

Abstract
Phosphopeptide-binding domains, including the FHA, SH2, WW, WD40, MH2, and Polo-box domains, as well as the 14-3-3 proteins, exert control functions in important processes such as cell growth, division, differentiation, and apoptosis. Structures and mechanisms of phosphopeptide binding are generally diverse, revealing few general principles. A computational method for analysis of phosphopeptide-binding domains was therefore developed to elucidate the physical and chemical nature of phosphopeptide binding, given this lack of structural similarity. The surfaces of nine phosphopeptide-binding proteins, representing seven distinct classes of phosphopeptide-binding modules, were discretized, and encoded with information about amino acid identity, surface curvature, and electrostatic potential at every point on the surface in order to identify local surface properties enriched in phosphoresidue contact sites. Cross-validation indicated that propensities corresponding to this enrichment calculated from a subset of the training data could be used to predict the phosphoresidue contact site on proteins not used in training with no false negative results, and with few unconfirmed positive predictions. The locations of phosphoresidue contact sites were then predicted on the surfaces of the checkpoint kinase Chk1 and the BRCA1 BRCT repeat domain, and these predictions are consistent with recent experimental evidence.
Keywords: phosphopeptide-binding domains, BRCA1, Chk1, functional site prediction
Many aspects of cellular biology, including cell cycle control, differentiation, and apoptosis, are regulated by the complex interplay of protein substrates with protein kinases, phosphatases, and phosphopeptide-binding domains (Zhou 2000; Yaffe and Elia 2001; Yaffe and Smerdon 2001; Yaffe 2002). Phosphopeptide-binding domains participate in signal transduction by recognizing and binding preferentially to the phosphorylated forms of specific proteins. In addition to binding directly to the phosphoserine, phosphothreonine, or phosphotyrosine residue, phosphopeptide-binding domains also recognize distinct linear sequence motifs surrounding the phospho-amino acid to achieve substrate specificity. To date, however, no comprehensive study has identified a unified set of physical-chemical, structural, or energetic requirements necessary and sufficient for phosphopeptide binding.
The structures of eight distinct classes of phosphopeptide-binding modules in complex with phosphorylated peptides or proteins have been solved (WW, PTB, SH2, MH2, FHA, WD40, Polo-box, 14-3-3). An examination of these structures reveals little structural similarity among the phosphopeptide-binding sites, apart from the evolutionary conservation seen among members of the same domain family (Yaffe and Smerdon 2001) (see Supplemental Material). We reasoned that, despite the lack of gross structural similarity, there should be some underlying chemical and physical characteristics that define the phosphopeptide-interacting surface. We therefore analyzed a representative collection of these domains in detail and evaluated a set of physical and chemical properties at discrete points along their molecular surfaces. These properties were used to calculate a propensity value for each property to occur within a phosphoresidue contact site.
We found that these propensity values were able to correctly identify the phosphoresidue contact site on phosphopeptide-binding domains for which the site was known, in a cross-validation procedure. We used these propensities to predict the location of phosphopeptide-binding sites on the surface of two domains for which there was no published phosphopeptide cocrystal structure; the BRCT-repeat domain of the protein BRCA1, and the kinase domain of the checkpoint protein Chk1. BRCA1 is a tumor-suppressing protein whose dysfunction predisposes women to breast and ovarian cancer. The BRCT-repeat domains of BRCA1 and several other proteins were recently shown to bind phosphopeptides as part of the DNA damage response (Manke et al. 2003; Yu et al. 2003). The checkpoint kinase Chk1 plays a critical role in the cell cycle response to DNA damage, and appears to be regulated by binding to phosphopeptides at a site distinct from that of its catalytic activity (Jeong et al. 2003). The resulting predictions are corroborated with experimental data identifying the sites of phosphopeptide interaction. We anticipate that this computational approach to identifying phosphopeptide-binding sites will find general utility in the functional annotation of the structural genome, in the characterization of the structure and function of new phosphopeptide-binding domains as they are discovered, and in the identification of sites to target with inhibitors of protein/phosphopeptide interaction.
Results
To investigate the unifying principles involved in phosphopeptide recognition, we examined nine X-ray crystal structures representing seven phosphoserine-, phosphothreonine-, and phosphotyrosine-binding domains (Table 1). We observed little, if any, identity in the amino acids or their three-dimensional arrangements within the phosphopeptide-binding sites (Yaffe and Smerdon 2001). Nevertheless, we felt that the physical and chemical requirements for phosphopeptide binding were in some manner encoded in these sites. We therefore built the phosphate-accessible molecular surfaces for each phosphopeptide-binding domain using a triangular mesh (Sanner et al. 1996), and a probe radius of 3 Å, corresponding to the approximate radius of a phosphate ion. Each vertex on the mesh was encoded with information corresponding to a set of characteristics including amino acid identity, local mean surface curvature, and solvated electrostatic potential (see Materials and Methods). For each characteristic, the likelihood of occurrence at the contact sites for phosphorylated side chains was calculated. This likelihood was normalized by comparison with the likelihood of finding that same characteristic over the total phosphate-accessible surface area of the nine proteins studied, to derive a propensity for that characteristic being found in a phosphoresidue contact site.
Table 1.
Structures used to calculate propensity data
| PDB ID | Protein | Domain type/Phosphorylated AA | Surface points | References |
| 1F8A | Pin1 | WW/2× pS | 35,582 | Verdecia et al. 2000 |
| 1G6G | Rad53 | FHA/pT | 24,372 | Durocher et al. 2000 |
| 1GXC | Chk2 | FHA/pT | 24,293 | Li et al. 2002 |
| 1KHX | Smad | MH2/2× pS | 40,832 | Wu et al. 2001 |
| 1LCJ | p56Lck | SH2/pY | 21,905 | Eck et al. 1993 |
| 1NEX | Cdc4 | WD40/pT | 76,798 | Orlicky et al. 2003 |
| 1QJB | 14-3-3ζ | 14-3-3/pS | 46,068 | Rittinger et al. 1999 |
| 1SPS | Src | SH2/pY | 21,954 | Waksman et al. 1993 |
| 1UMW | Pik1 | Polo-box/pT | 40,426 | Elia et al. 2003 |
Phosphoresidue contact site properties
Amino acid identity
For the set of phosphopeptide-binding domains studied, the distribution of amino acids at all surface points unsurprisingly shows large contributions from charged amino acids, with arginine, lysine, and glutamic acid having the highest percentages observed (Fig. 1A ▶, upper panel). In contrast, the highest percentages within the portion of the surface that contacts a phosphorylated side chain are contributed by arginine, lysine, serine, and tyrosine, while the acidic amino acids are almost never present (Fig. 1A ▶, middle panel). Propensities for each amino acid to contact the phosphorylated serine, threonine, or tyrosine side chains were calculated by normalizing the frequency of each amino acid at surface points in the phosphoresidue contact site by the frequency of that amino acid over the entire set of protein surfaces studied. This revealed the highest specific enrichment of tryptophan, histidine, tyrosine, and arginine, in that order, at phosphoresidue contact sites (Fig. 1A ▶, lower panel).
Figure 1.

Calculation of phosphoresidue contact propensities from global and phosphoresidue contact probability distributions. Probability distributions over the total protein surface (upper panels), over the phosphoresidue contact surface (middle panels), and the phosphoresidue contact propensity (lower panels) were calculated for the properties (A) amino acid identity, (B) mean surface curvature, and (C) solvated electrostatic potential. Error bars in lower panels indicate twice the standard deviation of the mean for removing each crystal structure from the data set, one at a time (N = 9). The horizontal lines in the bottom panels indicate the mean phosphoresidue contact propensity, which is always equal to 1.
While it might be expected that the positively charged amino acids lysine and arginine would be the most over-represented in sites that bind negatively charged phosphates, this appears not to be the case, since lysine and arginine are extremely common on the surface of proteins in general, while tryptophan is not. While it would be quite surprising to find a phosphopeptide-binding site without lysine or arginine in it, the mere presence of a lysine or arginine on the surface of a protein carries less predictive weight than the presence of a tryptophan. There are three tryptophan residues in phosphoresidue contact sites in our data set, one on each of the proteins Pin1, Cdc4, and Plk1. In addition to contacting the phosphoresidue, all three tryptophans contact proline residues to the C-terminal side of the phosphoresidue of the phosphopeptide. This indicates a strong possibility that the high incidence of phosphoresidue-contacting tryptophans in our data set may indicate the favorability of tryptophan/proline interaction in the context of the common phosphoresidue-proline motif. Interestingly, the contacts made between an arginine and a phosphorylated side chain typically involve a bidentate interaction with the guanadino group, while a tryptophan often stacks a large amount of its side-chain surface against a phosphoresidue. Based on this observation, we independently calculated propensities for points on the surface of the three guanadino nitrogen atoms of the arginine side chain, and for the points on the remainder of the arginine residue. This revealed that the points associated with the nitrogen atoms have a high contact propensity, second only to that of tryptophan, while points on the rest of the amino acid are unlikely to be contacted (data not shown). This indicates that calculating propensities based on chemical functional groups, rather than amino acid identity per se, may serve to improve this analysis in the future, particularly once more structures are available from which to derive propensities. Several amino acids, including cysteine, glutamine, and proline, were not observed to contact phosphorylated side chains, although this may be due to the relatively small size of the data set of known phosphopeptide-binding domain structures.
Surface curvature
A measure of the mean local curvature about each surface point was calculated (Meyer et al. 2003), and used to produce a propensity value related to surface curvature. There is a spike in the overall distribution of surface curvatures at approximately 0.3 Å−1, corresponding to the local concavity at any location where the 3 Å probe used to derive the molecular surface contacted three or more protein atoms (Fig. 1B ▶, upper panel). There is also a small shoulder in the distribution centered at a convex curvature of −0.5 Å−1, corresponding to regions where the probe touches only a single atom. The remainder of the distribution corresponds to saddle regions on the protein surface where the probe touches two atoms, and the surface has both concave and convex character.
Qualitatively, the distribution of surface points that bind to a phosphorylated side chain appears quite similar to the global distribution (Fig. 1B ▶, middle panel). Quantitatively, however, the propensity for phosphoresidue contact, obtained by dividing the phosphoresidue contact site frequency distribution by the overall frequency distribution, is enriched in two regions (Fig. 1B ▶, lower panel). One of these regions, with relatively high negative curvature values, is the ratio of sparsely populated regions of the contact site and global frequency distributions (Fig. 1B ▶, upper and middle panels), making the predictive validity of propensities in this region questionable. The second region of high propensity lies between curvature values of 0.1 and 0.6 Å−1 (Fig. 1B ▶, lower panel), and corresponds to regions of concavity in the protein surface that are highly populated in the global distribution. The data in this region quantifies the well accepted tendency of ligands to bind to concave regions of protein surface, in the specific context of phosphopeptide-binding domain:ligand interactions.
Electrostatic potential
To examine the effect of electrostatic potential on phosphopeptide binding, we used a continuum electrostatic model to calculate the solvated state potential of each phosphopeptide-binding domain in our data set in the absence of the cognate phosphopeptide ligand. The distribution of potentials on the phosphate-accessible surfaces of all proteins studied was bell shaped, and centered approximately at zero (Fig. 1C ▶, upper panel). As expected, the distribution of electrostatic potentials for the subset of the domain surfaces that contact a phosphorylated side chain is significantly shifted toward positive values (Fig. 1C ▶, middle panel). As a result, the propensity distribution over electrostatic potentials, calculated as the distribution of electrostatic potentials in phosphoresidue contact sites divided by the global distribution of electrostatic potentials, peaks in the range between +7 and +9 kT/e.
As might be expected, the propensity for binding to phosphorylated side chains trails off as the electrostatic potential at a surface point becomes more negative from this peak, falling to almost zero at neutral electrostatic potential. Interestingly, the propensity also falls off for surface points having the highest electrostatic potential. The implication, then, is that surface points with such high positive electrostatic potentials are not as well suited for binding phosphopeptides as points with more moderate potentials, despite the high negative charge of a phosphorylated amino acid side chain. This is likely due to the high energetic cost of desolvating a region of such extreme positive potential (Lee and Tidor 1997).
Predictive ability for known phosphoresidue contact sites
To determine whether the calculated propensities were unduly influenced by any single structure in the data set, a cross-validation procedure was used (“jack-knifing”) in which each structure was individually removed, and the propensities recalculated. The nine resulting sets of propensities were quite similar (shown by error bars in Fig. 1A ▶–C, lower panel), with individual propensity values in well populated regions of the distributions differing on average from those calculated for the full data set by less than 10% in the case of surface curvatures and electrostatic potentials, and by less than 25% for amino acid identities.
Of the three independent propensities calculated for amino acid identity, surface curvature, and electrostatic potential, none was sufficient on its own to unambiguously identify the site of known phosphoresidue contact on the set of phosphopeptide-binding domains studied here (Fig. 2 ▶, left panels). However, the scales of propensities encountered in this analysis provide a framework for understanding the contribution of each characteristic studied to phosphoresidue binding. The scales of propensity values encountered indicate the most favorable values of electrostatic potential are more predictive, with respect to phosphoresidue contact, than the most favorable values of amino acid identity or surface curvature. Nevertheless, unfavorable propensity values contributed by amino acid identity or surface curvature are capable of countering false-positive favorable contributions from positive electrostatic potential in order to improve the accuracy of our predictions, as shown in Figure 2 ▶.
Figure 2.

Calculation of joint propensity for phosphoresidue contact. Propensities were calculated independently for amino acid identity (upper left), local mean surface curvature (middle left), and solvated electrostatic potential (lower left). These propensities were combined multiplicatively to obtain a joint propensity for phosphoresidue contact (right). Two linear scales were used to depict unfavorable and favorable propensity. Unfavorable propensity values from 0 to 1 are colored from red to white. Favorable propensity values are colored from white to blue over the values 1 to 4 for amino acid identity, 1 to 2.5 for surface curvature, 1 to 12 for solvated electrostatic potential, and 1 to 20 for joint phosphoresidue contact propensity. In some regions, as with the area outlined in yellow, the three individually calculated propensities combine constructively to create a large region of favorable joint propensity. In other regions, such as the area outlined in green, an area that looks favorable for phosphoresidue contact by one measure, such as electrostatic potential, combines with the propensities generated by other characteristics to define a site that is less favorable, overall, for phosphoresidue contact.
We next investigated whether the amino acid identity, surface curvature, and electrostatic potential propensities could be combined in a prospective manner to identify phosphoresidue contact sites (Fig. 2 ▶; Materials and Methods) using cross-validation. For each structure in our set of known phosphopeptide-binding domains, the joint propensities calculated from every other member of the set were painted onto the surface of the domain of interest and visually inspected. As shown in Figure 3 ▶, the correct phosphate binding site was easily identified in every case as a contiguous region of mixed high and neutral joint propensity. No false negative prediction of a phosphoresidue contact site was made. In most cases, including that of the protein 14-3-3 ζ, the Smad MH2 domain, and both FHA and both SH2 domains studied, only a single site of significant size and propensity was observed. However, for Pin1, Cdc4, and the Polo-box domain of Plk1, a second site of comparable size and propensity to a known real phosphopeptide-binding site was also observed (Fig. 4 ▶). Intriguingly, the second predicted phosphoresidue contact region on the Pin1 surface lies at the catalytic site in Pin1’s proline isomerase domain. This site is known to bind specifically to, and isomerize, phosphopeptides containing the same motif as that recognized by the WW domain (Yaffe et al. 1997), and therefore corresponds to a phosphopeptide-binding site. In the case of Cdc4 and the Polo-box domain of Plk1, the second predicted phosphopeptide-binding site may represent false-positive predictions, or may indicate sites of further interaction with as-yet-unidentified phosphopeptides or other anionic ligands. It is also of interest to note that in most cases, the region of favorable propensity detected is quite a bit larger than the sites of phosphoresidue contact on which the method was trained. This indicates that the local properties most enriched in sites of phosphoresidue contact are also highly enriched in the surrounding regions. This may be reflective of a kinetic mechanism for attracting the phosphopeptide ligand to its binding site.
Figure 3.
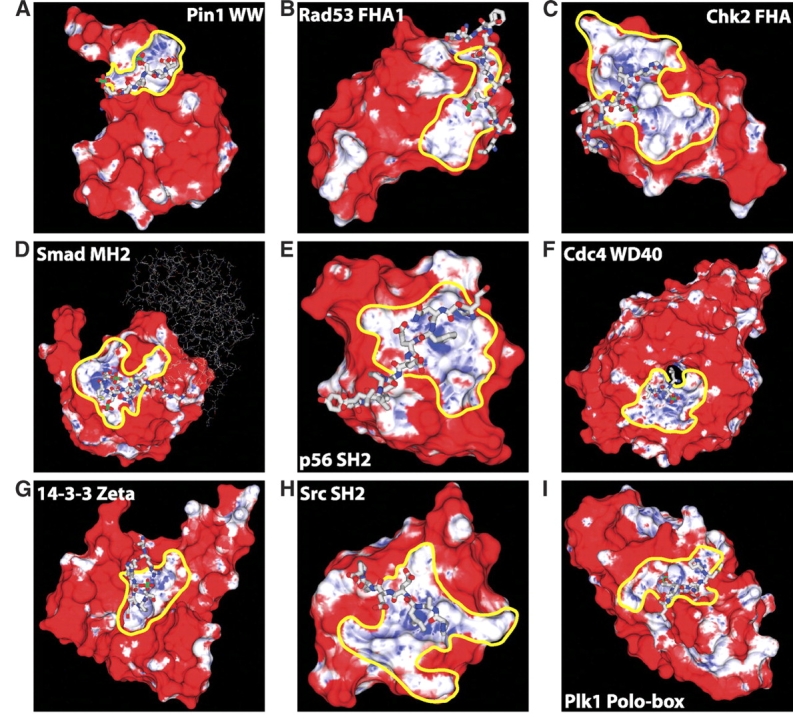
(All panels) Cross-validation of phosphoresidue contact site predictions on known phosphopeptide binding domains. Phosphopeptides are shown in a licorice representation, with the phosphate atom colored green. Surface coloring is linear from red to white for unfavorable propensity values from 0 to 1, and from white to blue for favorable propensity values of 1 to 30. Predicted phosphoresidue contact sites are outlined in yellow. The phosphopeptide-binding domain shown is indicated within each panel. The skinny sticks in panel D indicate the Smad monomer which contains the bound phosphopeptide. The phosphotyrosine phosphates in panels E and H are buried beneath the phosphate accessible surface.
Figure 4.

(All panels) Additional phosphoresidue contact site predictions. Additional site predictions were made on the surfaces of the indicated proteins. Surface coloring is linear from red to white for unfavorable propensity values of 0 to 1 and from white to blue for favorable propensity values from 1 to 30. Predicted phosphoresidue contact sites are outlined in yellow. The predictions for Cdc4 and Pin1 did not lie on the phosphopeptide-binding domains of those proteins.
Prediction of the phosphoresidue contact sites of Chk1 kinase and the BRCA1 BRCT-repeat domain
The method under development here is capable of predicting the location of phosphoresidue contact sites on the surface of phosphopeptide-binding domains whose unliganded structures are known. These predictions can then be investigated experimentally. Two such cases are currently available. The checkpoint kinase Chk1 has been found to be regulated by binding to the phosphorylated form of the protein claspin (Jeong et al. 2003) at a site within the kinase domain. The BRCT-repeat domains of several proteins, including BRCA1 and PTIP (Manke et al. 2003; Yu et al. 2003) have recently been identified as phosphopeptide-binding domains. One crystal structure of the Chk1 kinase domain, and three crystal structures of BRCA1 BRCT-repeat domains, in the absence of bound phosphopeptide are available. We therefore applied our method to these structures.
Application of local surface propensity analysis to the Chk1 kinase domain surface identified two possible sites for phosphopeptide binding (Fig. 5A ▶). These sites are connected by a small region of neutral propensity. The first site, located at the interface between the large and the small lobes of the kinase, but not in the kinase catalytic site, is made up of the amino acid side chains K54, R129, T153, R162, and N165 (Fig. 5A ▶, rightmost indicated site). The mutations K54A, R129A, and T153A, and R162A have all been shown to abrogate claspin binding in the frog Chk1 homolog Xchk1 (Jeong et al. 2003). Our results suggest that those residues are directly responsible for phosphoclaspin binding. The second site we identified, on the small lobe of the kinase domain, is adjacent to the first, and is made up of the Chk1 amino acid side chains K53, K60, H73 and R75 (Fig. 5A ▶, leftmost indicated site). While this site has not previously been identified as a site of phosphopeptide binding, it is known that phosphoclaspin binding to Xchk1 requires two separate claspin phosphorylation events, on residues S864 and S895. It is possible, therefore, that the two phosphopeptide residues pS864 and pS895, separated by 31 amino acids, are recognized by two distinct phosphopeptide-binding sites on the Chk1 surface.
Figure 5.

Predicted phosphoresidue contact sites on the surfaces of Chk1 and BRCA1. Surface phosphoresidue contact propensity plots (upper panels) and secondary structure (lower panels) with residues named in the text shown in licorice. (A) Chk1 kinase domain. On the surface plot, site one is outlined in yellow on the right, and site two is outlined in yellow on the left. In the secondary structure diagram, the small lobe of the Chk1 kinase domain is colored blue, and the large lobe is colored red. (B) BRCA1 BRCT repeat domain. The left panel indicates the first predicted site, which has been shown experimentally to be the site of phosphopeptide binding (Clapperton et al. 2004; Shiozaki et al. 2004; Williams et al. 2004), and the right panel indicates the second predicted site. The axis shown indicates the axis of rotation between the shown molecular faces. In the secondary structure diagrams, the first BRCT repeat, the linker, and the second repeat are colored blue, green, and red, respectively. Surface coloring is linear from red to white over the unfavorable propensity values 0 to 1 and from white to blue over favorable propensity values from 1 to 30. Portions of this figure were generated using the programs MOLSCRIPT (Kraulis 1991) and RASTER3D (Merritt and Bacon 1997).
Two predicted phosphopeptide-binding sites were also identified on the surface of the rat BRCA1 BRCT-repeat domain. The first of these is a bowl-shaped depression entirely within the first of the two BRCT repeats in the structure. The surface that composes the site is contributed by three amino acid side chains—K1648, S1601, and T1646 (Fig. 5B ▶, left panel). This triad of residues is conserved in the BRCA1 protein of humans. The other potential binding site is found in a channel composed of four amino acids at the interface between the second BRCT repeat and the helix linking the two repeats—R1697, R1791, H1692, and R1793 (Fig. 5B ▶, right panel). R1697 and H1692 are conserved in humans, while R1791 and R1793 are both glutamine in human BRCA1. Thus, the method presents two hypotheses for the site responsible for phosphopeptide-binding activity, which are readily tested by site-directed mutagenesis experiments.
During the preparation of this manuscript, the crystal structure of the human BRCA1 BRCT domain in complex with a phosphopeptide was solved (Clapperton et al. 2004; Shiozaki et al. 2004; Williams et al. 2004). In this structure, the phosphoresidue contact site was shown to correspond to the first of the two sites on the BRCA1 surface predicted by our method, indicating that for this site at least, our prediction was correct. This result, together with the experimentally corroborated prediction on the surface of the Chk1 kinase domain, indicates that the methodology described here has captured a large portion of the chemical and physical nature of phosphopeptide binding in a manner that is useful for predicting binding sites.
The phosphoresidue contact site predictions described here were originally made by visual inspection of the joint phosphoresidue contact potential on the surfaces of Chk1 and BRCA1 and selection of the largest site of favorable propensity. We are currently exploring a vertex clustering algorithm designed to identify large regions of favorable propensity in an automated fashion.
Discussion
We have developed a novel framework for phosphopeptide-binding site prediction. Our method is based on finely discretizing the surface of proteins, identifying physical and chemical properties that are overrepresented on those surfaces at sites of contact with phosphorylated amino acid side chains, and locating contiguous patches of those properties on the surfaces of proteins for which a prediction is to be made. Previous methods for the discovery of functional sites on proteins include patch analysis (Jones and Thornton 1997; Jones et al. 2003), in which properties are calculated for a number of large overlapping surface patches, and used in conjunction with heuristics to identify functional sites; and evolutionary trace analysis (Lichtarge et al. 2003), which depends on a large number of homologous protein sequences to find clusters of evolutionarily conserved residues. In contrast, the method described here, which uses discretized surface propensities, is capable of using a relatively small number of structures to determine local surface properties enriched in a functional site. The local nature of the surface properties analyzed appears to capture some of the physical and chemical properties required for phosphopeptide binding, despite the larger-scale dissimilarity of the binding sites used in training.
There are three important caveats to the computational method: First, we assume the independence of propensities calculated from a set of properties—amino acid identity, mean surface curvature, and electrostatic potential—which are not themselves independent. Given a large volume of data, it is possible to abandon this approximation by calculating an exact propensity value for every possible combination of property values. As more data become available, it should be possible to learn correct parameters for the combination of these propensity values.
Sites with the highest propensities for phosphoresidue contact have strong favorable propensity contributions from each of the three properties considered here. In the limit of currently available data, we find that all three properties considered here are necessary for accurate site prediction. Although strong favorable propensity for phosphoresidue contact is driven by the solvated electrostatic potential, false positive predictions that would be generated by the consideration of electrostatics alone are avoided by combining information about surface curvature and amino acid identity.
Second, we calculate and cross-validate propensity values from a set of crystal structures solved in the presence of phosphopeptide. These structures may involve some induced fit to their cognate peptides, whereas structures for which useful predictions can be made would be in their unliganded apo conformation. Despite this, we make predictions for the Chk1 kinase domain and the BRCA1 BRCT-repeat domain that are validated by experiment, indicating that the physical and chemical aspects of a phosphoresidue contact site which are captured by our model are not lost in the apo state.
Finally, the method described here is designed to identify the site of phosphoresidue contact on the surface of a known phosphopeptide-binding domain. It is clear that as novel phosphopeptide-binding domains are discovered, and as structural genomics efforts come to fruition, this approach will prove useful in rapidly identifying the functional sites on unliganded crystal structures without necessitating further crystallographic effort. Because the propensities calculated here are trained to differentiate phosphoresidue contact surface from the remainder of the surface of phosphopeptide-binding domains, this may be less useful in mining structural databases for novel phosphopeptide-binding domains. We expect, based on the emphasis given by our propensity scale to positive electrostatic potential, that this scale might score some anion- and phosphate-binding sites quite favorably. This has been confirmed by our examination of several nonphosphopeptide-binding proteins (data not shown). However, if the goal of future work is to differentiate among different types of anion-binding sites, appropriate propensity scales and other machine learning tools could certainly be developed, for example for the differentiation of phosphoresidue contact sites from such “decoy” sites.
The method described here is highly extendable, both in terms of the type of functional site examined, and in the characteristics for which propensities are calculated. Propensity calculations can be performed on continuous properties such as curvature and electrostatic potential, which have been discretized via binning, as well as on traditional discrete properties such as amino acid identity. Therefore, any property that can be assigned to the vertices of a protein surface can be applied to site predictions within this methodological framework. Moreover, predictions can be made within this framework for any functional categorization for which predictive physical surface properties can be found. Our successes in the identification of phosphoresidue contact sites on the surfaces of the Chk1 kinase domain and the BRCA1 BRCT-repeat domains indicate the utility of this methodology in functional site annotation.
Materials and methods
Structures
The structures used as a training set in this study (Table 1) were selected as being the best high-resolution crystal structures representative of the known phosphopeptide-binding domain/peptide interactions. Structures of one 14-3-3 protein, one group IV WW domain, one WD40 domain, one MH2 domain, two FHA domains, and two SH2 domains were used to gather propensity data. The single most well-resolved structures of the Chk1 kinase domain (PDB code 1IA8; Chen et al. 2000) and BRCA1 BRCT-repeat domain, from the rat BRCA1 protein, (PDB code 1L0B; Joo et al. 2002) were used for phosphoresidue contact site predictions.
Propensity calculation
For each property associated with a surface point—amino acid identity, surface curvature, and electrostatic potential—a propensity for phosphoresidue contact was calculated. The propensity of a property i was calculated as
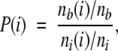 |
where nb(i) and nt(i) are the number of surface points with characteristic i contacting phosphoresidues and in total, respectively, and nb and nt are the number of surface points contacting phosphoresidues and total number of surface points in the data set, regardless of characteristic.
When attempting to predict the phosphoresidue contact site on a protein, the propensity assigned to each surface point was computed, under the simplifying assumption that propensities generated using amino acid identity, local mean surface curvature, and solvated electrostatic potential combine noncooperatively, as
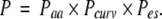 |
Figure 2 ▶ shows one example of the combination of these three individual propensities to derive a joint propensity.
Surface and contact calculation
The program MSMS (Sanner et al. 1996) was used to obtain a triangular surface mesh for each phosphopeptide-binding domain, using a probe radius of 3.0 Å, the approximate radius of a phosphate ion, and a surface density of 5.0 vertices/Å2. Calculations were performed on a monomer of each phosphopeptide-binding domain in the presence and the absence of only the phosphorylated side chain of the corresponding binding peptide. Surface points contacted by the phosphoresidue were identified as those that were surface accessible on the unliganded protein surface but buried in the protein/phosphoresidue complex surface such that they were further than 0.3 Å from the nearest point on the bound-state surface.
Amino acid identity assignment
The amino acid identity of each surface point was recorded as identified by MSMS, with points on the reentrant phosphate-accessible molecular surface assigned to the nearest atomic van der Waals sphere.
Mean surface curvature assignment
The mean surface curvature at each point was calculated according to the method of Meyer et al. (2003). In order to discretize the space of curvatures for propensity calculation, surface curvatures were binned with a bin width of 0.1 Å−1 between the values of −0.6 and 1.4 Å−1, with curvatures above and below the extrema placed in the highest and lowest bin, respectively. Calculated propensities were found to be insensitive to the bin size selected over a range of bin sizes from 0.05 Å−1 to 0.5 Å−1.
Solvated electrostatic potential assignment
The electrostatic potential at each surface point was calculated with a continuum electrostatic model with a locally modified version of the program DELPHI (Gilson et al. 1988; Sharp and Honig 1990a,b). The calculation used the phosphopeptide-binding domain alone, a solvent dielectric of 80, a salt concentration of 0.145 M, a protein dielectric of 4, and PARSE parameters (Sitkoff et al. 1994). Prior to calculating potentials, hydrogen atom positions and the titration and flip states of histidine, glutamine, and asparagine side chains were assigned to the protein structures using the program REDUCE (Word et al. 1999). Electrostatic potentials were discretized for propensity calculation by binning, with bin with 0.5 kT/e, with data below −15 kT/e or above +15 kT/e assigned to the lowest and the highest bin, respectively. Calculated propensities were found to be insensitive to the bin size selected over a range of bin sizes from 0.25 to 5.0 kT/e.
Electronic supplemental material
Images of several protein:phosphopeptide binding sites demonstrating the dissimilarity of contacts made to peptidyl-phosphates are available as a supplementary figure in the electronic edition.
Acknowledgments
We thank members of the Tidor and Yaffe laboratories for helpful discussion, and particularly Michael Altman for much of the development of our graphical analysis software. This work was partially supported by the NIH (GM060594 to M.B.Y. and GM065418 to B.T.) and by a Burroughs-Wellcome Career Development Award to M.B.Y.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04964705.
Supplemental material: see www.proteinscience.org
References
- Chen, P., Luo, C., Deng, Y.L., Ryan, K., Register, J., Margosiak, S., Tempczyk-Russell, A., Nguyen, B., Myers, P., Lundgren, K., et al. 2000. The 1.7 Å crystal structure of human cell cycle checkpoint kinase Chk1: Implications for Chk1 regulation. Cell 100 681–692. [DOI] [PubMed] [Google Scholar]
- Clapperton, J.A., Manke, I.A., Lowery, D.M., Ho, T., Haire, L.F., Yaffe, M.B., and Smerdon, S.J. 2004. Structure and mechanism of BRCA1 BRCT domain recognition of phosphorylated BACH1 with implications for cancer. Nat. Struct. Mol. Biol. 11 512–518. [DOI] [PubMed] [Google Scholar]
- Durocher, D., Taylor, I.A., Sarbassova, D., Haire, L.F., Westcott, S.L., Jackson, S.P., Smerdon, S.J., and Yaffe, M.B. 2000. The molecular basis of FHA domain: Phosphopeptide binding specificity and implications for phospho-dependent signaling mechanisms. Mol. Cell 6 1169–1182. [DOI] [PubMed] [Google Scholar]
- Eck, M.J., Shoelson, S.E., and Harrison, S.C. 1993. Recognition of a high-affinity phosphotyrosyl peptide by the Src homology-2 domain of P56(Lck). Nature 362 87–91. [DOI] [PubMed] [Google Scholar]
- Elia, A.E.H., Rellos, P., Haire, L.F., Chao, J.W., Ivins, F.J., Hoepker, K., Mohammad, D., Cantley, L.C., Smerdon, S.J., and Yaffe, M.B. 2003. The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo-box domain. Cell 115 83–95. [DOI] [PubMed] [Google Scholar]
- Gilson, M.K., Sharp, K.A., and Honig, B. 1988. Calculating the electrostatic potential of molecules in solution: Method and error assessment. J. Comp. Chem. 9 327–335. [Google Scholar]
- Jeong, S.Y., Kumagai, A., Lee, J., and Dunphy, W.G. 2003. Phosphorylated claspin interacts with a phosphate-binding site in the kinase domain of Chk1 during ATR-mediated activation. J. Biol. Chem. 278 46782–46788. [DOI] [PubMed] [Google Scholar]
- Jones, S. and Thornton, J.M. 1997. Prediction of protein-protein interaction sites using patch analysis. J. Mol. Biol. 272 133–143. [DOI] [PubMed] [Google Scholar]
- Jones, S., Shanahan, H.P., Berman, H.M., and Thornton, J.M. 2003. Using electrostatic potentials to predict DNA-binding sites on DNA-binding proteins. Nucleic Acids Res. 31 7189–7198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo, W.S., Jeffrey, P.D., Cantor, S.B., Finnin, M.S., Livingston, D.M., and Pavletich, N.P. 2002. Structure of the 53BP1 BRCT region bound to p53 and its comparison to the Brca1 BRCT structure. Genes & Dev. 16 583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. Molscript—A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Lee, L.P. and Tidor, B. 1997. Optimization of electrostatic binding free energy. J. Chem. Phys. 106 8681–8690. [Google Scholar]
- Li, J.J., Williams, B.L., Haire, L.F., Goldberg, M., Walker, E., Durocher, D., Yaffe, M.B., Jackson, S.P., and Smerdon, S.J. 2002. Structural and functional versatility of the FHA domain in DNA-damage signaling by the tumor suppressor kinase Chk2. Mol. Cell 9 1045–1054. [DOI] [PubMed] [Google Scholar]
- Lichtarge, O., Yao, H., Kristensen, D.M., Madabushi, S., and Mihalek, I. 2003. Accurate and scalable identification of functional sites by evolutionary tracing. J. Struct. Funct. Genomics 4 159–166. [DOI] [PubMed] [Google Scholar]
- Manke, I.A., Lowery, D.M., Nguyen, A., and Yaffe, M.B. 2003. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science 302 636–639. [DOI] [PubMed] [Google Scholar]
- Merritt, E.A. and Bacon, D.J. 1997. Raster3D: Photorealistic molecular graphics. Methods Enzymol. 277 505–524. [DOI] [PubMed] [Google Scholar]
- Meyer, M., Desbrun, M., Schröder, P., and Barr, A.H. 2003. Discrete differential-geometry operators for triangulated 2-manifolds. In Visualization and mathematics III, pp. 34–58. Springer Verlag, Heidelberg, Germany.
- Orlicky, S., Tang, X.J., Willems, A., Tyers, M., and Sicheri, F. 2003. Structural basis for phosphodependent substrate selection and orientation by the SCFCdc4 ubiquitin ligase. Cell 112 243–256. [DOI] [PubMed] [Google Scholar]
- Rittinger, K., Budman, J., Xu, J.A., Volinia, S., Cantley, L.C., Smerdon, S.J., Gamblin, S.J., and Yaffe, M.B. 1999. Structural analysis of 14-3-3 phosphopeptide complexes identifies a dual role for the nuclear export signal of 14-3-3 in ligand binding. Mol. Cell 4 153–166. [DOI] [PubMed] [Google Scholar]
- Sanner, M.F., Olson, A.J., and Spehner, J.C. 1996. Reduced surface: An efficient way to compute molecular surfaces. Biopolymers 38 305–320. [DOI] [PubMed] [Google Scholar]
- Shiozaki, E.N., Gu, L., Yan, N., and Shi, Y. 2004. Structure of the BRCT repeats of BRCA1 bound to a BACH1 phosphopeptide: Implications for signaling. Mol. Cell 14 405–412. [DOI] [PubMed] [Google Scholar]
- Sitkoff, D., Sharp, K.A., and Honig, B. 1994. Accurate calculation of hydration free energies using macroscopic solvent models. J. Phys. Chem. 98 1978–1988. [Google Scholar]
- Verdecia, M.A., Bowman, M.E., Lu, K.P., Hunter, T., and Noel, J.P. 2000. Structural basis for phosphoserine-proline recognition by group IV WW domains. Nat. Struct. Biol. 7 639–643. [DOI] [PubMed] [Google Scholar]
- Waksman, G., Shoelson, S.E., Pant, N., Cowburn, D., and Kuriyan, J. 1993. Binding of a high-affinity phosphotyrosyl peptide to the Src Sh2 domain—Crystal-structures of the complexed and peptide-free forms. Cell 72 779–790. [DOI] [PubMed] [Google Scholar]
- Williams, R.S., Lee, M.S., Hau, D.D., and Glover, J.N. 2004. Structural basis of phosphopeptide recognition by the BRCT domain of BRCA1. Nat. Struct. Mol. Biol. 11 519–525. [DOI] [PubMed] [Google Scholar]
- Word, J.M., Lovell, S.C., Richardson, J.S., and Richardson, D.C. 1999. Asparagine and glutamine: Using hydrogen atom contacts in the choice of side-chain amide orientation. J. Mol. Biol. 285 1735–1747. [DOI] [PubMed] [Google Scholar]
- Wu, J.W., Hu, M., Chai, J.J., Seoane, J., Huse, M., Li, C., Rigotti, D.J., Kyin, S., Muir, T.W., Fairman, R., et al. 2001. Crystal structure of a phosphorylated Smad2: Recognition of phosphoserine by the MH2 domain and insights on Smad function in TGF-β signaling. Mol. Cell 8 1277–1289. [DOI] [PubMed] [Google Scholar]
- Yaffe, M.B. 2002. Phosphotyrosine-binding domains in signal transduction. Nat. Rev. Mol. Cell Biol. 3 177–186. [DOI] [PubMed] [Google Scholar]
- Yaffe, M.B. and Elia, A.E. 2001. Phosphoserine/threonine-binding domains. Curr. Opin. Cell Biol. 13 131–138. [DOI] [PubMed] [Google Scholar]
- Yaffe, M.B. and Smerdon, S.J. 2001. Phosphoserine/threonine binding domains: You can’t pSERious? Structure 9 R33–R38. [DOI] [PubMed] [Google Scholar]
- Yaffe, M.B., Schutkowski, M., Shen, M.H., Zhou, X.Z., Stukenberg, P.T., Rahfeld, J.U., Xu, J., Kuang, J., Kirschner, M.W., Fischer, G., et al. 1997. Sequence-specific and phosphorylation-dependent proline isomerization: A potential mitotic regulatory mechanism. Science 278 1957–1960. [DOI] [PubMed] [Google Scholar]
- Yu, X.C., Chini, C.C.S., He, M., Mer, G., and Chen, J.J. 2003. The BRCT domain is a phospho-protein binding domain. Science 302 639–642. [DOI] [PubMed] [Google Scholar]
- Zhou, M.M. 2000. Phosphothreonine recognition comes into focus. Nat. Struct. Biol. 7 1085–1087. [DOI] [PubMed] [Google Scholar]