense with amine groups on Aβ peptides. The resulting covalently modified Aβ peptides are more hydrophobic, and appear to form exceptionally potent nuclei for fibril formation, thereby accelerating fibrillogenesis (Zhang et al. 2004).
IAPP and insulin
Fibrils of islet amyloid polypeptide (IAPP) form at peptide concentrations 100-fold lower than those found in the secretory granules of the pancreatic islet β-cells, where IAPP is made. This raises the question of how fibril formation is prevented in normal individuals. Recent reports indicate that insulin, stored together with IAPP in secretory cells, inhibits IAPP amyloid formation (Jaikaran et al. 2003; Larson and Miranker 2004). Insulin binds to the surface of IAPP fibrils without causing their disassembly (Jaikaran et al. 2003; Larson and Miranker 2004). Binding of insulin appears to inhibit IAPP fibril elongation and may prevent aggregation of fibers into higher order arrays (Larson and Miranker 2004).
Prion conversion by RNA
The central event in prion pathology is the conversion of normal PrPC to a pathological protease-resistant form of the protein PrPSc. This conformational switch occurs ineffectively in vitro, requiring a large 50-fold excess of the pro-tease-resistant form of the prion protein (Deleault et al. 2003). Recent results show that the conversion to protease resistant prion reaches stoichiometric efficiency in the presence of RNA (Deleault et al. 2003). Conversion is stimulated by mammalian but not by invertebrate RNA, suggesting that RNA is a host-specific cofactor (Deleault et al. 2003). Conversion to the protease resistant form of the prion protein is also stimulated by heparan sulfate, and is inhibited by thiol blockers (Supattapone 2004).
Glycosaminoglycans
Together with collagens, glycosaminoglycans (GAGs) and proteoglycans are the principal components of the extracellular matrix—the connective meshwork that contributes to the stability, development, communication, and migration of cells (Bosman and Stamenkovic 2003). The extracellular matrix includes specialized structures such as basement membranes to which cells adhere. Amyloid deposits are often localized to these structures, and often contain tightly bound components of basement membranes (Sipe 1992; Fukuchi et al. 1998; Ancsin 2003; Hirschfield and Hawkins 2003).
The GAGs are a group of negatively charged heterogeneous polysaccharides built up from repeating disaccharide units (Fig. 1 ▶). Proteoglycans are formed when GAG chains are covalently attached to the serine residues of various core proteins. Compared to glycoproteins, proteoglycans are much larger and have a higher proportion of polysaccharide (up to 95% by weight). Proteoglycans often have multiple GAG chains attached; these are unbranched, and have from 10 to more than 100 carbohydrate residues. Glycosaminoglycans and proteoglycans are found associated with all types of amyloid deposits (Sipe 1992; Fukuchi et al. 1998; Ancsin 2003; Hirschfield and Hawkins 2003), including the neurofibrillary tangles formed by the microtubule-associated protein tau, which are specific to Alzheimer’s disease (Fukuchi et al. 1998; Cohlberg et al. 2002). Of the seven different types of GAGs (Fig. 1 ▶), heparan sulfate and dermatan sulfate are the most prominent in amyloid deposits (Hirschfield and Hawkins 2003). Interestingly, GAGs also associate with deposits of α-synuclein in vitro (Cohlberg et al. 2002). The protein α-synuclein is the primary component of the Lewy bodies observed in the substantia nigra of patients with Parkinson’s disease (Cohlberg et al. 2002). In contrast to extracellular amyloids, α-synuclein deposits are intracellular.
Figure 1.
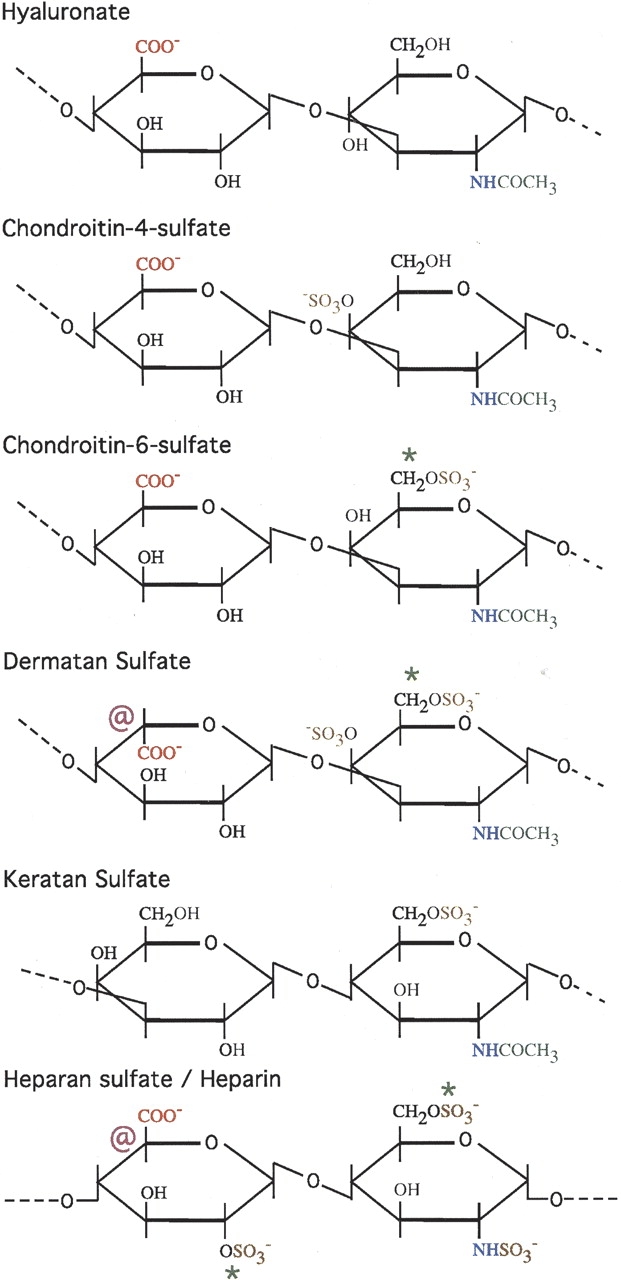
Repeating disaccharide units found in different types of glycosaminoglycans. The disaccharide units consist of a hexuronic acid such as gluconorate or iduronate, and a hexosamine such as N-acetylglucosamine (GlcNAc) or N-acetylgalactosamine (GalNAc) (Garret and Grisham 1999; Ancsin 2003). The symbol “@” indicates that the carboxyl group on the 5-carbon can exist in multiple epimerization states. The symbol “*” indicates that the substituent can be either a hydrogen (—H) or a sulfate group (—SO3−). Negatively charged groups are shown in red (—COO−) and brown (—SO3−). After synthesis, GAGs are extensively processed in the Golgi by enzymes that catalyze their sulfonation and epimerization. Hyaluronate is the only GAG that is not sulfated, and the only GAG that does not occur in proteoglycans. Heparin and heparan sulfate are shown together. Heparan sulfate is less extensively modified than heparin, has fewer sulfate groups, is less negatively charged, and is more difficult to obtain than heparin. Heparin occurs mainly in the cytoplasmic granules of mast cells, while heparan sulfate is found on cell surfaces and in the extracellular matrix.
Proteoglycans and GAGs have been implicated in the nucleation of fibrils. They can also stabilize mature fibrils against dissociation (Yamaguchi et al. 2003) and proteolytic degradation (Gupta-Bansal et al. 1995). Heparan sulfate and HSPGs have been shown to bind to a number of amyloid precursor proteins including β2-microglobulin (Ohashi et al. 2002) and Alzheimer’s APP. Recent progress has been made in identifying HSPG binding sites in Alzheimer’s amyloid precursor protein (APP) by X-ray crystallography (Rossjohn et al. 1999; Wang and Ha 2004). Heparan sulfate and HSPGs also bind to the fibrillar forms of amyloid-forming proteins, typically with much stronger affinity than to the monomeric forms. They have been shown to facilitate the formation of fibrils of IAPP (Castillo et al. 1998; Potter-Perigo et al. 2003), Aβ (Castillo et al. 1999), SAA (Ancsin and Kisilevsky 1999), tau protein (Goedert et al. 1996), α-synuclein (Cohlberg et al. 2002), and prion protein (Supattapone 2004). It has been proposed that GAGs may have a scaffolding role, promoting fibrilization-prone conformations of the amyloid precursor proteins (Cohlberg et al. 2002; Ancsin 2003). Although the morphologies and tinctorial properties properties of amyloids from a variety of sources appear uniform (Dobson 1999, 2002), EM studies suggest differences between fibrils formed in vitro and in situ. In EM images of deposits isolated from patients with amyloid disease, proteoglycans, and SAP appear to form the cores of the structures, whereas the specific “misfolded” protein characteristic of the amyloid type is found on the surface of the fibrils (Inoue et al. 1997; Inoue and Kisilevsky 1999).
GAG chains bind amyloid fibrils noncovalently, and with high affinity. Complexes of GAGs with Aβ (1–40) fibrils, for example, are sufficiently stable to protect the glycan chains from digestion with the enzyme heparanase (Bame et al. 1997). This raises the mechanistic question of how the polar and flexible GAG polyanions come to associate with the presumably hydrophobic and rigid interior of amyloid fibrils. Heparin binds to Aβ, IAPP, and α-synuclein, with a much higher affinity for the fibril than for the soluble states. These observations indicate that recognition depends on the quaternary structure of the aggregates (Watson et al. 1997; Cohlberg et al. 2002). Binding of GAGs to Aβ and to α-synuclein fibrils is strongly correlated with sulfate content, and hence, with the number of negative charges on the glycans (Castillo et al. 1999; Cohlberg et al. 2002). Chemically desulfated heparin fails to enhance Aβ fibril formation (Castillo et al. 1999). Oddly, it has been shown that α-synuclein fibril formation can be stimulated both by negatively charged GAGs and by positively charged poly-lysine (Cohlberg et al. 2002).
Heparin and heparan sulfate bind to a number of native proteins through basic amino acid clusters. Some of these proteins contain consensus sequence motifs of the type XBBXBX and XBBBXXBBX, where B are the basic residues Arg, Lys, His (Cardin and Weintraub 1989; Ancsin and Kisilevsky 1999; Cohlberg et al. 2002). Other heparin-binding proteins lack consensus sequence motifs, and recognize epitopes defined by the arrangements of basic residues in the protein’s three-dimensional structure. The lack of sequence conservation between different types of amyloid precursor proteins suggests that GAG-binding epitopes are determined by the organization of basic amino acids in the quaternary structures of amyloid fibrils. In the case of proteoglycans, both the GAG chains and the core protein can potentially contribute to association with amyloid. Experiments with Alzheimer’s Aβ peptides in vitro suggested that heparan sulfate alone can facilitate the conversion of Aβ peptides into fibrils (Fraser et al. 1992). By contrast, both heparan sulfate and the core protein of perlecan were needed to form amyloid deposits in vivo (Snow et al. 1994).
Accessory proteins and proteoglycans
SAP
Perhaps the best understood amyloid cofactor is serum amyloid P component (SAP). This glycoprotein is present in all amyloid deposits formed in situ, where it stabilizes fibrils and lowers their susceptibility to proteolytic degradation (Hind et al. 1984; Emsley et al. 1994; Pepys et al. 1994; Hamazaki 1995; Tennent et al. 1995; Pepys 2001). When found in amyloid, the protein is sometimes called “AP” for amyloid P component (Pepys et al. 1994), although it has been shown that at least the protein core of AP is identical to SAP. The normal function of SAP is not clearly established, but one potential function of the glycoprotein is to bind plasma chromatin (Pepys 2001) released from necrotic and apoptotic cells (Paul and Carroll 1999). SAP knockout studies in a mouse model suggest that the glycoprotein offers protection against lupus—an autoimmune disease whose manifestations include the formation of antibodies against nuclear components such as DNA (Paul and Carroll 1999). SAP has additional procoagulant activity (Zahedi 1997), binds to components of the immune system (Gewurz et al. 1995), has chaperone activity in vitro (Coker et al. 2000), and is a component of a number of basement membranes (Zahedi 1997).
The three-dimensional structure of SAP belongs to the pentraxin family, which also includes C-reactive protein. The SAP monomers adopt an LNS-like β-jellyroll fold (Fig. 2 ▶). Native SAP glycoprotein in serum has a quaternary structure arranged in a homo-pentameric ring (Emsley et al. 1994). By contrast, isolated SAP in the presence of Ca2+ undergoes an aggregation and precipitation reaction. Aggregation of SAP is believed to be promoted by an (AB)n face-to-face stacking of pentamers, where the A face corresponds to the side of the molecule that includes the longest α-helix (α-helix face), and the B-face (binding face) to the side of the monomer that includes the two Ca2+ ions (Fig. 2 ▶). The aggregation reaction is blocked by ligands that bind to SAP through its two Ca2+ ions, and by high salt. In the presence of small molecule ligands that bind to the double calcium site and inhibit aggregation, SAP gives decameric crystal structures resulting from pentameric rings with B-to-B (dAMP) or A-to-B (MOβDG) arrangements (Thompson et al. 2002). Although a high-resolution structure of the apo protein is not yet available, SAP in the absence of Ca2+ is believed to have a decameric quaternary structure with an A-to-A arrangement of pentamers(Thompson et al. 2002).
Figure 2.

Examples of LNS/pentraxin motifs from SAP (Emsley et al. 1994), agrin (Stetefeld et al. 2004), and laminin (Hohenester and Engel 2002). The fold is a β-sandwich comprised of a convex (blue) and a concave (red) β-sheet. The LNS/pentraxin fold is distantly related to those of the C-type lectins. LNS-fold proteins have a broad range of binding partners ranging from proteins to carbohydrates to steroids (Rudenko et al. 2001). Despite similar folding topologies, metal and substrate binding sites are poorly conserved (Rudenko et al. 2001). For example, the double Ca2+ binding site in the SAP monomer structure occurs in an entirely different location than the Ca2+ binding sites in agrin and laminin. Note that the LNS motif of SAP is part of a homopentameric ring structure.
SAP exists as a glycosylated protein in vivo. Its biantennary glycostructure is unusual in that its sequence is highly conserved (Pepys et al. 1994). SAP shows Ca2+-dependent binding to glycosaminoglycans such as heparan sulfate and dermatan sulfate (Hamazaki 1987), and to laminin (Zahedi 1997). Binding of SAP to amyloids also requires Ca2+ (Hamazaki 1987, 1995; Pepys 2001).
Agrin
The best understood function of the heparan sulfate proteoglycan agrin is its orchestration of acetylcholine receptor clustering on postsynaptic membranes (Sanes and Lichtman 2001; Bezakova and Ruegg 2003). The formation of these structures is a hallmark of neuromuscular junction development. Agrin is a mosaic protein comprised of ~22 domains, including nine protease inhibitor domains (Verbeek et al. 1999; Stetefeld et al. 2004). Splicing of agrin mRNA results in variants that include a globular N-terminal domain, which binds to laminin and anchors agrin to basement membranes. Alternatively the N-terminal domain can be swapped for a transmembrane segment that results in a lipid-membrane bound form of agrin (Bezakova and Ruegg 2003). Splice sites in agrin’s globular LNS2 domain modulate binding to α-dystroglycan, an interaction thought to serve as a structural bridge between the basal lamina and muscle (Sanes and Lichtman 2001; Bezakova and Ruegg 2003). Additional mRNA splice sites generate neuron-specific isoforms of agrin, with inserts of eight to 18 residues in the last globular domain, LNS3. These insert containing forms are necessary for the acetylcholine receptor clustering activity of agrin (Bezakova and Ruegg 2003; Stetefeld et al. 2004). Besides agrin’s functions in neuromuscular junction development, the proteoglycan is involved in synapse remodeling (Sanes and Lichtman 2001), in the formation of central nervous system synapses, and in the organization of “immunological synapses” (Khan et al. 2001; Bezakova and Ruegg 2003). Agrin is normally found in several areas of the brain, including neurons and in the basement membranes of brain microvasculature. Recently, an insoluble form of agrin was found to be highly concentrated in both diffuse and neuritic plaques of brains from patients with Alzheimer’s disease (AD) (Donahue et al. 1999; Verbeek et al. 1999; van Horssen et al. 2001). Moreover, agrin was also associated with the neurofibrillary tangles characteristic of the disease (Donahue et al. 1999). The brains of AD patients showed basement membrane abnormalities, which may be related to the sequestration of agrin by amyloid (Donahue et al. 1999). Agrin in a B0 isoform is expressed in a variety of tissues other than nerve cells (Bezakova and Ruegg 2003), and it remains to be seen if agrin-B0 associates with other types of amyloid fibrils besides those involved in AD.
In vitro studies (Cotman et al. 2000) indicate that agrin binds to fibrillar, but not to soluble Aβ. Binding of agrin accelerates Aβ-fibril formation, stabilizes the fibrils against proteolytic degradation (Cotman et al. 2000), and may indirectly be involved in Aβ toxicity through the abrogation of agrin’s normal functions in synapse remodeling and microvasculature function (Donahue et al. 1999). Pretreatment of agrin with nitrous acid (conditions, and acid concentration were not given) almost completely diminished binding to fibrillar Aβ (Cotman et al. 2000). Retention of tenascin and merosin binding, suggested that only the GAG chains and not the protein core are affected by treatment of agrin with nitrous acid; and consequently, that agrin binds to Aβ fibrils through its GAG chains (Cotman et al. 2000). It is important to note, however, that agrin has ~22 protein domains that could also potentially be affected by the nitrous acid treatment. These domains include globular LNS domains (Fig. 2 ▶) that do not interact with tenascin or merosin, but that bind to GAGs in a Ca2+-dependent manner.
Perlecan
Perlecan is a basement membrane HSPG with roles in the adhesion, development, differentiation, and proliferation of cells (Hartmann and Maurer 2001; Jiang and Couchman 2003). In addition, perlecan interacts with a number of growth factors and participates in forming a semipermeable barrier in kidney glomerular basement membranes. Perlecan coimmunolocalizes with all types of amyloids (Snow and Wight 1989). Perlecan accelerates fibril formation (Castillo et al. 1998, 1999), stabilizes fibrils once formed (Castillo et al. 1997), and protects fibrils from proteolytic degradation (Gupta-Bansal et al. 1995).
Laminin
The large multidomain glycoprotein laminin is an integral component of basement membranes. In the presence of Ca2+, laminins polymerize into gel-like structures. These structures serve as a scaffold and recruit other components needed for basement membrane assembly during development. Studies using a mouse model suggest that laminin is the only factor absolutely required to form basement membranes during early stages of embryo development (Sasaki et al. 2004; Yurchenco et al. 2004). Additional functions of laminins include maintenance of basement membrane structure, and acting as anchors for proteins that associate with the basement membrane. Laminins are involved in a range of specific biological processes such as chemotaxis, mitosis, induction of tumor metastasis, and development of the peripheral nervous system (Engel 1992). The molecules are heterotrimers composed of α, β, and γ chains connected through a three-stranded heterotrimeric coiled coil. There are five α, three β, and three γ chains that can form at least 15 different types of laminins (Sasaki et al. 2004). Electron microscopy of laminin (EHS) reveals a cross-like shape, with the longest α chain forming the central stalk (Engel 1992). At the end of this stalk, is a string of five globular LNS-type domains (Fig. 2 ▶; Engel 1992).
In contrast to the molecules discussed above, laminin (EHS) has been shown to inhibit amyloid formation (Castillo et al. 2000). Laminin-1 can also dissociate fibrils into protofibrils and further into amorphous aggregates, and has been reported to protect cultured neurons from Aβ-amyloid neurotoxicity (Morgan et al. 2002). The inhibitory effects appear to be specific to Aβ-amyloid, and were not observed with IAPP fibrils (Castillo et al. 2000). Digestion of laminin (EHS) with the protease elastase was used to map the binding site for Aβ fibrils to the last three globular domains of the α-chain (Castillo et al. 2000). Short peptides derived from laminin-α chain sequences upstream of the LNS domain also inhibit fibril formation, and may constitute a second binding domain for Aβ fibrils (Castillo et al. 2000; Morgan et al. 2002).
Apolipoproteins E and J
Apolipoprotein E (apoE) is a 34 kDa protein involved in lipid transport and metabolism. Together with plasma lipids, apoE assembles into lipoprotein complexes that are recognized by low-density lipoprotein (LDL) receptors (Strittmatter et al. 1993; Bales et al. 2002). Interestingly, the first step in the uptake of low-density lipoproteins by hepatocytes involves binding of ApoE to HSPGs (Fukuchi et al. 1998). In the central nervous system apoE is involved in the redistribution of lipids and cholesterol during membrane repair and synapse remodeling, and in the transport of lipids through the cerebrospinal fluid (Bales et al. 2002). Isoforms of apoE are genetic markers for late-onset Alzheimer’s disease with alleles ɛ4 and ɛ2 increasing and decreasing the risk for the disease, respectively (Bales et al. 2002). The isoforms differ in only two amino acids (ɛ2–Cys112, Cys 158; ɛ 4–Arg112, Arg 158), which affect the binding preferences for high-density lipoproteins (ɛ2) versus low-density lipoproteins (ɛ4). Carriers of the ɛ4 allele have an additional predisposition for hypercholesterolemia (Bales et al. 2002).
ApoE (ɛ3 or ɛ4 isoform) tightly binds to soluble Aβ peptide forming complexes that resist dissociation after boiling for 5 min in 2% SDS (Strittmatter et al. 1993). ApoE also binds to Aβ in its fibril form; hence, antibodies to apoE colocalize with nearly all neuritic plaques in Alzheimer’s disease brains (Bales et al. 2002). The importance of apoE in Alzheimer pathology is highlighted by the observation that transgenic mice carrying the gene for human APP, but lacking the gene for ApoE form only diffuse plaques but not the neuritic plaques and cerebral blood vessels amyloid deposits characteristic of the disease (Bales et al. 2002; Holtzman 2003). In addition to its role in Aβ fibrillogenesis, apoE has a role in Aβ peptide clearance from the brain (Bales et al. 2002; Holtzman 2003). The second major brain apolipoprotein, apoJ (also called clusterin), binds soluble Aβ with a 2 nM Kd, and increases amyloid toxicity. Owing to its chaperone-like properties, apoJ solubilizes a large variety of proteins with exposed hydrophobic patches. The toxicity of ApoJ may arise from its ability to shift the equilibrium between fibrillar and soluble forms of the Aβ peptide toward the soluble forms, thereby unmasking toxic epitopes (DeMattos et al. 2002b).
Shared themes among the major proteins that interact with amyloid
All of the proteins described above are extracellular. Functionally, they can be loosely classified as proteins involved in the formation of supramolecular assemblies. ApoE forms lipoprotein complexes involved in lipid transport; the other four proteins participate in various aspects of basement membrane structure. Laminin is the main component of basement membranes. Together with laminin, agrin and perlecan share roles in basement membrane structure. As previously described, SAP in addition to circulating in the blood (Hind et al. 1984) is found associated with a number of basement membranes (Zahedi 1997). The link between amyloid deposits and the extracellular matrix (Kisilevsky 2000; Ancsin 2003; Bush 2003) is intriguing, given the importance of the latter to the function, association, and development of cells.
SAP, agrin, laminin, and perlecan share LNS/pentraxin β-jellyroll folding motifs as parts of their three-dimensional structures (Fig. 2 ▶). The LNS motif of SAP forms a pentameric ring structure. In agrin, laminin, and perlecan, LNS domains occur at the C-terminal ends of the multidomain proteins. LNS motifs occur in a number of functional contexts. The calcium- and substrate-binding sites of LNS domains are poorly conserved, making it difficult to draw functional inferences based on the presence of this folding motif (Rudenko et al. 2001). Nevertheless, SAP, agrin, laminin, and perlecan share at least one common functional aspect—binding to the GAGs heparin/heparan sulfate. Heparin binds to LNS domains 4 of laminin α1, domains 3 and 5 of laminin α2 (Talts et al. 1999), LNS4 of laminin α4 (Yamashita et al. 2004), LNS2 of agrin (Burgess et al. 2002), and the three LNS domains within perlecan domain V (Brown et al. 1997). Affinity for heparin indicates a potential to bind to matrix bound heparan sulfate proteoglycans (Talts et al. 1999). Heparan sulfate binding has not been studied as extensively because it is harder to obtain (Kisilevsky 2000), but has been localized to the LNS domain pairs LG4–5 of laminin α1 (Sasaki et al. 1998), and to the agrin LNS2 domain (Burgess et al. 2002). The N-terminal portions of laminin α3B and α5 chains, which do not contain LNS domains, have additional binding sites for heparan sulfate (Garbe et al. 2002). SAP has been shown to bind to heparin, heparan sulfate, and dermatan sulfate (Hamazaki 1987). ApoE, whose structure unlike the other four proteins above does not include LNS motifs, binds to heparan sulfate proteoglycans as part of its function in lipid transport (Fukuchi et al. 1998; Libeu et al. 2001). These molecules, through additional interactions with components of basement membranes (Yurchenco et al. 2004), could anchor fibrils to the extracellular matrix, and provide a nidus for further amyloid growth. SAP, which associates with basement membranes and has shown to bind laminin with high affinity (Zahedi 1997) could conceivably play a similar role. Agrin and perlecan, which are heparan sulfate proteoglycans as well as having the capacity to bind heparan sulfate, could potentially further aggravate amyloid growth by forming intermolecular cross-links.
Other proteins associated with amyloids
In addition to the cofactors described in detail above, other molecules have been reported to interact with amyloid, specifically with amyloid formed by the Alzheimer’s Aβ peptide. Predominant among these are basement membrane molecules including collagen IV and entactin (Kiuchi et al. 2002), the proteoglycans decorin, biglycan, versican (Snow et al. 1995), the heparan sulfate proteoglycans glypican-1, and syndecans 1 through 3 (van Horssen et al. 2002). It is interesting to note that Alzheimer’s APP itself is classified as a “part time” proteoglycan (Bosman and Stamenkovic 2003) since it is sometimes found in forms with GAG chains attached. APP also has binding sites for HSPGs (Rossjohn et al. 1999; Wang and Ha 2004). Additional proteins reported to interact with Aβ amyloid that do not share readily apparent relationships, include the serpin antichy-motrypsin (Lukacs and Christianson 1996), C1q A complement collagen-like domain (Wang et al. 2000), α 2 macro-globulin (Hughes et al. 1998), p75 neutrophin receptor (Yaar et al. 1997), and α 7 nicotinic acetylcholine receptor (Wang et al. 2000).
Cofactors in theraputic approaches
Cofactors can affect the assembly, stability, and toxicity of amyloid fibrils. Each of these facets of amyloid etiology is a potential therapeutic target, and knowledge about the roles of cofactors in amyloid diseases has been exploited to develop new strategies for treatment. Efforts include the development of improved diagnostics and therapies for disease.
Diagnostics
Until recently, diagnosis of amyloid pathology depended on the analysis of samples from tissue biopsies or autopsies (Pepys 2001; Bacskai et al. 2003). The development of non-invasive diagnostics would allow better monitoring of disease progression and evaluation of treatments. Current diagnostics include small molecule probes, derived from amyloid-binding dyes such as Congo Red and Thioflavin (Lee 2002; Bacskai et al. 2003). Cofactors involved in the progression of the disease by comparison, offer the advantage of higher selectivity. The group of Pepys, has pioneered scintigraphy of amyloid using radioactively labeled 123I-SAP tracers (Pepys 2001; Pepys et al. 2002). This approach takes advantage of the fact that SAP is associated with amyloid deposits. Following intravenous injection, 123I-SAP is catabolized rapidly and cleared in the urine if no amyloid is present (Pepys 2001). When amyloid is present, 123I- SAP from the blood exchanges through a dynamic equilibrium with the preexisting amyloid-bound SAP. The radioactive tracer provides quantitative information on the location and amount of amyloid (Pepys 2001). In turn, 123I-SAP scintigraphy can be used to monitor dynamic turnover of amyloid, and can assess the efficacy of treatments aimed at regressing amyloid deposits (Pepys et al. 2002). For some applications, it would clearly be desirable to have multiple diagnostic probes. Radioactive or spin-labeled analogs of agrin that cross the blood–brain barrier may have applications for detecting amyloid deposits in Alzheimer’s disease patients (Gurwitz 2000).
Interference with amyloid facillitators
Highly sulfated GAGs are ubiquitous in amyloid deposits, and participate in the assembly of fibrils. Compounds that compete with the binding of GAGs or proteoglycans to the amyloid precursor proteins have shown effectiveness in blocking fibril assembly (Kiselevsky et al. 2003). These compounds include chain terminators that once incorporated into growing GAG chains block further elongation, and carbohydrate precursors that cannot be properly sulfated thus abrogating the requirement for highly negatively charged polyanions for efficient amyloid binding (Kiselevsky et al. 2003). In another approach, sulfated polyanion mimetics of heparan sulfate such as poly(vinylsulfonate) or derivatives of dextran sulfate were used to interfere with SAA fibril assembly (Inoue et al. 1996) and to block prion protein propagation (Adjou et al. 2003). Additional strategies for interfering with amyloid assembly include mimetics of epitopes on proteins such as laminin that promote fibril disassembly (Castillo et al. 2000), and inhibition of the expression of molecules such as apoE which promote fibril formation (Bales et al. 2002).
Targeting the amyloid enforcer SAP
A different strategy targets molecules that stabilize amyloids to degradation in vivo. Early studies showed that a trace component of agarose, methyl 4,6-O-(1-carboxyethylidene)- β-D-galactopyranoside (MOβDG) inhibited the Ca2+-dependent binding reactions of SAP, including the binding of SAP to amyloid (Hind et al. 1984). Presumably this compound mimics the Ca2+-dependent affinity of SAP for glycosaminoglycans such as heparan sulfate and dermatan sulfate (Hamazaki 1987). At millimolar concentrations, MOβDG displaces SAP from amyloid and returns it to the blood where it can be transported to the liver for catabolic clearance (Hind et al. 1984; Pepys 2001). Amyloids regress as the fibril-stabilizing and protease-resistance-conferring SAP molecules are removed (Pepys 2001). A high-throughput screen of a compound library was used to identify CPHPC, a new competitive inhibitor of SAP that interferes with amyloid binding (Pepys et al. 2002). Compared to the 50-μM affinity constant for MOβDG, the apparent affinity constant for the CPHPC compound is 10 nM. This value is comparable to the 10-nM dissociation constant estimated for binding of SAP to isolated amyloid fibrils. X-ray structures of the SAP complexes with MOβDG (Thompson et al. 2002) and CPHPC (Pepys et al. 2002) show that the compounds bind near the double-Ca2+ ligation site of the protein, and thus probably work by interfering with the Ca2+-dependent binding of SAP to amyloid (Thompson et al. 2002). Both compounds stabilize decameric forms of SAP, which are rapidly cleared from circulation by the liver (Pepys et al. 2002).
Shifting the partition of Aβ across the blood–brain barrier
Antibodies raised against Aβ peptides offer a potential therapeutic approach to Alzheimer’s disease by reducing the load of Aβ in the central nervous system (Schenk et al. 1999). Aβ synthesized in the brain can be catabolized locally, or transported into the plasma and cleared peripherally (Bales et al. 2002; DeMattos et al. 2002a). Using a mouse model it has been shown that a peripherally administered antibody to Aβ can act as a “peripheral sink” that by binding Aβ in the blood markedly increases the efflux of Aβ from the brain to the plasma (DeMattos et al. 2002a). The “peripheral sink” approach could find more general utility with other molecules that bind to plasma Aβ (Matsuoka et al. 2003) and lack some of the adverse effects associated with Aβ-directed antibodies.
Alleviation of metal-induced toxicity
The mechanism by which amyloid deposits exert their pathologic effects remains unresolved, but evidence suggests a role for metals in at least some aspects of amyloid toxicity (Bush and Tanzi 2002; Bush 2003). Nonspecific high-affinity chelators that reduce the overall concentration of metals in the body are unlikely to be successful in treating amyloidoses, because toxicity occurs in the specific context of amyloid-bound metals (Bush 2002, 2003). Moreover, toxicity is localized to the tissues neighboring amyloid deposits, such as the brain in AD. Success has been reported in a mouse model for AD with clioquinil, a retired antibiotic that serves as an orally deliverable Cu–Zn chelator. The hydrophobic character of Clioquinil enables it to cross the blood–brain barrier, in contrast to more common hydrophilic metal chelators. The compound reduces Aβ deposits by ~50% in mice, and is thought to work by simultaneously facilitating the dissolution of Zn2+-bound amyloid and inhibiting copper and iron-mediated H2O2 production. Inhibition of H2O2 further depletes Aβ by an indirect clearance mechanism (Bush 2003). Once resolubilized, Aβ peptides can be removed in the blood or taken up by cells for degradation (Bush 2003). Because clioquinil has a relatively weak nanomolar affinity for zinc and copper, it does not induce systemic metal depletion as would be the case with stronger chelators (Bush 2002).
Open questions regarding the biophysics of amyloids
This review has summarized the roles of accessory molecules in the biology of amyloid diseases. Much remains to be understood about the biophysics of amyloid–cofactor interactions: How large of an effect do cofactors have on the structures and stabilities of amyloids? To what extent do the core protein and carbohydrate components of glycoproteins and proteoglycans participate in amyloid recognition? What are the structural determinants of cofactor binding to amyloids? Addressing these questions should lead to a better appreciation of the role of cofactors in the development of amyloid disease and will undoubtedly contribute to a better understanding of the mechanisms of amyloid toxicity.
Acknowledgments
This work was supported by the NSF (MCB 0236316).
Abbreviations
Aβ, Alzheimerβ-peptide
AD, Alzheimer’s disease
apoE, apolipoprotein E
APP, (Alzheimer) amyloid precursor protein
IAPP, islet amyloid polypeptide
GAG, glycosaminoglycan
HSPG, heparan sulfate proteoglycan
LNS, laminin neurexin and SHBG-like domain
PrPC, soluble form of prion protein
PrPSc, protease-resistant form of prion protein
SAA, serum amyloid A
SAP, serum amyloid P component
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04887005.
References
- Adjou, K.T., Simoneau, S., Sales, N., Lamoury, F., Dormont, D., Papy-Garcia, D., Barritault, D., Deslys, J.P., and Lasmezas, C.I. 2003. A novel generation of heparan sulfate mimetics for the treatment of prion diseases. J. Gen. Virol. 84 2595–2603. [DOI] [PubMed] [Google Scholar]
- Alexandrescu, A.T. 2001. An NMR-based quenched hydrogen exchange investigation of model amyloid fibrils formed by cold shock protein A. Pac. Symp. Biocomput. 67–78. [DOI] [PubMed]
- Alexandrescu, A.T. and Rathgeb-Szabo, K. 1999. An NMR investigation of solution aggregation reactions preceding the misassembly of acid-denatured cold shock protein A into fibrils. J. Mol. Biol. 291 1191–1206. [DOI] [PubMed] [Google Scholar]
- Ancsin, J.B. 2003. Amyloidogenesis: Historical and modern observations point to heparan sulfate proteoglycans as a major culprit. Amyloid 10 67–79. [DOI] [PubMed] [Google Scholar]
- Ancsin, J.B. and Kisilevsky, R. 1999. The heparin/heparan sulfate-binding site on apo-serum amyloid A. Implications for the therapeutic intervention of amyloidosis. J. Biol. Chem. 274 7172–7181. [DOI] [PubMed] [Google Scholar]
- Anfinsen, C.B. 1973. Principles that govern the folding of protein chains. Science 181 223–230. [DOI] [PubMed] [Google Scholar]
- Bacskai, B.J., Hickey, G.A., Skoch, J., Kajdasz, S.T., Wang, Y., Huang, G.F., Mathis, C.A., Klunk, W.E., and Hyman, B.T. 2003. Four-dimensional multiphoton imaging of brain entry, amyloid binding, and clearance of an amyloid-β ligand in transgenic mice. Proc. Natl. Acad. Sci. 100 12462–12467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bales, K.R., Dodart, J.C., DeMattos, R.B., Holtzman, D.M., and Paul, S.M. 2002. Apolipoprotein E, amyloid, and Alzheimer disease. Mol. Intervent. 2 363–375. [DOI] [PubMed] [Google Scholar]
- Bame, K.J., Danda, J., Hassall, A., and Tumova, S. 1997. Aβ(1–40) prevents heparanase-catalyzed degradation of heparan sulfate glycosaminoglycans and proteoglycans in vitro. A role for heparan sulfate proteoglycan turnover in Alzheimer’s disease. J. Biol. Chem. 272 17005–17011. [DOI] [PubMed] [Google Scholar]
- Barnham, K.J., McKinstry, W.J., Multhaup, G., Galatis, D., Morton, C.J., Curtain, C.C., Williamson, N.A., White, A.R., Hinds, M.G., Norton, R.S., et al. 2003. Structure of the Alzheimer’s disease amyloid precursor protein copper binding domain. A regulator of neuronal copper homeostasis. J. Biol. Chem. 278 17401–17407. [DOI] [PubMed] [Google Scholar]
- Bezakova, G. and Ruegg, M.A. 2003. New insights into the roles of agrin. Nat. Rev. Mol. Cell Biol. 4 295–308. [DOI] [PubMed] [Google Scholar]
- Booth, D.R., Sunde, M., Bellotti, V., Robinson, C.V., Hutchinson, W.L., Fraser, P.E., Hawkins, P.N., Dobson, C.M., Radford, S.E., Blake, C.C., et al. 1997. Instability, unfolding and aggregation of human lysozyme variants underlying amyloid fibrillogenesis. Nature 385 787–793. [DOI] [PubMed] [Google Scholar]
- Bosman, F.T. and Stamenkovic, I. 2003. Functional structure and composition of the extracellular matrix. J. Pathol. 200 423–428. [DOI] [PubMed] [Google Scholar]
- Brown, J.C., Sasaki, T., Gohring, W., Yamada, Y., and Timpl, R. 1997. The C-terminal domain V of perlecan promotes β1 integrin-mediated cell adhesion, binds heparin, nidogen and fibulin-2 and can be modified by glycosaminoglycans. Eur. J. Biochem. 250 39–46. [DOI] [PubMed] [Google Scholar]
- Burgess, R.W., Dickman, D.K., Nunez, L., Glass, D.J., and Sanes, J.R. 2002. Mapping sites responsible for interactions of agrin with neurons. J. Neurochem. 83 271–284. [DOI] [PubMed] [Google Scholar]
- Bush, A.I. 2002. Metal complexing agents as therapies for Alzheimer’s disease. Neurobiol. Aging 23 1031–1038. [DOI] [PubMed] [Google Scholar]
- ———. 2003. The metallobiology of Alzheimer’s disease. Trends Neurosci. 26 207–214. [DOI] [PubMed] [Google Scholar]
- Bush, A.I. and Tanzi, R.E. 2002. The galvanization of β-amyloid in Alzheimer’s disease. Proc. Natl. Acad. Sci. 99 7317–7319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush, A.I., Pettingell, W.H., Multhaup, G., d Paradis, M., Vonsattel, J.P., Gusella, J.F., Beyreuther, K., Masters, C.L., and Tanzi, R.E. 1994. Rapid induction of Alzheimer A β amyloid formation by zinc. Science 265 1464–1467. [DOI] [PubMed] [Google Scholar]
- Cardin, A.D. and Weintraub, H.J. 1989. Molecular modeling of protein–glycosaminoglycan interactions. Arteriosclerosis 9 21–32. [DOI] [PubMed] [Google Scholar]
- Castillo, G.M., Ngo, C., Cummings, J., Wight, T.N., and Snow, A.D. 1997. Perlecan binds to the β-amyloid proteins (A β) of Alzheimer’s disease, accelerates A β fibril formation, and maintains A β fibril stability. J. Neurochem. 69 2452–2465. [DOI] [PubMed] [Google Scholar]
- Castillo, G.M., Cummings, J.A., Yang, W., Judge, M.E., Sheardown, M.J., Rimvall, K., Hansen, J.B., and Snow, A.D. 1998. Sulfate content and specific glycosaminoglycan backbone of perlecan are critical for perlecan’s enhancement of islet amyloid polypeptide (amylin) fibril formation. Diabetes 47 612–620. [DOI] [PubMed] [Google Scholar]
- Castillo, G.M., Lukito, W., Wight, T.N., and Snow, A.D. 1999. The sulfate moieties of glycosaminoglycans are critical for the enhancement of β-amyloid protein fibril formation. J. Neurochem. 72 1681–1687. [DOI] [PubMed] [Google Scholar]
- Castillo, G.M., Lukito, W., Peskind, E., Raskind, M., Kirschner, D.A., Yee, A.G., and Snow, A.D. 2000. Laminin inhibition of β-amyloid protein (Aβ) fibrillogenesis and identification of an Aβ binding site localized to the globular domain repeats on the laminin a chain. J. Neurosci. Res. 62 451–462. [DOI] [PubMed] [Google Scholar]
- Cherny, R.A., Legg, J.T., McLean, C.A., Fairlie, D.P., Huang, X., Atwood, C.S., Beyreuther, K., Tanzi, R.E., Masters, C.L., and Bush, A.I. 1999. Aqueous dissolution of Alzheimer’s disease Aβ amyloid deposits by biometal depletion. J. Biol. Chem. 274 23223–23228. [DOI] [PubMed] [Google Scholar]
- Chiti, F., Taddei, N., Bucciantini, M., White, P., Ramponi, G., and Dobson, C.M. 2000. Mutational analysis of the propensity for amyloid formation by a globular protein. EMBO J. 19 1441–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohlberg, J.A., Li, J., Uversky, V.N., and Fink, A.L. 2002. Heparin and other glycosaminoglycans stimulate the formation of amyloid fibrils from α-synuclein in vitro. Biochemistry 41 1502–1511. [DOI] [PubMed] [Google Scholar]
- Coker, A.R., Purvis, A., Baker, D., Pepys, M.B., and Wood, S.P. 2000. Molecular chaperone properties of serum amyloid P component. FEBS Lett. 473 199–202. [DOI] [PubMed] [Google Scholar]
- Cotman, S.L., Halfter, W., and Cole, G.J. 2000. Agrin binds to β-amyloid (Aβ), accelerates aβ fibril formation, and is localized to Aβ deposits in Alzheimer’s disease brain. Mol. Cell. Neurosci. 15 183–198. [DOI] [PubMed] [Google Scholar]
- Dawson, P.E. and Kent, S.B. 2000. Synthesis of native proteins by chemical ligation. Annu. Rev. Biochem. 69 923–960. [DOI] [PubMed] [Google Scholar]
- Deleault, N.R., Lucassen, R.W., and Supattapone, S. 2003. RNA molecules stimulate prion protein conversion. Nature 425 717–720. [DOI] [PubMed] [Google Scholar]
- DeMattos, R.B., Bales, K.R., Cummins, D.J., Paul, S.M., and Holtzman, D.M. 2002a. Brain to plasma amyloid-β efflux: A measure of brain amyloid burden in a mouse model of Alzheimer’s disease. Science 295 2264–2267. [DOI] [PubMed] [Google Scholar]
- DeMattos, R.B., O’Dell, M.A., Parsadanian, M., Taylor, J.W., Harmony, J.A., Bales, K.R., Paul, S.M., Aronow, B.J., and Holtzman, D.M. 2002b. Clusterin promotes amyloid plaque formation and is critical for neuritic toxicity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. 99 10843–10848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson, C.M. 1999. Protein misfolding, evolution and disease. Trends Biochem. Sci. 24 329–332. [DOI] [PubMed] [Google Scholar]
- ———. 2002. Protein misfolding and human disease. Sci. World J. 2 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2003. Protein folding and misfolding. Nature 426 884–890. [DOI] [PubMed] [Google Scholar]
- Donahue, J.E., Berzin, T.M., Rafii, M.S., Glass, D.J., Yancopoulos, G.D., Fallon, J.R., and Stopa, E.G. 1999. Agrin in Alzheimer’s disease: Altered solubility and abnormal distribution within microvasculature and brain parenchyma. Proc. Natl. Acad. Sci. 96 6468–6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunker, A.K. and Obradovic, Z. 2001. The protein trinity—Linking function and disorder. Nat. Biotechnol. 19 805–806. [DOI] [PubMed] [Google Scholar]
- Emsley, J., White, H.E., O’Hara, B.P., Oliva, G., Srinivasan, N., Tickle, I.J., Blundell, T.L., Pepys, M.B., and Wood, S.P. 1994. Structure of pentameric human serum amyloid P component. Nature 367 338–345. [DOI] [PubMed] [Google Scholar]
- Engel, J. 1992. Laminins and other strange proteins. Biochemistry 31 10643–10651. [DOI] [PubMed] [Google Scholar]
- Fandrich, M., Fletcher, M.A., and Dobson, C.M. 2001. Amyloid fibrils from muscle myoglobin. Nature 410 165–166. [DOI] [PubMed] [Google Scholar]
- Fraser, P.E., Nguyen, J.T., Chin, D.T., and Kirschner, D.A. 1992. Effects of sulfate ions on Alzheimer β/A4 peptide assemblies: Implications for amyloid fibril–proteoglycan interactions. J. Neurochem. 59 1531–1540. [DOI] [PubMed] [Google Scholar]
- Fukuchi, K., Hart, M., and Li, L. 1998. Alzheimer’s disease and heparan sulfate proteoglycan. Front. Biosci. 3 d327–d337. [DOI] [PubMed] [Google Scholar]
- Garbe, J.H., Gohring, W., Mann, K., Timpl, R., and Sasaki, T. 2002. Complete sequence, recombinant analysis and binding to laminins and sulphated ligands of the N-terminal domains of laminin α3B and α5 chains. Biochem. J. 362 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garret, R.H. and Grisham, C.M. 1999. Biochemistry, 2nd ed. Saunders, New York.
- Gewurz, H., Zhang, X.H., and Lint, T.F. 1995. Structure and function of the pentraxins. Curr. Opin. Immunol. 7 54–64. [DOI] [PubMed] [Google Scholar]
- Goedert, M., Jakes, R., Spillantini, M.G., Hasegawa, M., Smith, M.J., and Crowther, R.A. 1996. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 383 550–553. [DOI] [PubMed] [Google Scholar]
- Gupta-Bansal, R., Frederickson, R.C., and Brunden, K.R. 1995. Proteoglycan-mediated inhibition of A β proteolysis. A potential cause of senile plaque accumulation. J. Biol. Chem. 270 18666–18671. [DOI] [PubMed] [Google Scholar]
- Gurwitz, D. 2000. New imaging techniques for early diagnosis of Alzheimer’s disease. Mol. Med. Today 6 340. [DOI] [PubMed] [Google Scholar]
- Hamazaki, H. 1987. Ca2+-mediated association of human serum amyloid P component with heparan sulfate and dermatan sulfate. J. Biol. Chem. 262 1456–1460. [PubMed] [Google Scholar]
- ———. 1995. Ca(2+)-dependent binding of human serum amyloid P component to Alzheimer’s β-amyloid peptide. J. Biol. Chem. 270 10392–10394. [DOI] [PubMed] [Google Scholar]
- Hartmann, U. and Maurer, P. 2001. Proteoglycans in the nervous system—The quest for functional roles in vivo. Matrix Biol. 20 23–35. [DOI] [PubMed] [Google Scholar]
- Hind, C.R., Collins, P.M., Caspi, D., Baltz, M.L., and Pepys, M.B. 1984. Specific chemical dissociation of fibrillar and non-fibrillar components of amyloid deposits. Lancet 2 376–378. [DOI] [PubMed] [Google Scholar]
- Hirschfield, G.M. and Hawkins, P.N. 2003. Amyloidosis: New strategies for treatment. Int. J. Biochem. Cell Biol. 35 1608–1613. [DOI] [PubMed] [Google Scholar]
- Hohenester, E. and Engel, J. 2002. Domain structure and organisation in extra-cellular matrix proteins. Matrix Biol. 21 115–128. [DOI] [PubMed] [Google Scholar]
- Holtzman, D.M. 2003. Potential role of endogenous and exogenous amyloid-β binding molecules in the pathogenesis, diagnosis, and treatment of Alzheimer disease. Alzheimer Dis. Assoc. Disord. 17 151–153. [DOI] [PubMed] [Google Scholar]
- Huang, T.H.J., Fraser, P.E., and Chakrabartty, A. 1997. Fibrillogenesis of Alzheimer Aβ peptides studied by fluorescence energy transfer. J. Mol. Biol. 269 214–224. [DOI] [PubMed] [Google Scholar]
- Huang, X., Atwood, C.S., Hartshorn, M.A., Multhaup, G., Goldstein, L.E., Scarpa, R.C., Cuajungco, M.P., Gray, D.N., Lim, J., Moir, R.D., et al. 1999. The A β peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry 38 7609–7616. [DOI] [PubMed] [Google Scholar]
- Hughes, S.R., Khorkova, O., Goyal, S., Knaeblein, J., Heroux, J., Riedel, N.G., and Sahasrabudhe, S. 1998. α2-macroglobulin associates with β-amyloid peptide and prevents fibril formation. Proc. Natl. Acad. Sci. 95 3275–3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue, S. and Kisilevsky, R. 1999. In situ electron microscopy of amyloid deposits in tissues. Methods Enzymol. 309 496–509. [DOI] [PubMed] [Google Scholar]
- Inoue, S., Hultin, P.G., Szarek, W.A., and Kisilevsky, R. 1996. Effect of poly-(vinylsulfonate) on murine AA amyloid: A high-resolution ultrastructural study. Lab. Invest. 74 1081–1090. [PubMed] [Google Scholar]
- Inoue, S., Kuroiwa, M., Ohashi, K., Hara, M., and Kisilevsky, R. 1997. Ultra-structural organization of hemodialysis-associated β 2-microglobulin amyloid fibrils. Kidney Int. 52 1543–1549. [DOI] [PubMed] [Google Scholar]
- Jaikaran, E.T., Nilsson, M.R., and Clark, A. 2004. Pancreatic β-cell granule peptides form heteromolecular complexes which inhibit islet amyloid polypeptide fibril formation. Biochem. J. 377 709–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, X. and Couchman, J.R. 2003. Perlecan and tumor angiogenesis. J. Histochem. Cytochem. 51 1393–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobling, M.F., Huang, X., Stewart, L.R., Barnham, K.J., Curtain, C., Volitakis, I., Perugini, M., White, A.R., Cherny, R.A., Masters, C.L., et al. 2001. Copper and zinc binding modulates the aggregation and neurotoxic properties of the prion peptide PrP106–126. Biochemistry 40 8073–8084. [DOI] [PubMed] [Google Scholar]
- Khan, A.A., Bose, C., Yam, L.S., Soloski, M.J., and Rupp, F. 2001. Physiological regulation of the immunological synapse by agrin. Science 292 1681–1686. [DOI] [PubMed] [Google Scholar]
- Kisilevsky, R. 2000. Review: Amyloidogenesis—Unquestioned answers and unanswered questions. J. Struct. Biol. 130 99–108. [DOI] [PubMed] [Google Scholar]
- Kiselevsky, R., Szarek, W.A., Ancsin, J., Bhat, S., Li, Z., and Marone, S. 2003. Novel glycosaminoglycan precursors as anti-amyloid agents, Part III. J. Mol. Neurosci. 20 291–297. [DOI] [PubMed] [Google Scholar]
- Kiuchi, Y., Isobe, Y., Fukushima, K., and Kimura, M. 2002. Disassembly of amyloid β-protein fibril by basement membrane components. Life Sci. 70 2421–2431. [DOI] [PubMed] [Google Scholar]
- Larson, J.L. and Miranker, A.D. 2004. The mechanism of insulin action on islet amyloid polypeptide fiber formation. J. Mol. Biol. 335 221–231. [DOI] [PubMed] [Google Scholar]
- Lee, V.M.-Y. 2002. Amyloid binding ligands as Alzheimer’s disease therapies. Neurobiol. Aging 23 1039–1042. [DOI] [PubMed] [Google Scholar]
- Lee, J.Y., Cole, T.B., Palmiter, R.D., Suh, S.W., and Koh, J.Y. 2002. Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc. Natl. Acad. Sci. 99 7705–7710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libeu, C.P., Lund-Katz, S., Phillips, M.C., Wehrli, S., Hernaiz, M.J., Capila, I., Linhardt, R.J., Raffai, R.L., Newhouse, Y.M., Zhou, F., et al. 2001. New insights into the heparan sulfate proteoglycan-binding activity of apolipo-protein E. J. Biol. Chem. 276 39138–39144. [DOI] [PubMed] [Google Scholar]
- Liu, S.T., Howlett, G., and Barrow, C.J. 1999. Histidine-13 is a crucial residue in the zinc ion-induced aggregation of the A β peptide of Alzheimer’s disease. Biochemistry 38 9373–9378. [DOI] [PubMed] [Google Scholar]
- Lopez De La Paz, M., Goldie, K., Zurdo, J., Lacroix, E., Dobson, C.M., Hoenger, A., and Serrano, L. 2002. De novo designed peptide-based amyloid fibrils. Proc. Natl. Acad. Sci. 99 16052–16057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs, C.M. and Christianson, D.W. 1996. Is the binding of β-amyloid protein to antichymotrypsin in Alzheimer plaques mediated by a β-strand insertion? Proteins 25 420–424. [DOI] [PubMed] [Google Scholar]
- Maggio, J.E., Stimson, E.R., Ghilardi, J.R., Allen, C.J., Dahl, C.E., Whitcomb, D.C., Vigna, S.R., Vinters, H.V., Labenski, M.E., and Mantyh, P.W. 1992. Reversible in vitro growth of Alzheimer disease β-amyloid plaques by deposition of labeled amyloid peptide. Proc. Natl. Acad. Sci. 89 5462–5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka, Y., Saito, M., LaFrancois, J., Saito, M., Gaynor, K., Olm, V., Wang, L., Casey, E., Lu, Y., Shiratori, C., et al. 2003. Novel therapeutic approach for the treatment of Alzheimer’s disease by peripheral administration of agents with an affinity to β-amyloid. J. Neurosci. 23 29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard, C.J., Cappai, R., Volitakis, I., Cherny, R.A., White, A.R., Beyreuther, K., Masters, C.L., Bush, A.I., and Li, Q.X. 2002. Overexpression of Alzheimer’s disease amyloid-β opposes the age-dependent elevations of brain copper and iron. J. Biol. Chem. 277 44670–44676. [DOI] [PubMed] [Google Scholar]
- Miura, T., Suzuki, K., Kohata, N., and Takeuchi, H. 2000. Metal binding modes of Alzheimer’s amyloid β-peptide in insoluble aggregates and soluble complexes. Biochemistry 39 7024–7031. [DOI] [PubMed] [Google Scholar]
- Morgan, C.J., Gelfand, M., Atreya, C., and Miranker, A.D. 2001. Kidney dialysis-associated amyloidosis: A molecular role for copper in fiber formation. J. Mol. Biol. 309 339–345. [DOI] [PubMed] [Google Scholar]
- Morgan, C., Bugueno, M.P., Garrido, J., and Inestrosa, N.C. 2002. Laminin affects polymerization, depolymerization and neurotoxicity of Aβ peptide. Peptides 23 1229–1240. [DOI] [PubMed] [Google Scholar]
- Ohashi, K., Kisilevsky, R., and Yanagishita, M. 2002. Affinity binding of glycosaminoglycans with β (2)-microglobulin. Nephron 90 158–168. [DOI] [PubMed] [Google Scholar]
- Paul, E. and Carroll, M.C. 1999. SAP-less chromatin triggers systemic lupus erythematosus. Nat. Med. 5 607–608. [DOI] [PubMed] [Google Scholar]
- Pepys, M.B. 2001. Pathogenesis, diagnosis and treatment of systemic amyloidosis. Philos. Trans. R. Soc. Lond. B 356 203–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepys, M.B., Rademacher, T.W., Amatayakul-Chantler, S., Williams, P., Noble, G.E., Hutchinson, W.L., Hawkins, P.N., Nelson, S.R., Gallimore, J.R., Herbert, J., et al. 1994. Human serum amyloid P component is an invariant constituent of amyloid deposits and has a uniquely homogeneous glycostructure. Proc. Natl. Acad. Sci. 91 5602–5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepys, M.B., Herbert, J., Hutchinson, W.L., Tennent, G.A., Lachmann, H.J., Gallimore, J.R., Lovat, L.B., Bartfai, T., Alanine, A., Hertel, C., et al. 2002. Targeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosis. Nature 417 254–259. [DOI] [PubMed] [Google Scholar]
- Potter-Perigo, S., Hull, R.L., Tsoi, C., Braun, K.R., Andrikopoulos, S., Teague, J., Bruce Verchere, C., Kahn, S.E., and Wight, T.N. 2003. Proteoglycans synthesized and secreted by pancreatic islet β-cells bind amylin. Arch. Biochem. Biophys. 413 182–190. [DOI] [PubMed] [Google Scholar]
- Rochet, J.C. and Lansbury Jr., P.T. 2000. Amyloid fibrillogenesis: Themes and variations. Curr. Opin. Struct. Biol. 10 60–68. [DOI] [PubMed] [Google Scholar]
- Rogers, J.T., Randall, J.D., Cahill, C.M., Eder, P.S., Huang, X., Gunshin, H., Leiter, L., McPhee, J., Sarang, S.S., Utsuki, T., et al. 2002. An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J. Biol. Chem. 277 45518–45528. [DOI] [PubMed] [Google Scholar]
- Rossjohn, J., Cappai, R., Feil, S.C., Henry, A., McKinstry, W.J., Galatis, D., Hesse, L., Multhaup, G., Beyreuther, K., Masters, C.L., et al. 1999. Crystal structure of the N-terminal growth factor-like domain of Alzheimer amyloid precursor protein. Nat. Struct. Biol. 6 327–331. [DOI] [PubMed] [Google Scholar]
- Rudenko, G., Hohenester, E., and Muller, Y.A. 2001. LG/LNS domains: Multiple functions—One business end? Trends Biochem. Sci. 26 363–368. [DOI] [PubMed] [Google Scholar]
- Sanes, J.R. and Lichtman, J.W. 2001. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat. Rev. Neurosci. 2 791–805. [DOI] [PubMed] [Google Scholar]
- Sasaki, T., Costell, M., Mann, K., and Timpl, R. 1998. Inhibition of glycosaminoglycan modification of perlecan domain I by site-directed mutagenesis changes protease sensitivity and laminin-1 binding activity. FEBS Lett. 435 169–172. [DOI] [PubMed] [Google Scholar]
- Sasaki, T., Fassler, R., and Hohenester, E. 2004. Laminin: The crux of basement membrane assembly. J. Cell Biol. 164 959–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk, D., Barbour, R., Dunn, W., Gordon, G., Grajeda, H., Guido, T., Hu, K., Huang, J., Johnson-Wood, K., Khan, K., et al. 1999. Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400 173–177. [DOI] [PubMed] [Google Scholar]
- Sipe, J.D. 1992. Amyloidosis. Annu. Rev. Biochem. 61 947–975. [DOI] [PubMed] [Google Scholar]
- Snow, A.D. and Wight, T.N. 1989. Proteoglycans in the pathogenesis of Alzheimer’s disease and other amyloidoses. Neurobiol. Aging 10 481–497. [DOI] [PubMed] [Google Scholar]
- Snow, A.D., Sekiguchi, R., Nochlin, D., Fraser, P., Kimata, K., Mizutani, A., Arai, M., Schreier, W.A., and Morgan, D.G. 1994. An important role of heparan sulfate proteoglycan (Perlecan) in a model system for the deposition and persistence of fibrillar A β-amyloid in rat brain. Neuron 12 219–234. [DOI] [PubMed] [Google Scholar]
- Snow, A.D., Kinsella, M.G., Parks, E., Sekiguchi, R.T., Miller, J.D., Kimata, K., and Wight, T.N. 1995. Differential binding of vascular cell-derived proteoglycans (perlecan, biglycan, decorin, and versican) to the β-amyloid protein of Alzheimer’s disease. Arch. Biochem. Biophys. 320 84–95. [DOI] [PubMed] [Google Scholar]
- Stetefeld, J., Alexandrescu, A.T., Maciejewski, M.W., Jenny, M., Rathgeb-Szabo, K., Schulthess, T., Landwehr, R., Frank, S., Ruegg, M.A., and Kammerer, R.A. 2004. Modulation of agrin function by alternative splicing and Ca2+ binding. Structure (Camb.) 12 503–515. [DOI] [PubMed] [Google Scholar]
- Strittmatter, W.J., Weisgraber, K.H., Huang, D.Y., Dong, L.-M., Salvesen, G.S., Pericak-Vance, M., Schmechel, D., Saunders, A.M., Goldgaber, D., and Roses, A.D. 1993. Binding of human apolipoprotein E to synthetic amyloid β peptide: Isoform-specific effects and implications for late-onset Alzheimer disease. Proc. Natl. Acad. Sci. 90 8098–8102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supattapone, S. 2004. Prion protein conversion in vitro. J. Mol. Med. 82 348–356. [DOI] [PubMed] [Google Scholar]
- Talts, J.F., Andac, Z., Gohring, W., Brancaccio, A., and Timpl, R. 1999. Binding of the G domains of laminin α1 and α2 chains and perlecan to heparin, sulfatides, α-dystroglycan and several extracellular matrix proteins. EMBO J. 18 863–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennent, G.A., Lovat, L.B., and Pepys, M.B. 1995. Serum amyloid P component prevents proteolysis of the amyloid fibrils of Alzheimer disease and systemic amyloidosis. Proc. Natl. Acad. Sci. 92 4299–4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, D., Pepys, M.B., Tickle, I., and Wood, S. 2002. The structures of crystalline complexes of human serum amyloid P component with its carbohydrate ligand, the cyclic pyruvate acetal of galactose. J. Mol. Biol. 320 1081–1086. [DOI] [PubMed] [Google Scholar]
- Uversky, V.N., Li, J., and Fink, A.L. 2001. Metal-triggered structural transformations, aggregation, and fibrillation of human α-synuclein. A possible molecular link between Parkinson’s disease and heavy metal exposure. J. Biol. Chem. 276 44284–44296. [DOI] [PubMed] [Google Scholar]
- van Horssen, J., Otte-Holler, I., David, G., Maat-Schieman, M.L., van den Heuvel, L.P., Wesseling, P., de Waal, R.M., and Verbeek, M.M. 2001. Heparan sulfate proteoglycan expression in cerebrovascular amyloid β deposits in Alzheimer’s disease and hereditary cerebral hemorrhage with amyloidosis (Dutch) brains. Acta Neuropathol. (Berl.) 102 604–614. [DOI] [PubMed] [Google Scholar]
- van Horssen, J., Kleinnijenhuis, J., Maass, C.N., Rensink, A.A., Otte-Holler, I., David, G., van den Heuvel, L.P., Wesseling, P., de Waal, R.M., and Verbeek, M.M. 2002. Accumulation of heparan sulfate proteoglycans in cerebellar senile plaques. Neurobiol. Aging 23 537–545. [DOI] [PubMed] [Google Scholar]
- Verbeek, M.M., Otte-Holler, I., van den Born, J., van den Heuvel, L.P., David, G., Wesseling, P., and de Waal, R.M. 1999. Agrin is a major heparan sulfate proteoglycan accumulating in Alzheimer’s disease brain. Am. J. Pathol. 155 2115–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadsworth, J.D., Hill, A.F., Joiner, S., Jackson, G.S., Clarke, A.R., and Collinge, J. 1999. Strain-specific prion–protein conformation determined by metal ions. Nat. Cell Biol. 1 55–59. [DOI] [PubMed] [Google Scholar]
- Walsh, D.M., Klyubin, I., Fadeeva, J.V., Cullen, W.K., Anwyl, R., Wolfe, M.S., Rowan, M.J., and Selkoe, D.J. 2002. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416 535–539. [DOI] [PubMed] [Google Scholar]
- Wang, Y. and Ha, Y. 2004. The X-ray structure of an antiparallel dimer of the human amyloid precursor protein E2 domain. Mol. Cell 15 345–353. [DOI] [PubMed] [Google Scholar]
- Wang, H.Y., Lee, D.H., Davis, C.B., and Shank, R.P. 2000. Amyloid peptide Aβ (1–42) binds selectively and with picomolar affinity to α7 nicotinic acetylcholine receptors. J. Neurochem. 75 1155–1161. [DOI] [PubMed] [Google Scholar]
- Watson, D.J., Lander, A.D., and Selkoe, D.J. 1997. Heparin-binding properties of the amyloidogenic peptides Aβ and amylin. Dependence on aggregation state and inhibition by Congo red. J. Biol. Chem. 272 31617–31624. [DOI] [PubMed] [Google Scholar]
- Watt, N.T. and Hooper, N.M. 2003. The prion protein and neuronal zinc homeostasis. Trends Biochem. Sci. 28 406–410. [DOI] [PubMed] [Google Scholar]
- Yaar, M., Zhai, S., Pilch, P.F., Doyle, S.M., Eisenhauer, P.B., Fine, R.E., and Gilchrest, B.A. 1997. Binding of β-amyloid to the p75 neurotrophin receptor induces apoptosis. A possible mechanism for Alzheimer’s disease. J. Clin. Invest. 100 2333–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi, I., Suda, H., Tsuzuike, N., Seto, K., Seki, M., Yamaguchi, Y., Hasegawa, K., Takahashi, N., Yamamoto, S., Gejyo, F., et al. 2003. Glycosaminoglycan and proteoglycan inhibit the depolymerization of β2-microglobulin amyloid fibrils in vitro. Kidney Int. 64 1080–1088. [DOI] [PubMed] [Google Scholar]
- Yamashita, H., Beck, K., and Kitagawa, Y. 2004. Heparin binds to the laminin α 4 chain LG4 domain at a site different from that found for other laminins. J. Mol. Biol. 335 1145–1149. [DOI] [PubMed] [Google Scholar]
- Yurchenco, P.D., Amenta, P.S., and Patton, B.L. 2004. Basement membrane assembly, stability and activities observed through a developmental lens. Matrix Biol. 22 521–538. [DOI] [PubMed] [Google Scholar]
- Zahedi, K. 1997. Characterization of the binding of serum amyloid P to laminin. J. Biol. Chem. 272 2143–2148. [PubMed] [Google Scholar]
- Zhang, Q., Powers, E.T., Nieva, J., Huff, M.E., Dendle, M.A., Bieshke, J., Glabe, C.G., Eschenmoser, A., Wentworth Jr., P., Lerner, R.A., et al. 2004. Metabolite-initiated protein misfolding may trigger Alzheimer’s disease. Proc. Natl. Acad. Sci. 101 4752–4757. [DOI] [PMC free article] [PubMed] [Google Scholar]