

Introduction
The title I have chosen for my personal recollections describes, in a nutshell, the direction my scientific endeavors from the time of my Ph.D. thesis, which I began in 1970, to the present day. Over the years, I have changed fields a number of times. There were periods when I was preoccupied with the development of methods; at other times, the focus was on biological problems. Science can be advanced by new hypotheses about how things work, which can be tested and proven right or wrong, and by new methods which enable us to tackle questions that we were unable to address with the existing methods. Or, as Richard Feynman put it, “Science means, sometimes, a special method of finding things out. Sometimes it means the body of knowledge arising from the things found out. It may also mean the new things you can do when you have found something out, or the actual doing of new things. This last field is usually called technology. . . .” (R.P. Feynman in the John Danz Lectures, 1963 [Feynman 1998]).
Apprenticeship with great freedom
After graduating from the University of Bonn in late 1969, I joined the Institute of Biophysics and Electron Microscopy at the University of Düsseldorf in January 1970. The director of the Institute at the time was Helmut Ruska, who became my Ph.D. supervisor. Helmut Ruska, a medical doctor, was the younger brother of Ernst Ruska, the electrical engineer who, in 1932, at the age of 26, had published his calculations on the theoretical resolving power of an electron microscope and, in the face of strong skepticism, had completed the development of a commercial instrument by 1939 (Ruska 1979). Rarely has a scientific instrument had such an impact on so many branches of science, and yet it took more than 50 years before Ernst Ruska was rewarded with the Nobel Prize in Physics in 1986 for his fundamental work in electron optics and his design of the first electron microscope. Helmut Ruska, who was very close to his brother, realized immediately the potential of such an instrument for the biomedical sciences, in particular the visualization of hitherto invisible infectious agents and for ultrastructural studies of cells (Fig. 1 ▶). Helmut Ruska played a very important role in the early days of electron microscopy, not only by raising awareness and support—his clinical mentor at the Charité in Berlin, Richard Siebeck, became a decisive advocate at a critical time—but also by his achievements in the visualization of viruses, bacteria, and blood cells (for review, see Kruger et al. 2000; for relevant references, see also Ruska 1979).
Figure 1.

Pioneers of electron microscopy. (Left) Ernst Ruska (1906–1988). (Right) Helmut Ruska (1908–1973).
At the time I joined Helmut Ruska’s laboratory, the performance of transmission electron microscopes had reached a level that allowed the imaging of single heavy atoms. Several groups in Europe, the United States, and Japan tried to take advantage of this capability and to use heavy atoms as site-specific labels, e.g., for mapping the bases in strands of DNA. In the same vein, Helmut Ruska gave me the task of exploring the use of heavy atom labels to study membrane topology. I decided to begin with well-defined model membranes before tackling membranes of biological relevance. I never got that far! Using Langmuir–Blodgett techniques, I prepared monomolecular layers at the water–air interface and transferred them under precisely controlled conditions to specimen supports, but when I exposed my carefully designed lipid layers to the electron beam, they faded away before I was able to take a picture. Occasionally, I obtained images of remnants of them with the heavy atoms coalesced into clusters. Eventually, with an unusually radiation-resistant organometallic compound of no relevance to biology, thorium-hexafluoracetylacetonate, I succeeded in obtaining images showing a heavy atom pattern that was consistent with my design plan (Baumeister and Hahn 1972).
Helmut Ruska was preoccupied with administrative duties during my time as a graduate student in his laboratory and, as a consequence, his supervision of me was very casual. Nevertheless, he was very supportive and he gave me all the resources I needed for my work. In late August 1973, only a few months after receiving my Ph.D., Helmut Ruska died after a short illness.
I had offers from other places, but decided to stay in Düsseldorf, and since it took several years until a successor for Helmut Ruska was found, I enjoyed complete freedom during my postdoctoral years. Thanks to benevolent reviewers, I obtained my first grant in 1974 and I began to work on radiation damage—the electron microscopist’s greatest foe. I used a variety of methods for a quantitative assessment of radiation damage in lipids and proteins under the conditions encountered in electron microscopy (Baumeister et al. 1976; Hahn et al. 1976). My hope was that a better understanding of the underlying radiation chemistry might enable us to find a remedy—a vain hope as it turned out (Baumeister 1978).
Heading into new directions
Having realized that I was on an unproductive path, I had to change direction. While the electron microscopy community in Germany with its strong tradition in electron optics was preoccupied with “high resolution,” others, driven more strongly by their desire to obtain insights into biomolecular architectures, took more pragmatic approaches. Already in 1968, De Rosier and Klug had formulated the principles for the three-dimensional reconstruction of objects from projection images and applied them to the tail of bacteriophage T4, taking advantage of its helical symmetry (De Rosier and Klug 1968). When working with periodic or repetitive structures, one can minimize radiation damage by underexposing the samples; the information is retrieved from the statistically noisy images by averaging over many identical structures. Averaging can be performed by direct superposition (see, e.g., Markham et al. 1964) or by Fourier transform filtering (see, e.g., De Rosier and Klug 1972) using both optical and digital methods (Aebi et al. 1973). Using the aforementioned stratagem and applying it to unstained, glucose-embedded purple membrane, Henderson and Un-win succeeded in 1975 in obtaining a 7 Å structure of bacteriorhodopsin (Henderson and Unwin 1975), which became the paradigm of a membrane protein structure.
Having read about a bacterium of legendary radiation resistance, Micrococcus radiodurans (now Deinococcus radiodurans), and knowing from the literature that a regular protein layer was a component of its cell wall, I focused my work on this structure. Before long, I obtained decent micrographs of this structure (Fig. 2A ▶), which I called the hexagonally-packed-intermediate (HPI)-layer, but I ran into a dilemma with the image processing. After having done some initial experiments with optical filtration, I became convinced that computer methods were the future; however, with the notable exception of Walter Hoppe in Munich, optical methods were preferred to computer methods in Germany at the time. The arguments in favor of optical image processing (averaging and correction of contrast transfer function) were the size of the images that could be processed and the speed. The downside was lack of flexibility, and the fabrication of suitable masks became a serious bottleneck. Since I had neither access to the necessary infrastructure nor the know-how for computer-based processing, I started a collaboration with Olaf Kübler at the ETH in Zürich who was in an inverse position: He had the software and the hardware that was needed but no data. In the following years, we made substantial progress in elucidating the molecular architecture of the D. radiodurans cell envelope (Baumeister and Kübler 1978; Kübler and Baumeister 1978; Baumeister et al. 1981, 1982).
Figure 2.

(A) Electron micrograph of a metal-shadowed HPI-layer as obtained by detergent extraction of the Deinococcus radiodurans cell envelope. Areas marked R show the rough inner surface; areas marked S show the smoother outer surface. For details, see Baumeister et al. (1981). (B) An 8 Å projection map of the HPI-layer embedded in aurothioglucose. (For details, see Rachel et al. 1986.)
In 1979, I organized a meeting entitled “Electron Microscopy at Molecular Dimensions” held at Burg Gemen near Münster, Germany, which in retrospect became quite influential (see, e.g., Deisenhofer and Michel 1989). Besides state-of-the-art applications, it covered many developments in technology from image recording and processing to low-temperature electron microscopy (EM), or strategies for making regular 2-D arrays (Baumeister and Vogell 1980). The intrinsic disorder in the 2-D protein arrays limits the resolution one can attain by Fourier filtering, and during the Gemen meeting, I became convinced that there are better ways of dealing with imperfect 2-D crystals. The emerging methods for averaging of single molecules offered an alternative, and I decided to join forces with Joachim Frank. A few months later when I visited him in Albany, we explored the application of correlation-based averaging to the micrographs of the HPI-layer, but to our disappointment we failed to obtain meaningful results during this short period of time. A year later, when I spent several months at the Cavendish Laboratory in Cambridge, England, working with Owen Saxton, we were able to overcome the problems and obtained the first correlation-averaged images of the HPI-layer with a significantly improved resolution. In trying to get this work published, we faced unusual problems; it took several rounds of reviewing and several steps down the ladder of (journal) prestige, until our manuscript was finally published (Saxton and Baumeister 1982). In retrospect, however, it is gratifying to see that more than 20 years later, this paper is still cited frequently and, in the guise of “lattice unbending” (Baldwin et al. 1988) our strategem for overcoming the limitations due to lattice disorder became part of the standard repertoire used for processing images of 2-D crystals. Shortly thereafter, we applied correlation averaging to Scanning Transmission Electron Microscopy (STEM) images of unstained preparations of the HPI-layer and obtained the first quantitative mass maps (Engel et al. 1982), the beginning of a long-standing and successful collaboration with Andreas Engel at the Biocenter in Basel, Switzerland.
The first decade in Martinsried: Studying protein architecture on prokaryotic cell surfaces
At the beginning of 1982, I moved to the Max-Planck-Institute of Biochemistry where, after a short overlap period, I was appointed successor of Walter Hoppe. Hoppe was a microscopist-turned-X-ray crystallographer of remarkable originality and theoretical ability but with limited interest in the practical aspects of structural biology. No humble man, he insisted that his “nonconventional” approach to the structural analysis of individual macromolecules, as he liked to call it, was superior to other strategies (for review, see Hoppe 1983).
I remained unconvinced, and felt that the clever tactics of “single particle analysis” as pioneered by Joachim Frank, a former student of Hoppe, and Marin van Heel at the Fritz-Haber-Institute in Berlin held greater promise. Not only did their approach greatly simplify data acquisition, the combination of intelligent image classification procedures and extensive averaging had the great advantage of yielding significant and interpretable structural data (for a recent review, see Frank 2002). In all fairness, I must add that in spite of a fierce public dispute I had with Walter Hoppe a few years earlier (Baumeister and Hahn 1975; Hoppe et al. 1975) and divergent views on the course to take, he was, in general, supportive when I arrived in Martinsried and began to set up my laboratory. We continued our work with the HPI-layer; a 3-D model was generated in due course and, using cryomicroscopy, an 8 Å projection map was also obtained (Baumeister et al. 1986; Rachel et al. 1986; Fig. 2B ▶).
With the plentiful resources now at our disposal, we not only extended our structural studies to several other bacterial surface layers, we also widened our repertoire of methods. Our comparative structural studies revealed some common architectural principles (Baumeister et al. 1986, 1988) and sequence analyses led to the identification of new motifs (Peters et al. 1987, 1989; Lupas et al. 1994) such as the S-layer homology domain (for a recent review, see Engelhardt and Peters 1998) but the biological function of Slayers remained an enigma. Intuitively, I still feel that there must be some function beyond mediating adhesion to animate or inanimate surfaces or protecting underlying components of the cell envelope, but this remains pure speculation.
Colleagues in Martinsried (Wolfram Zillig) and in Regensburg (Karl-Otto Stetter) introduced me to the exciting world of extremophiles. Most hyperthermophiles belong to the archaeal domain of life where (glyco)protein surface layers are common. They represent the main macromolecular component of the cell envelope and are intimately associated with the plasma membrane. Some show a high degree of order and have a role in maintaining and possibly determining cell shape (Wildhaber and Baumeister 1987; Phipps et al. 1991b) while others form poorly ordered and flexible surface networks on pleomorphic cells (Wildhaber et al. 1987; Peters et al. 1995). In spite of their apparent diversity, archaeal surface layers have some common structural principles: A stalk of variable length (10–70 nm) emanates from a membrane-anchoring domain and connects to a highly variable (filiform or bulky) domain that forms a canopy-like layer by means of end-to-end contacts enclosing a quasi-periplasmic space (Baumeister and Lembcke 1992). A periplasmic space of unusual width and maintained by a rod-shaped spacer protein (Omp α) is also found in the hyperthermophilic ancestral bacterium Thermotoga maritima (Engel et al. 1992; Lupas et al. 1995).
The structural principles of archaeal surface layer proteins is exemplified particularly clearly by tetrabrachion, the giant glycoprotein found on the surface of Staphylothermus marinus, where it forms a poorly ordered, branched network (Peters et al. 1995). This filiform molecule is anchored in the cell membrane at the C-terminal end of a 70-nm-long stalk and branches at the other end into four arms, each of 24 nm length, which form the canopy-like meshwork. A hybrid approach, which used EM and biochemical data as well as molecular biology and bioinformatics, led to a very detailed model structure (Fig. 3 ▶; Peters et al. 1996), the salient features of which, in the meantime, have been confirmed by X-ray crystallography (Stetefeld et al. 2000). The C-terminal part is formed by a right-handed, coiled coil of four α-helices; the almost flawless pattern of aliphatic residues, mainly leucine and isoleucine, throughout the hydrophobic core of the stalk provides an explanation for its exceptional stability. At a proline residue, the stalk switches from a right-handed supercoil to a left-handed one. At a flexible glycine-rich hinge region, the stalk branches into four arms, each formed by a “heavy chain” and a “light chain”, which in turn are each derived from the translated 1524-residue polypeptide by internal proteolytic cleavage. The most likely topology of the arms is a three-stranded coil of antiparallel β-sheets. There is a patch of negative charges on the outer face of the coiled coil near the middle of the stalk, which serves as an anchoring device for a large, hyperthermostable protease of the subtilisin family; in the stalk-bound form the protease is resistant to heat inactivation up to a temperature of 125°C (Mayr et al. 1996), while the stalk withstands heating up to 130°C. Obviously, one function of the Staphylothermus surface layer is to provide an extracellular holding compartment for a protease that could otherwise cause havoc.
Figure 3.

(A) The Staphylothermus marinus surface layer as revealed by freeze-etching (left). In the absence of MgCl2 the detergent extracted surface layer dissociates into micelles formed by the tetrabrachion-protease complexes (right). (B) The tetrabrachion-protease complex. (Left) Model showing the mode of interaction of the tetrabrachion-protein complexes in the layer structure. (Center) Electron micrograph of the negatively stained complex released from the surface layer meshwork by SDS-heat treatment (for details, see Peters et al. 1995). (Right) Folding topology of tetrabrachion. The location of N-terminal residues, cysteine residues, and the unique proline residue separating the left- and right-handed supercoiled domains are marked by circles. Putative disulfide bridges are indicated. The flexible hinge segment, the protease-binding region, and the membrane anchor are marked by rectangles. (For details, see Peters et al. 1996.)
The next decade: Proteasomes, thermosomes, and other elements of intracellular protein quality control
In 1989, my laboratory became interested in studying the structure and function of a large (20S) protein complex, at the time known as the multicatalytic proteinase (Dahlmann et al. 1989). Already in 1980, a large, multisubunit protease had been isolated and characterized (Hase et al. 1980; Wilk and Orlowski 1980; Orlowski and Wilk 1981). Initially, the multicatalytic proteinase was believed to be composed of 3–5 subunits, ranging from 24 kDa to 28 kDa in size; it displayed three distinct proteolytic activities (trypsin-like, chymotrypsin-like, and peptidylglutamylpeptide-hydrolyzing) when assays were performed with small synthetic peptides, and it was noted that the integrity of the 20S complex was essential for all proteolytic activities. Attempts were made to assign specific activities to distinct subunits, but in spite of the efforts of many groups, the nature of the active sites remained enigmatic. Along a different line, a particle named “prosome” was under intensive investigation in the mid-1980’s (for review, see Scherrer et al. 1990). Reminiscent in size and subunit composition of the multicatalytic protease complex, it appeared to be associated with RNA and it was suggested to have a role in the regulation of gene expression. In 1988, it was established beyond doubt that the prosome and the multicatalytic proteinase complex were one and the same particle (Arrigo et al. 1988; Falkenburg et al. 1988) and the name “proteasome” was coined by Alfred L. Goldberg (Harvard Medical School) to highlight its only established function, the proteolytic one, and its complex structure. In the following years, evidence began to accumulate that the 20S proteasome was part of an even larger complex, the 26S proteasome, which was implicated in the ATP-dependent degradation of ubiquitin-conjugated proteins (Eytan et al. 1989; Driscoll and Goldberg 1990; Rechsteiner et al. 1993).
By 1990, the 20S proteasome was structurally rather featureless and its subunit composition and stoichiometry were ill-defined. Reports that proteasomes could undergo changes in subunit composition during development (Haass and Kloetzel 1989) made its structural analysis a daunting challenge, since structural methods rely, in one guise or another, on averaging and, therefore, on homogeneous preparations of molecules. This led us to search for proteasomes of hopefully simpler subunit composition in prokaryotic cells. While our initial attempts to find proteasomes in bacteria were unsuccessful, we found them in the archaeon Thermoplasma acidophilum (Dahlmann et al. 1989). The Thermoplasma proteasome turned out to be very similar in size and shape to proteasomes from eukaryotic cells, but much simpler in subunit composition; it comprises only two subunits, α (25.8 kDa) and β (22.3 kDa). The two subunits have significant sequence similarity, suggesting that they arose from a common ancestor via gene duplication (Zwickl et al. 1991, 1992a). Due to its relative simplicity, the ensuing years saw the Thermoplasma proteasome play a pivotal role in elucidating the structure and enzymatic mechanism of this intriguing protein degradation machine.
In 1991, a first, three-dimensional structure of the Thermoplasma proteasome was obtained by EM single particle analysis, showing with remarkable clarity the organization of the barrel-shaped complex with its tripartite inner compartment (Hegerl et al. 1991). Immunoelectron microscopy studies allowed us to assign the α-subunits to the two outer rings of the barrel, and the β-subunits to the inner rings (Grziwa et al. 1991). Mass measurements by STEM helped us to establish the stoichiometry (α7β7β7α7), and metal decoration studies of proteasome crystals (not yet good enough for high resolution X-ray crystallography) clearly revealed the symmetry of the 20S complex. The structural model we put forward on the basis of these data stood the test of time and it recurred in all proteasomes, eukaryotic and prokaryotic (Pühler et al. 1992).
Another important advance was the expression of fully assembled and functional 20S proteasomes in Escherichia coli (Zwickl et al. 1992b; Fig. 4A ▶). It not only allowed us to perform systematic mutagenesis studies aimed at identifying the active site, it also greatly facilitated the growth of crystals diffracting to high resolution (Jap et al. 1993). In 1995, the crystal structure analysis was completed in a collaboration with the group of Robert Huber (Löwe et al. 1995; Fig. 4B ▶). The long-sought catalytic nucleophile of the 20S proteasome, the N-terminal threonine of the mature β-subunit was identified independently and almost simultaneously by site-directed mutagenesis and crystal structure analysis (Löwe et al. 1995; Seemüller et al. 1995). As anticipated from their sequence similarity the (noncatalytic) α-and the (catalytic) β-type subunits showed the same fold: a four-layer α + β structure with two antiparallel five-stranded β-sheets, flanked on one side by two, and on the other side by three α-helices. In the β-type subunits, the β-sheet sandwich is closed at one end by four hairpin loops and opens at the opposite end to form the active-site cleft; the cleft is oriented toward the inner surface of the central cavity. In the α-type subunits, an additional helix formed by an N-terminal extension crosses the top of the β-sheet sandwich and fills this cleft. Initially, the proteasome fold was believed to be unique; however, it turned out to be common to a new superfamily of proteins referred to as Ntn (N-terminal nucleophile) hydrolases (Brannigan et al. 1995). Beyond the common fold, members of this family share the mechanisms of the nucleophilic attack and self-processing (for reviews, see Baumeister et al. 1998; Dodson and Wlodawer 1998; Seemüller et al. 2001; Zwickl et al. 2002).
Figure 4.
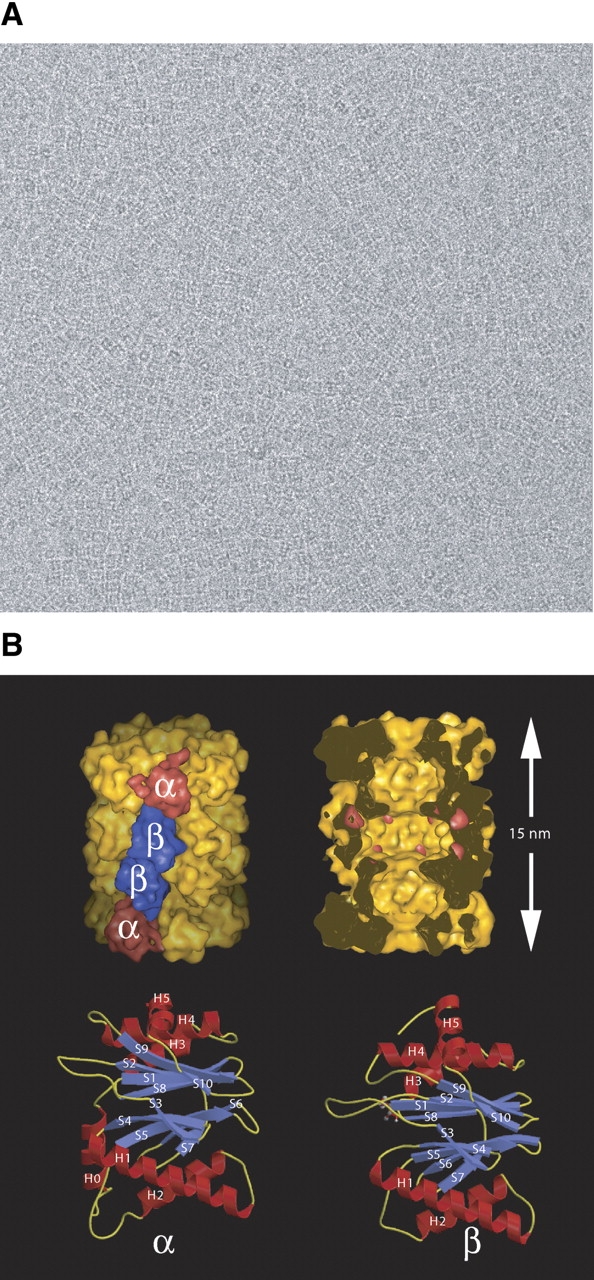
The 20S proteasome from Thermoplasma acidophilum. (A) Electron micrograph of recombinant 20S proteasomes in vitreous ice. (B,top left) Structure of the 20S proteasome in surface representation, low-pass filtered to 1 nm resolution. The α- and β-subunits are located in the outer and the inner rings, respectively. (Top right) The same structure cut open along the sevenfold axis to display the inner compartments with the active sites of the β-subunits in the central chamber marked in red. (Bottom left and right) Similar fold of the α- (left) and β- subunits (right). Both subunits contain a sandwich of two, five-stranded antiparallel β-sheets flanked by helices. (For details, see Löwe et al. 1995; Zwickl et al. 2002.)
The crystal structure revealed that access to the inner cavity that harbors the active sites is controlled by four constrictions. The constrictions in the α-rings which give access to the two “antechambers” are narrow and partially obstructed, while the constrictions which regulate access to the central cavity are wider. We were able to show with Nanogold-labeled substrates, visible in electron micrographs, that polypeptides indeed enter the proteasome via the orifice at the center of the α-rings. Bulky additions to the polypeptide chain, such as a gold cluster, prevent passage into the interior, suggesting that the discrimination between folded and unfolded substrates is based on a size-exclusion mechanism (Wenzel and Baumeister 1995). Thus the 20S proteasome is a molecular nano-compartment that confines the proteolytic reaction to its interior and sequesters it from the crowded environment of the cell. Interestingly, formation of the active sites by the posttranslational removal of the propeptides of the β-subunits (Seemüller et al. 1996) is coupled to the assembly of the 20S proteasome in such a manner that activation is delayed until the assembly is complete (for review, see Seemüller et al. 2001). This led us to propose the concept of self-compartmentalization as a regulatory principle (Lupas et al. 1997; Baumeister et al. 1998).
As mentioned earlier, it began to transpire in the early 1990s that the 20S proteasome of eukaryotes associates with regulatory complexes, in an ATP-dependent manner, to form the 26S proteasome. Now it is firmly established that this 2.5 MDa complex altogether comprising more than 30 different subunits acts downstream in the ubiquitin–proteasome pathway and is the central player in intracellular proteolysis. Proteins destined for degradation are marked by covalent attachment of Ub chains, which mediate recognition by the 26S proteasome (for recent reviews, see Hershko and Ciechanover 1998; Voges et al. 1999). In 1993, we were able to provide the first detailed description of the 26S complex, based on electron microscopy and image analysis (Peters et al. 1993). The averages showing the regulatory (19S) particles attached to one or both ends of the 20S proteasome core particle (the “dragon-head” or “double dragon-head” motif) became the classical textbook images of the 26S proteasome. Since then, however, progress has been embarrassingly slow; the notorious instability of the complex and its dynamics have made it very difficult to achieve more than gradual improvements of the structural model (Glickmann et al. 1998; Walz et al. 1998; Hölzl et al. 2000). While it is clear that the role of the 19S regulatory complexes is the preparation of substrates for degradation in the 20S core particle—involving the recognition of ubiquitinated substrates, the removal of the polyubiquitin chains, the unfolding of substrates, and assistance in translocation across the gates of the 20S complex—the precise topology and role of the 19S subunits is hitherto only dimly understood (Zwickl et al. 1999).
In 1991, we found, in a serendipitous manner, a novel ATPase complex. During the lysis of accidentally heat-shocked Pyridictium cells on electron microscopy grids, a massive release of toroidal particles composed of the stacked octameric rings was observed (Phipps et al. 1991a). Not only the shape, but also the heat-shock induction of this complex were reminiscent of the GroEL/Hsp60 family, and therefore raised the possibility that it represented an archaeal chaperonin. Subsequently, we named it “thermosome” to highlight its heat induction and extreme thermostability (Phipps et al. 1993). Independently, a closely related complex (TF55) was discovered in the laboratory of Art Horwich in Yale (Trent et al. 1991). The thermosome or TF55 were the first representatives of the Group II chaperonins found in archaea and in the eukaryotic cytosol. The main structural feature distinguishing the Group II from the Group I chaperonins is, in the absence of a co-chaperonin, a built-in lid provided by the protrusions of the apical domains which can seal the folding chamber by an iris-type closure mechanism (Klumpp et al. 1997; Gutsche et al. 1999).
In 1996, in our quest for a more comprehensive understanding of the protein quality control machinery in Thermoplasma we found a fascinating, large proteolytic complex that works in conjunction with an array of aminopeptidases (Tamura et al. 1996). In view of the shape of the hexamer, we named it “tricorn protease”; soon thereafter we were able to show that tricorn protease exists in the cell as a giant icosahedral complex of approximately 15 MDa, which in addition to its peptide-cleaving activity, appears to serve as an organizing center for the more downstream elements of the protein degradation pathway (Walz et al. 1997). Tricorn protease converts the oligo-peptides (typically about 8 amino acid residues) released by the proteasome into smaller (2–4 residue) peptides which are degraded further by aminopeptidases (Tamura et al. 1998). These findings stimulated the search for “functional homologs” of tricorn protease in eukaryotic cells; one of the candidates is tripeptidylpeptidase (TPP) II, another giant protein complex with an intriguing structure (Geier et al. 1999; Rockel et al. 2002).
In 2000, we completed the sequencing of the genome of Thermoplasma acidophilum, an endeavor we had undertaken with modest resources (Ruepp et al. 2000). It not only served to further establish Thermoplasma as a model system for studying cellular protein quality control, it also provided the platform for a very ambitious project, namely the mapping of its cellular proteome by cryoelectron tomography; this, in turn, can be expected to shed new light on the pathways of intracellular protein quality control (Fig. 5 ▶).
Figure 5.
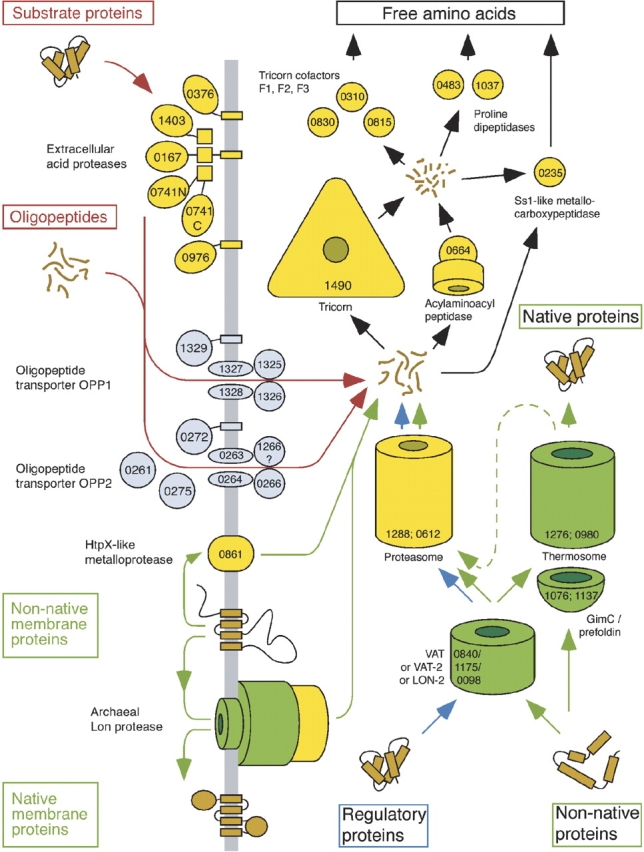
The protein quality control system in Thermoplasma acidophilum. Components of the proteolytic pathway are shown in yellow; chaperones, in green. The numbers refer to the ORF code. (For details, see Ruepp et al. 2000.)
The latest frontier: Charting molecular landscapes inside cells by cryoelectron tomography
The foundations of electron tomography were laid already in the late 1960s. In their landmark paper, De Rosier and Klug outlined very clearly and in general terms the principles of 3D reconstruction from electron micrographs (De Rosier and Klug 1968). Being aware of the practical problems in recording 3-D data sets, they took advantage of the helical symmetry of the bacteriophage T4 tail in a very pragmatic manner. Walter Hoppe, guided by his background in X-ray crystallography, also realized the potential of 3-D electron microscopy. Diverging from the approaches taken by most others, he focused on the development of methods suitable for studying individual structures (“Crystallography of crystals consisting of a single unit cell,” Hoppe 1978). In fact, his group presented as early as 1974 a 3-D reconstruction of single fatty acid synthetase molecules obtained by tomography (Hoppe et al. 1974). As mentioned earlier, the “brute force” approach they used provoked some criticism. Besides doubts that negative staining can portray details of the underlying structure to the resolution they claimed, the main concern was the enormous electron dose to which the specimen was exposed during recording of the data. There was much discussion in the following years as to whether it might ultimately be possible to do electron tomography with acceptable electron doses. Also in 1968, R.G. Hart at the Lawrence Livermore Laboratory published a paper entitled “Electron microscopy of unstained biological material: The polytropic montage” (Hart 1968). Despite its vision, the Hart paper had negligible impact. For a vision to materialize, timing is a crucial element; if it is too early, the necessary technologies might not yet exist.
The key problem in electron tomography, which for many years was a formidable obstacle and a deterrent, is to reconcile two requirements that are in conflict with each other: To obtain a reconstruction that is detailed and largely undistorted, one has to collect data over as wide a tilt range as possible with increments as small as possible (for review, see Baumeister et al. 1999). At the same time, the electron dose must be minimized. Above a critical dose, the specimen undergoes structural degradation that, in the worst case, can render a reconstruction meaningless. In principle, one could fractionate the dose over as many projections as an optimized tilt geometry might require. However, there is a practical limitation; the signal-to-noise ratio of the 2-D images has to be sufficient to permit their accurate alignment by cross-correlation. This problem is further aggravated by the far-from-perfect mechanical accuracy of the tilting devices that causes image shifts and changes of focus. Therefore, following each change of tilt angle, the specimen (or its image) has to be realigned and refocused. Doing this manually and with minimal exposure to the electron beam is utterly impossible.
In the late 1980s when computer-controlled electron microscopes and large-area charge-coupled device (CCD) cameras became available, we saw an opportunity to automate tomographic data acquisition (Typke 1991; Dierksen et al. 1992, 1995; Koster et al. 1992). This made the recording of data sets not only less cumbersome, but first and foremost it allowed the cumulative electron dose to be kept within tolerable limits. The fraction of the dose that is spent on overhead (search, recentering, [auto-]focusing) can be kept as low as 3% of the total dose; in other words, almost all electrons are used for gaining information (Koster et al. 1997). As is evident from the recent resurgence, this has changed the perspectives of electron tomography in a most profound manner; electron tomography had been used from time to time for ultrastructural studies, mostly of plastic embedded biological material, but it has gathered momentum only recently.
As demonstrated originally with “phantom cells,” that is, lipid vesicles encapsulating specific sets of macromolecules, automated tomography in a “low-dose mode” has enabled us to combine the potential of 3-D imaging with the best possible preservation of biological samples, that is, embedded in vitreous ice (Dierksen et al. 1995; Grimm et al. 1997). Vitrification by rapid freezing ensures not only a close-to-life preservation of molecular and cellular structures, but it also allows the capture of dynamic events (Dubochet et al. 1988). It avoids the risks of artifacts traditionally associated with chemical fixation and staining or with the dehydration of cellular structures. Equally important, tomograms of frozen-hydrated structures represent their natural density distribution, whereas staining reactions tend to produce intricate mixtures of positive and negative staining. As a consequence the interpretation of such tomograms in molecular terms may be very problematic if not impossible (Baumeister 2002).
With the use of automated procedures and user-friendly software, meanwhile, the recording of tilt series and their processing has become routine. It is in fact now less cumbersome and less time-consuming to obtain a cryotomogram than going through the conventional procedures of plastic embedding and sectioning the material. With smaller structures (e.g., bacteriophages docked onto proteoliposomes) a resolution of 2.5 nm has been obtained (Böhm et al. 2001). With whole prokaryotic cells or thin eukaryotic cells grown directly on EM grids, resolution is usually in the range of 4–5 nm, but prospects for further improvements are good (Plitzko et al. 2002). Better detectors, in particular, will allow a finer 3-D sampling, which, in turn, will improve resolution (see above) and allow tomography to enter the realm of molecular resolution (2–3 nm).
Even at the present practical level of resolution, cryotomograms of organelles or cells contain an imposing amount of information. They are, essentially, 3-D images of entire proteomes, and they should ultimately enable us to map the spatial relationships of the full complement of macromolecules in an unperturbed cellular context; however, new strategies and innovative image analysis techniques are needed for “mining” this information. Retrieving it is confronted with two major problems: Cryotomograms are “contaminated” by residual noise, and they are distorted by missing data—in spite of optimized image acquisition schemes. Moreover, the cytoplasm of most cells is densely packed (“crowded”) with molecules literally touching each other. It is therefore often impossible to perform a segmentation and to extract features, based on visual inspection of the tomograms. Denoising procedures (Frangakis and Hegerl 2001) can facilitate the visualization of features, but advanced pattern recognition techniques are needed for detecting and identifying specific macromolecules by their respective structural signatures.
The most powerful method for improving the signal-to-noise ratio is averaging. Although averaging can obviously not be applied to tomograms of pleomorphic structures in a first instance, such tomograms may nevertheless contain repetitive elements which can be extracted in silico, and the subtomograms containing them can be subjected to classification and averaging. These averages can be used subsequently for replacing the original data in the tomograms, resulting in “synthetic” tomograms with a locally improved signal-to-noise ratio. This strategy was used, for example, to obtain a density map of whole Herpes simplex virions (Grünewald et al. 2003).
In spite of the low signal-to-noise ratio of tomograms, continuous structures such as membranes of cytoskeletal filaments are easy to recognize. Cryotomograms of Dictyostelium discoideum cells grown directly on carbon support films have provided unprecedented insights into the organization of actin filaments in an unperturbed cellular environment (Medalia et al. 2002). The tomograms show, on the level of individual filaments, their modes of interaction (isotropic networks, bundles, etc.), they allow us to determine the branching angles precisely (in 3-D), and they reveal the structure of membrane attachment sites. For the quantitative analysis of large data sets, as is needed for extracting statistically significant quantitative data, it will be necessary to develop algorithms for automated segmentation, to establish connectivity of filaments in noisy data sets—a notoriously difficult problem—and measure structural parameters of filaments (Fig. 6A ▶).
Figure 6.

Cryoelectron tomography of Dictyostelium discoideum cell. (A) Visualization of the actin network and cytoplasmic complexes in a Dictyostelium cell grown directly on an EM grid and embedded in vitreous ice (for details, see Medalia et al. 2002). (B) Visualization of a 26S proteasome within an intact Dictyostelium cell. (Left) Slice from a tomogram. Dominant features are ribosomes, some of them attached to the endoplasmic reticulum (lower left corner), and actin filaments. The encircled particle is a 26S proteasome. (Right) enlarged contour plot of the single (unaveraged) 26S proteasome (projection of a stack of slices from tomogram).
Cryoelectron tomography enables us to obtain images of single macromolecules inside intact cells as is exemplified by Figure 6B ▶, which shows a single 26S proteasome within the cytoplasm of a Dictyostelium cell. Although in this case the detection and identification was facilitated by the large size (~2.5 MDa) and the peculiar shape of this complex, it indicates that a molecular signature-based approach to mapping cellular proteomes should become feasible.
Alternatively, one could envisage strategies for introducing electron-dense labels marking the spatial distribution of the molecules of interest. Such an approach, however, would no longer be noninvasive—unless it is based entirely on genetic manipulations—and it would be difficult, if not impossible, to achieve quantitative detection. Moreover, it is hard to imagine how this approach could be parallelized such that it becomes a high-throughput technology capable of mapping entire proteomes. For every molecule of interest it would be necessary to repeat the whole procedure, the labeling of cells, the recording of tilt series, and the tomographic reconstruction. Even if this could be accomplished, it would be a daunting challenge to interrelate the individual maps and to reveal the structure of molecular networks, owing to the stochastic nature of cellular supramolecular architecture. Therefore, there is a strong incentive to exploit the information content of cryotomograms by means of intelligent pattern recognition algorithms. With this approach, a tomogram needs to be produced only once, and it is then interpreted in a sequential manner in terms of its molecular architecture. The strategy we are pursuing is “template matching” (Böhm et al. 2000; Frangakis et al. 2002). Provided that high- or medium-resolution structures of the macromolecules of interest are available, they can be used for a systematic interrogation of the tomograms (Fig. 7 ▶). Image simulations have shown that template matching is indeed a feasible approach for identifying macromolecules in “noisy” tomograms. Experimental studies with “phantom cells”, i.e., lipid vesicles encapsulating known sets of proteins provide a means of validating the results of the template matching (Fig. 8 ▶). At the present resolution of 4–5 nm, only very large complexes (ribosomes, 26S proteasomes) can be mapped with high fidelity (>95%); an improvement in resolution to 2 nm will allow the mapping of medium-sized complexes (~200–400 kDa, depending on shape). While tomograms with a resolution of 2 nm are a realistic prospect, major technical innovations will be required to go beyond.
Figure 7.

Strategy for the detection and identification of macromolecules in cellular volumes. Because of the crowded nature of cells and the high noise levels in tomograms (left), an interactive segmentation and feature extraction is, in most cases, not feasible. It requires automated pattern recognition techniques to exploit the rich information content of such tomograms. An approach that has been demonstrated to work is based on the recognition of the structural signature (size, shape) of molecules by template matching. Templates of the macromolecules under scrutiny are obtained by high- or medium-resolution techniques. Theses templates are then used to search the volume of the tomograms (Vin) systematically for matching structures by cross-correlation. The tomogram has to be scanned for all possible Eulerian angles around three different axes, with templates of all the different protein structures in which one is interested. The search is computationally demanding, but can be parallelized efficiently. The output information (Vout) is a set of coordinates that describes the positions and orientation of all the molecules found in the tomogram. (For details, see Frangakis et al. 2002.)
Figure 8.

Mapping molecular landscapes by pattern recognition. Volume-rendered representation of an ice-embedded “phantom cell” containing thermosomes (blue) and 20S proteasomes (yellow) with a 1:1 molar ratio. The two protein species were identified by template matching and are represented by averages derived from the tomogram. (For details, see Frangakis et al. 2002.)
Once the challenges of obtaining a sufficiently good resolution are met, the next challenge will be to create comprehensive libraries of templates. A whole array of methods can be used to this end. Worldwide structural genomics efforts will increase the pace with which high resolution structures of domains, subunits, and larger molecular entities become available and eventually provide a comprehensive structural dictionary. In integrative hybrid approaches, combining information gathered by a variety of techniques, computational methods will play a crucial role (for a recent review, see Sali et al. 2003). EM-based “single” particle analysis will undoubtedly become a major player in furnishing medium-resolution (~1 nm) structures of complexes. Currently, this technique is slow and cumbersome, but great strides have been made in recent years toward improving throughput by automating data acquisition and analysis (Carragher et al. 2004).
With cryoelectron tomography providing 3-D images at molecular resolution of cells in a close-to-life state, and with the availability of image analysis tools for interpreting the tomograms, we are poised now to integrate structural information gathered at multiple levels—from atoms to cells—into pseudoatomic maps of organelles or cells. The move from proteomics parts lists to precise maps of supramolecular landscapes will provide unprecedented insights into the network structures that underlie higher cellular functions and the structural principles that orchestrate them.
Epilogue
An epilogue is the place for reflections and also for acknowledgments. I have deliberately changed fields a few times. In doing so, the decision to leave a field was usually more difficult than the decision to embark on a new one. After working on a problem for a significant period of time, one becomes emotionally attached to it or even obsessed by it, but there is also an element of convenience: One knows the field with all of its ramifications, and one becomes established and is recognized by his/her peers. On the other hand, a change of fields can be rejuvenating. One is less inhibited by the knowledge of problems or obstacles, and more willing to take new approaches. By looking at a problem from a different angle, new opportunities arise and it is often the interface between fields and disciplines where the sparks fly. I believe, for example, that our work in cyoelectron tomography will eventually enable us to address problems in intracellular protein quality control in a new manner.
The work I have described in this essay would not have been accomplished without the support and the great efforts of many coworkers and colleagues. I have been fortunate to work with generations of talented and motivated students and postdoctoral fellows and I greatly enjoyed the collaboration with fine colleagues, with several of them over long periods of time to this day. I mentioned a few of them in the main text, but for the sake of the space it was impossible to acknowledge them all; the names of most of them appear as coauthors in the list of references. I wish to thank them all. Finally, I had the privilege to work in environments that were very supportive and allowed me to undertake the projects I liked to do, irrespective of the chances of success.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041148605.
References
- Aebi, U., Smith, P.R., Dubochet, J., Henry, C., and Kellenberger, E. 1973. A study of the structure of the T-layer of Bacillus brevis. J. Supramol. Struct. 1 498–522. [DOI] [PubMed] [Google Scholar]
- Arrigo, A.P., Tanaka, K., Goldberg, A.L., and Welch, W.J. 1988. Identity of the 19S “prosome” particle with the large multifunctional protease complex of mammalian cells (the proteasome). Nature 331 192–194. [DOI] [PubMed] [Google Scholar]
- Baldwin, J.M., Henderson, R., Beckman, E., and Zemlin, F. 1988. Images of purple membrane at 2.8 Å resolution obtained by cryo-electron microscopy. J. Mol. Biol. 202 585–591. [DOI] [PubMed] [Google Scholar]
- Baumeister, W. 1978. Biological horizons in molecular microscopy. Eur. J. Cell Biol. 17 246–297. [PubMed] [Google Scholar]
- ———. 2002. Electron tomography: Towards visualizing the molecular organization of the cytoplasm. Curr. Opin. Struct. Biol. 12 679–684. [DOI] [PubMed] [Google Scholar]
- Baumeister, W. and Hahn, M. 1972. Electron microscopy of thorium atoms in monomolecular layers. Nature 241 445–447. [Google Scholar]
- ———. 1975. Relevance of three-dimensional reconstructions of stain distributions for structural analysis of biomolecules. Hoppe-Seyler’s Z. Physiol. Chem. 356 1313–1316. [DOI] [PubMed] [Google Scholar]
- Baumeister, W. and Kübler, O. 1978. Topographic study of the cell surface of Micrococcus radiodurans. Proc. Natl. Acad. Sci. 75 5525–5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumeister, W. and Lembcke, G. 1992. Structural features of archaebacterial cell envelopes. J. Bioenerg. Biomembr. 24 567–575. [DOI] [PubMed] [Google Scholar]
- Baumeister, W. and Vogell, W. 1980. Electron microscopy at molecular dimensions. Springer Verlag, Berlin.
- Baumeister, W., Fringeli, U.P., Hahn, M., Kopp, F., and Seredynski, J. 1976. Radiation-damage in tripalmitin layers studied by means of infrared spectroscopy and electron-microscopy. Biophys. J. 16 791–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumeister, W., Kübler, O., and Zingsheim, H.P. 1981. The structure of the cell envelope of Micrococcus radiodurans as revealed by metal shadowing and decoration. J. Ultrastruct. Res. 75 60–71. [DOI] [PubMed] [Google Scholar]
- Baumeister, W., Karrenberg, F., Rachel, R., Engel, A., Ten Heggeler, B., and Saxton, W.O. 1982. The major cell envelope protein of Micrococcus radiodurans (R1): structural and chemical characterization. Eur. J. Biochem. 725 535–544. [DOI] [PubMed] [Google Scholar]
- Baumeister, W., Barth, M., Hegerl, R., Guckenberger, R., Hahn, M., and Saxton, W.O. 1986. Three-dimensional structure of the regular surface layer (HPI-layer) of Deinococcus radiodurans. Appendix: W.O. Saxton and W. Baumeister: Principles of organization in S layers. J. Mol. Biol. 187 241–253. [DOI] [PubMed] [Google Scholar]
- Baumeister, W., Wildhaber, I., and Engelhardt, H. 1988. Bacterial surface proteins. Some structural, functional and evolutionary aspects. Biophys. Chem. 29 39–49. [DOI] [PubMed] [Google Scholar]
- Baumeister, W., Walz, J., Zühl, F., and Seemüller, E. 1998. The proteasome: Paradigm of a self-compartmentalizing protease. Cell 92 367–380. [DOI] [PubMed] [Google Scholar]
- Baumeister, W., Grimm, R., and Walz, J. 1999. Electron tomography of molecules and cells. Trends Cell Biol. 9 81–85. [DOI] [PubMed] [Google Scholar]
- Böhm, J., Frangakis, A., Hegerl, R., Nickell, S., Typke, D., and Baumeister, W. 2000. Toward detecting and identifying macromolecules in a cellular context: Template matching applied to electron tomograms. Proc. Natl. Acad. Sci. 97 14245–14250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böhm, J., Lambert, O., Frangakis, A., Letellier, L., Baumeister, W., and Rigaud, J.L. 2001. FhuA-mediated phage genome transfer into liposomes: A cryo-electron tomography study. Curr. Biol. 11 1168–1175. [DOI] [PubMed] [Google Scholar]
- Brannigan, J.A., Dodson, G., Duggleby, H.J., Moody, P.C.E., Smith, J.L., Tomchick, D.R., and Murzin, A.G. 1995. A protein catalytic framework with an N-terminal nucleophile is capable of self-activation. Nature 378 416–419. [DOI] [PubMed] [Google Scholar]
- Carragher, B., Fellmann, D., Guerra, F., Milligan, R.A., Mouche, F., Pulokas, J., Sheehan, B., Quispe, J., Suloway, C., Zhu, Y., et al. 2004. Rapid routine structure determination of macromolecular assemblies using electron microscopy: Current progress and further challenges. J. Synchrotron Radiat. 11 83–85. [DOI] [PubMed] [Google Scholar]
- Dahlmann, B., Kopp, F., Kuehn, L., Niedel, B., Pfeifer, G., Hegerl, R., and Baumeister, W. 1989. The multicatalytic proteinase (prosome) is ubiquitous from eukaryotes to archaebacteria. FEBS Lett. 251 125–131. [DOI] [PubMed] [Google Scholar]
- De Rosier, D.J. and Klug, A. 1968. Reconstruction of three dimensional structures from electron micrographs. Nature 217 130–134. [DOI] [PubMed] [Google Scholar]
- ———. 1972. Structure of the tubular variants of the head of bacteriophage T4 (polyheads). J. Mol. Biol. 65 469–488. [DOI] [PubMed] [Google Scholar]
- Deisenhofer, J. and Michel, H. 1989. The photosynthetic reaction center from the purple bacterium rhodopseudomonas viridis (Nobel lecture). Angew. Chemie 28 872–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierksen, K., Typke, D., Hegerl, R., Koster, A.J., and Baumeister, W. 1992. Towards automatic electron tomography. Ultramicroscopy 40 71–87. [Google Scholar]
- Dierksen, K., Typke, D., Hegerl, R., Walz, J., Sackmann, E., and Baumeister, W. 1995. Three-dimensional structure of lipid vesicles embedded in vitreous ice and investigated by automated electron tomography. Biophys. J. 68 1416–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson, G. and Wlodawer, A. 1998. Catalytic triads and their relatives. Trends Biochem. Sci. 23 347–352. [DOI] [PubMed] [Google Scholar]
- Driscoll, J. and Goldberg, A.L. 1990. The proteasome (multicatalytic protease) is a component of the 1500 kDa proteolytic complex which degrades ubiquitin-conjugated proteins. J. Biol. Chem. 265 4789–4792. [PubMed] [Google Scholar]
- Dubochet, J., Adrian, M., Chang, J.J., Homo, J.C., Lepault, J., McDowall, A.W., and Schulz, P. 1988. Cryo-electron microscopy of vitrified specimens. Q. Rev. Biophys. 21 129–228. [DOI] [PubMed] [Google Scholar]
- Engel, A., Baumeister, W., and Saxton, W.O. 1982. Mass mapping of a protein complex with the scanning transmission electron microscope. Proc. Natl. Acad. Sci. 79 4050–4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel, A.M., Cejka, Z., Lupas, A., Lottspeich, F., and Baumeister, W. 1992. Isolation and cloning of Omp α, a coiled-coil protein spanning the periplasmic space of the ancestral eubacterium Thermotoga maritima. EMBO J. 11 4369–4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt, H. and Peters, J. 1998. Structural research on surface layers: A focus on stability, surface layer homology domains, and surface layer-cell wall interactions. J. Struct. Biol. 124 276–302. [DOI] [PubMed] [Google Scholar]
- Eytan, E., Ganoth, D., Armon, T., and Hershko, A. 1989. ATP-dependent incorporation of 20S protease into the 26S complex that degrades proteins conjugated to ubiquitin. Proc. Natl. Acad. Sci. 86 7751–7755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkenburg, P.E., Haass, C., Kloetzel, P.M., Niedel, B., Kopp, F., Kuehn, L., and Dahlmann, B. 1988. Drosophila small cytoplasmic 19S ribonucleoprotein is homologous to the rat multicatalytic proteinase. Nature 331 190–192. [DOI] [PubMed] [Google Scholar]
- Feynman, R.P. 1998. The meaning of it all. Addison-Wesley, Reading, MA.
- Frangakis, A. and Hegerl, R. 2001. Noise reduction in electron tomographic reconstructions using nonlinear anisotropic diffusion. J. Struct. Biol. 135 239–250. [DOI] [PubMed] [Google Scholar]
- Frangakis, A., Böhm, J., Förster, F., Nickell, S., Nicastro, D., Typke, D., Hegerl, R., and Baumeister, W. 2002. Identification of macromolecular complexes in cryoelectron tomograms of phantom cells. Proc. Natl. Acad. Sci. 99 14153–14158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank, J. 2002. Single-particle imaging of macromolecules by cryo-electron microscopy. Annu. Rev. Biomol. Struct. 31 303–319. [DOI] [PubMed] [Google Scholar]
- Geier, E., Pfeifer, G., Wilm, M., Lucchiari-Hartz, M., Baumeister, W., Eichmann, K., and Niedermann, G. 1999. A giant protease with potential to substitute for some functions of the proteasome. Science 283 978–981. [DOI] [PubMed] [Google Scholar]
- Glickman, M.H., Rubin, D.M., Coux, O., Wefes, I., Pfeifer, G., Cejka, Z., Baumeister, W., Fried, V.A., and Finley, D. 1998. A subcomplex of the proteasome regulatory particle required for ubiquitin-conjugate degradation and related to the COP9-signalosome and elF3. Cell 94 615–623. [DOI] [PubMed] [Google Scholar]
- Grimm, R., Bärmann, M., Häckl, W., Typke, D., Sackmann, E., and Baumeister, W. 1997. Energy filtered electron tomography of ice-embedded actin and vesicles. Biophys. J. 72 482–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grünewald, K., Desai, P., Winkler, D.C., Heymann, J.B., Belnap, D.M., Baumeister, W., and Steven, A.C. 2003. Three-dimensional structure of Herpes simplex virus from cryo-electron tomography. Science 302 1396–1398. [DOI] [PubMed] [Google Scholar]
- Grziwa, A., Baumeister, W., Dahlmann, B., and Kopp, F. 1991. Localization of subunits in proteasomes from Thermoplasma acidophilum by immunoelectron microscopy. FEBS Lett. 290 186–190. [DOI] [PubMed] [Google Scholar]
- Gutsche, I., Essen, L.-O., and Baumeister. W. 1999. Group II chaperonins: New TRiC(k)s and turns of a protein folding machine. J. Mol. Biol. 293 295–312. [DOI] [PubMed] [Google Scholar]
- Haass, C. and Kloetzel, P.M. 1989. The Drosophila proteasome undergoes changes in its subunit pattern during development. Exp. Cell Res. 180 243–252. [DOI] [PubMed] [Google Scholar]
- Hahn, M., Seredynski, J., and Baumeister, W. 1976. Inactivation of catalase monolayers by irradiation with 100 keV electrons. Proc. Natl. Acad. Sci. 73 823–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart, R.G. 1968. Electron microscopy of unstained biological material: The polytropic montage. Science 159 1464–1467. [DOI] [PubMed] [Google Scholar]
- Hase, J., Kobashi, K., Nakai, N., Mitsui, K., Iwata, K., and Takadera, T. 1980. The quaternary structure of carp muscle alkaline protease. Biochim. Biophys. Acta 611 205–213. [DOI] [PubMed] [Google Scholar]
- Hegerl, R., Pfeifer, G., Pühler, G., Dahlmann, B., and Baumeister, W. 1991. The three-dimensional structure of proteasomes from Thermoplasma acidophilum as determined by electron microscopy using random conical tilting. FEBS Lett. 283 117–121. [DOI] [PubMed] [Google Scholar]
- Henderson, R. and Unwin, P.N.T. 1975. Three-dimensional model of purple membrane obtained by electron microscopy. Nature 257 28–32. [DOI] [PubMed] [Google Scholar]
- Hershko, A. and Ciechanover, A. 1998. The ubiquitin system. Ann. Rev. Biochem. 67 425–479. [DOI] [PubMed] [Google Scholar]
- Hölzl, H., Kapelari, B., Kellermann, J., Seemüller, E., Sümegi, M., Udvardy, A., Medalia, O., Sperling, J., Müller, J.A., Engel, A., et al. 2000. The regulatory complex of Drosophila melangolaster 26S proteasomes: Subunit composition and localization of a deubiquitylating enzyme. J. Cell Biol. 150 119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe, W. 1978. Three-dimensional electron microscopy of individual structures: Crystallography of “crystals” consisting of a single unit cell. Chem. Scripta 79 227–243. [Google Scholar]
- ———. 1983. Electron diffraction with the transmission electron microscope as a phase-determining diffractometer—From spatial frequency filtering to the three-dimensional structure analysis of ribosomes. Angew. Chem. Int. Ed. Engl. 22 456–485. [Google Scholar]
- Hoppe, W., Gassmann, J., Hunsmann, N., Schramm, H.J., and Sturm, M. 1974. Three-dimensional reconstruction of individual negatively stained yeast fatty-acid synthetase molecules from tilt series in the electron microscope. H.-S. Z. Physiol. Chem. 355 1483–1487. [PubMed] [Google Scholar]
- ———. 1975. Comments on the paper “Relevance of three-dimensional reconstructions of stain distributions for structural analysis of biomolecules.” Hoppe-Seyler’s Z. Physiol. Chem. 356 1317–1320. [DOI] [PubMed] [Google Scholar]
- Jap, B., Pühler, G., Lücke, H., Typke, D., Löwe, J., Stock, D., Huber, R., and Baumeister, W. 1993. Preliminary x-ray crystallography study of the proteasome from Thermoplasma acidophilum. J. Mol. Biol. 234 881–884. [DOI] [PubMed] [Google Scholar]
- Klumpp, M., Baumeister, W., and Essen, L.-O. 1997. Structure of the substrate binding domain of the thermosome, an archaeal group II chaperonin. Cell 91 263–270. [DOI] [PubMed] [Google Scholar]
- Koster, A.J., Chen, H., Sedat, J.W., and Agard, D.A. 1992. Automated microscopy for electron tomography. Ultramicroscopy 46 207–227. [DOI] [PubMed] [Google Scholar]
- Koster, A.J., Grimm, R., Typke, D., Hegerl, R., Stoschek, A., Walz, J., and Baumeister, W. 1997. Perspectives of molecular and cellular electron tomography. J. Struct. Biol. 120 276–308. [DOI] [PubMed] [Google Scholar]
- Kruger, D.H., Schneck, P., and Gelderblom, H.R. 2000. Helmut Ruska and the visualization of viruses. Lancet 355 1713–1717. [DOI] [PubMed] [Google Scholar]
- Kübler, O. and Baumeister, W. 1978. The structure of periodic cell wall component (HPI-layer) of Micrococcus radiodurans. Eur. J. Cell Biol. 17 1–9. [PubMed] [Google Scholar]
- Löwe, J., Stock, D., Jap, B., Zwickl, P., Baumeister, W., and Huber, R. 1995. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 Å resolution. Science 268 533–539. [DOI] [PubMed] [Google Scholar]
- Lupas, A., Engelhardt, H., Peters, J., Santarius, U., Volker, S., and Baumeister, W. 1994. Domain structure of the Acetogenium kivui surface layer revealed by electron crystallography and sequence analysis. J. Bacteriol. 176 1224–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupas, A., Müller, S., Goldie, K., Engel, A.M., Engel, A., and Baumeister, W. 1995. Model structure of the Omp α rod, a parallel four-stranded coiled coil from the hyperthermophilic eubacterium Thermotoga maritima. J. Mol. Biol. 248 180–189. [DOI] [PubMed] [Google Scholar]
- Lupas, A., Flanagan, J.M., Tamura, T., and Baumeister, W. 1997. Self-compartmentalizing proteases. Trends Biochem. Sci. 22 399–404. [DOI] [PubMed] [Google Scholar]
- Markham, R., Hitchborn, J.H., Hills, G.J., and Frey, S. 1964. The anatomy of the tobacco mosaic virus. Virology 22 342–359. [DOI] [PubMed] [Google Scholar]
- Mayr, J., Lupas, A., Kellermann, J., Eckerskorn, C., Baumeister, W., and Peters, J. 1996. A hyperthermostable protease of the subtilisin family bound to the surface layer of the archaeon Staphylothermus marinus. Curr. Biol. 6 739–749. [DOI] [PubMed] [Google Scholar]
- Medalia, O., Weber, I., Frangakis, A., Nicastro, D., Gerisch, G., and Baumeister, W. 2002. Macromolecular architecture in eukaryotic cells visualized by cryoelectron tomography. Science 298 1209–1213. [DOI] [PubMed] [Google Scholar]
- Orlowski, M. and Wilk, S. 1981. A multicatalytic protease complex from pituitary that forms enkephalin and enkephalin containing peptides. Biochim. Biophys. Res. Commun. 101 814–822. [DOI] [PubMed] [Google Scholar]
- Peters, J., Peters, M., Lottspeich, F., Schäfer, W., and Baumeister, W. 1987. Nucleotide sequence analysis of the gene encoding the Deinococcus radiodurans surface protein, derived amino acid sequence and complementary protein chemical studies. J. Bacteriol. 169 5216–5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, J., Peters, M., Lottspeich, F., and Baumeister, W. 1989. S-layer protein gene of Acetogenium kivui: Cloning and expression in Escherichia coli and determination of the nucleotide sequence. J. Bacteriol. 171 6307–6315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, J.-M., Cejka, Z., Harris, J.R., Kleinschmidt, J.A., and Baumeister, W. 1993. Structural features of the 26S proteasome complex. J. Mol. Biol. 234 932–937. [DOI] [PubMed] [Google Scholar]
- Peters, J., Nitsch, M., Kühlmorgen, B., Golbik, R., Lupas, A., Kellermann, J., Engelhardt, H., Pfander, J.-P., Müller, S., Goldie, K., et al. 1995. Tetrabrachion: A filamentous archaebacterial surface protein assembly of unusual structure and extreme stability. J. Mol. Biol. 245 385–401. [DOI] [PubMed] [Google Scholar]
- Peters, J., Baumeister, W., and Lupas, A. 1996. Hyperthermostable surface layer protein tetrabrachion from the archaebacterium Staphylothermus marinus: Evidence for the presence of a right-handed coiled coil derived from the primary structure. J. Mol. Biol. 257 1031–1041. [DOI] [PubMed] [Google Scholar]
- Phipps, B.M., Hoffmann, A., Stetter, K.O., and Baumeister, W. 1991a. A novel ATPase complex selectively accumulated upon heat shock is a major cellular component of thermophilic archaebacteria. EMBO J. 10 1711–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phipps, B.M., Huber, R., and Baumeister, W. 1991b. The cell envelope of the hyperthermophilic archaebacterium Pyrobaculum organotrophum consists of two regularly arrayed protein layers: Three-dimensional structure of the outer layer. Mol. Microbiol. 5 253–265. [DOI] [PubMed] [Google Scholar]
- Phipps, B.M., Typke, D., Hegerl, R., Volker, S., Hoffmann, A., Stetter, K.O., and Baumeister, W. 1993. Structure of a molecular chaperone from a thermophilic archaebacterium. Nature 361 475–477. [Google Scholar]
- Plitzko, J., Frangakis, A.S., Nickell, S., Förster, F., Gross, A., and Baumeister, W. 2002. In vivo veritas: Electron cryotomography of cells. Trends Biotechnol. 20 S40–S44. [Google Scholar]
- Pühler, G., Weinkauf, S., Bachmann, L., Müller, S., Engel, A., Hegerl, R., and Baumeister, W. 1992. Subunit stoichiometry and three-dimensional arrangement in proteasomes from Thermoplasma acidophilum. EMBO J. 11 1607–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachel, R., Jakubowski, U., Tietz, H., Hegerl, R., and Baumeister, W. 1986. Projected structure of the surface protein of Deinococcus radiodurans determined to 8 Å resolution by cryomicroscopy. Ultramicroscopy 20 305–316. [Google Scholar]
- Rechsteiner, M., Hoffman, L., and Dubiel, W. 1993. The multicatalytic and 26S proteases. J. Biol. Chem. 268 6065–6068. [PubMed] [Google Scholar]
- Rockel, B., Peters, J., Kühlmorgen, B., Glaeser, R.M., and Baumeister, W. 2002. A giant protease with a twist: The TPP II complex from Drosophila studied by electron microscopy. EMBO J. 21 5979–5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruepp, A., Graml, W., Santos-Martinez, M.-L., Koretke, K.K., Volker, C., Mewes, H.W., Frishman, D., Stocker, S., Lupas, A.N., and Baumeister, W. 2000. The genome sequence of the thermoacidophilic scavenger Thermoplasma acidophilum. Nature 407 508–513. [DOI] [PubMed] [Google Scholar]
- Ruska, E. 1979. Die frühe Entwicklung der Elektronenlinsen und der Elektronenmikroskopie. Acta Hist. Leopoldina 12 7–136. [PubMed] [Google Scholar]
- Sali, A., Glaeser, R., Earnest, T., and Baumeister, W. 2003. From words to literature in structural proteomics. Nature 422 216–225. [DOI] [PubMed] [Google Scholar]
- Saxton, W.O. and Baumeister, W. 1982. The correlation averaging of a regularly arranged bacterial cell envelope protein. J. Microsc. 127 127–138. [DOI] [PubMed] [Google Scholar]
- Scherrer, K., Nothwang, H.G., Silva, P., Bey, F., Olin-Coux, M., Huesca, M., Coux, O., Arcangeletti, C., Chezzi, C., Buri, J.F., et al. 1990. The prosomes: Molecular and cellular biology. Mol. Biol. Rep. 14 75. [DOI] [PubMed] [Google Scholar]
- Seemüller, E., Lupas, A., Stock, D., Löwe, J., Huber, R., and Baumeister, W. 1995. Proteasome from Thermoplasma acidophilum: A threonine protease. Science 268 579–582. [DOI] [PubMed] [Google Scholar]
- Seemüller, E., Lupas, A., and Baumeister, W. 1996. Autocatalytic processing of the 20S proteasome. Nature 382 486–470. [DOI] [PubMed] [Google Scholar]
- Seemüller, E., Zwickl, P., and Baumeister, W. 2001. Self-processing of subunits of the proteasome. In Co- and posttranslational proteolysis of proteins, The enzymes, Vol. XXII, 3rd ed. (eds. R.E. Dalbey and D.S. Sigman), pp. 335–371. Academic Press.
- Stetefeld, J., Jenny, M., Schulthess, T., Landwehr, R., Engel, J., and Kammerer, R.A. 2000. Crystal structure of a naturally occurring parallel right-handed coiled coil tetramer. Nat. Struct. Biol. 7 772–775. [DOI] [PubMed] [Google Scholar]
- Tamura, T., Tamura, N., Cejka, Z., Hegerl, R., Lottspeich, F., and Baumeister, W. 1996. Tricorn protease—The core of a modular proteolytic system. Science 274 1385–1389. [DOI] [PubMed] [Google Scholar]
- Tamura, N., Lottspeich, F., Baumeister, W., and Tamura, T. 1998. The role of tricorn protease in its aminopeptidase-interacting factors in cellular protein degradation. Cell 95 637–648. [DOI] [PubMed] [Google Scholar]
- Trent, J.D., Nimmesgern, E., Wall, J.S., Hartl, F.U., and Horwich, A.L. 1991. A molecular chaperone from a thermophilic archaebacterium is related to the eukaryotic protein t-complex polypeptide-1. Nature 354 490–493. [DOI] [PubMed] [Google Scholar]
- Typke, D., Dierksen, K., and Baumeister, W. 1991. Automatic electron tomography. In Proceedings of the 49th Annual Meeting of the Electron Microscopy Society of America (ed. W. Bailey), pp. 544–545. San Francisco Press, San Francisco, CA.
- Voges, D., Zwickl, P., and Baumeister, W. 1999. The 26S proteasome: A molecular machine designed for controlled proteolysis. Ann. Rev. Biochem. 68 1015–1068. [DOI] [PubMed] [Google Scholar]
- Walz, J., Tamura, T., Tamura, N., Grimm, R., Baumeister, W., and Koster, A.J. 1997. Tricorn protease exists as an icosahedral supermolecule in vivo. Mol. Cell 1 59–65. [DOI] [PubMed] [Google Scholar]
- Walz, J., Erdmann, A., Kania, M., Typke, D., Koster, A.J., and Baumeister, W. 1998. 26S proteasome structure revealed by three-dimensional electron microscopy. J. Struct. Biol. 121 19–29. [DOI] [PubMed] [Google Scholar]
- Wenzel, T. and Baumeister, W. 1995. Conformational constraints in protein degradation by the 20S proteasome. Nat. Struct. Biol. 2 199–204. [DOI] [PubMed] [Google Scholar]
- Wildhaber, I. and Baumeister, W. 1987. The cell envelope of Thermoproteus tenax: Three-dimensional structure of the surface layer and its role in shape maintenance. EMBO J. 6 1475–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildhaber, I., Santarius, U., and Baumeister, W. 1987. The three-dimensional structure of the surface protein of Desulfurococcus mobilis. J. Bacteriol. 169 5563–5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilk, S. and Orlowski, M. 1980. Cation-sensitive neutral endopeptidase: Isolation and specificity of the bovine pituitary enzyme. J. Neurochem. 35 1172–1182. [DOI] [PubMed] [Google Scholar]
- Zwickl, P., Lottspeich, F., Dahlmann, B., and Baumeister, W. 1991. Cloning and sequencing of the gene encoding the large (α-) subunit of proteasome from Thermoplasma acidophilum. FEBS Lett. 278 217–221. [DOI] [PubMed] [Google Scholar]
- Zwickl, P., Grziwa, A., Pühler, G., Dahlmann, B., Lottspeich, F., and Baumeister, W. 1992a. Primary structure of the Thermoplasma proteasome and its implications for the structure, function and evolution of the multicatalytic proteinase. Biochemistry 31 964–972. [DOI] [PubMed] [Google Scholar]
- Zwickl, P., Lottspeich, F., and Baumeister, W. 1992b. Expression of functional Thermoplasma acidophilum proteasomes in Escherichia coli. FEBS Lett. 312 157–160. [DOI] [PubMed] [Google Scholar]
- Zwickl, P., Voges, D., and Baumeister, W. 1999. The proteasome: A macro-molecular assembly designed for controlled proteolysis. Philos. T. Roy. Soc. B. 354 1501–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwickl, P., Seemüller, E., Kapelari, B., and Baumeister, W. 2002. The proteasome: A supramolecular assembly designed for controlled proteolysis. In Protein folding in the cell (eds. F.M. Richards et al.), pp. 187–222. Academic Press, San Diego, CA. [DOI] [PubMed]