

Abstract
Evidence from proteins and peptides supports the conclusion that intrapeptide hydrogen bonds stabilize the folded form of proteins. Paradoxically, evidence from small molecules supports the opposite conclusion, that intrapeptide hydrogen bonds are less favorable than peptide–water hydrogen bonds. A related issue—often lost in this debate about comparing peptide–peptide to peptide– water hydrogen bonds—involves the energetic cost of an unsatisfied hydrogen bond. Here, experiment and theory agree that breaking a hydrogen bond costs between 5 and 6 kcal/mol. Accordingly, the likelihood of finding an unsatisfied hydrogen bond in a protein is insignificant. This realization establishes a powerful rule for evaluating protein conformations.
Keywords: protein hydrogen bonds, protein stability, hydrogen bond satisfaction, protein folding
The contribution that hydrogen bonds make to protein stability has been an ongoing topic of research since Pauling’s seminal models of protein secondary structure (Pauling and Corey 1951; Pauling et al. 1951). Energetic questions about hydrogen-bonding in proteins are usually formulated in terms of a comparison between peptide–water and peptide–peptide hydrogen bonds. Here we examine a related question: Are all hydrogen bond donors and acceptors in proteins satisfied? This question prompted us to reanalyze earlier data and to suggest a hydrogen bonding hypothesis.
In their description of the α-helix, Pauling et al. (1951) asserted that the energy of the peptide N–H• • •O=C hydrogen bond was of order −8 kcal/mol, and that “such instability would result from the failure to form these bonds that we may be confident of their presence.” Pauling’s earlier estimate of the total protein hydrogen bond energy was −5 kcal/mol (Mirsky and Pauling 1936). From solution studies of urea dimers, Schellman estimated that an intrapeptide hydrogen bond would be enthalpically favored over a peptide–water hydrogen bond by ~1.5 kcal/mol (Schellman 1955). These and similar early studies led to the conclusion that the peptide hydrogen bond is a significant factor in stabilizing protein conformations.
This view was to change dramatically following a famous review by Kauzmann (1959), who invoked the thermodynamics of small model compounds to argue that stabilization of the folded state of a protein is due almost exclusively to the hydrophobic effect. Soon after Kauzmann’s proposal, Klotz and Franzen (1962) determined that the enthalpy of the interamide hydrogen bond of N-methyl acetamide in water was zero, and concluded that “the intrinsic stability of interpeptide hydrogen bonds in aqueous solution is small.” Similarly, hydrogen bonding involving another small molecule, ɛ-caprolactam, in dilute solution was shown to be negligible (Susi and Ard 1969). Kauzmann’s proposal, bolstered by these later studies, led to the widely held view that the hydrophobic effect makes the major energetic contribution to protein stability, with hydrogen bonds contributing little, or perhaps even opposing, the folding process. See Baldwin (2003) for a recent discussion of these issues.
The accumulation of high-resolution X-ray crystal structures of proteins in the 1980s prompted several major surveys of hydrogen bonding in proteins (Baker and Hubbard 1984; Stickle et al. 1992; Savage et al. 1993; McDonald and Thornton 1994). These studies concur in finding that most buried polar groups (~90%) in globular proteins are hydrogen-bonded and that most intrapeptide hydrogen bonds are within elements of secondary structure: α-helices, β-sheets, and β-turns.
Concurrent with these surveys, Scholtz et al. (1991) determined that the enthalpy of helix formation for polyalanine in water is favorable by ~1 kcal/mol/hydrogen bond, measured using calorimetry, and this value can be further enhanced by burial and dehydration (Baldwin and Rose 1999; Fernandez et al. 2002). Similar helix formation enthalpies of peptides containing different amino acid residues have recently been reported (Richardson et al. 2005). Makhatadze and Privalov (1993) estimated that the enthalpy of an intrapeptide hydrogen bond buried in the protein interior could be as large as −12 kcal/mol (see also Fig. 1 ▶ in Rose and Wolfenden 1993).
Figure 1.

Unsatisfied main chain hydrogen bonds during molecular dynamics simulations. Data are shown for molecular dynamics simulations of the 82 residue syntenin PDZ2 domain (1r6j) (162 main chain donors and acceptors and two PRO residues). The X-ray crystal structure of this protein (Kang et al. 2004) has one internal main chain unsatisfied hydrogen bond donor, LEU233 N.
The observed ubiquity of intrapeptide hydrogen bonds in X-ray structures and the experimental demonstration of their favorable enthalpy of formation in peptides and proteins are consistent with the view that intrapeptide hydrogen bonds are favored over peptide–water hydrogen bonds. In contrast to this view, Honig and colleagues (BenTal et al. 1997) used finite difference Poisson- Boltzmann methods to calculate the energetics of hydrogen bonding of N-methyl acetamide in water and organic solvent. A thermodynamic cycle using these energy values indicates that the formation of N–H• • •O=C in water and subsequent transfer to a nonpolar solvent is disfavored by several kcal/mol (cf. Fig. 1 ▶ in BenTal et al. 1997). The investigators concluded “that the formation and burial of a hydrogen bond opposes protein folding” (BenTal et al. 1997). These conflicting ideas provoked controversy over whether or not such bonds contribute to overall protein stability (Fersht and Serrano 1993; Honig and Yang 1995; Lazaridis et al. 1995; BenTal et al. 1997).
Adding fuel to the fire, Myers and Pace used experimentally determined free energy differences from numerous single-residue polar to apolar mutations to argue that “hydrogen bonds stabilize proteins and that the average net stabilization is −1 to −2 kcal/mol per intramolecular hydrogen bond” (Myers and Pace 1996) with buried residues contributing as much as −3.5 kcal/mol (Shirley et al. 1992). Their conclusion was later corroborated in a study of lysozyme mutants (Takano et al. 1999).
Summarizing this ongoing discussion, the weight of present evidence from peptides and proteins favors the conclusion that an intrapeptide hydrogen bond stabilizes a protein by 1–2 kcal/mol. However, this conclusion has yet to be reconciled with small molecule experiments and calculations, perhaps owing to failure of group additivity (Roseman 1988; Avbelj et al. 2000).
Hydrogen bond satisfaction
An important realization is often overlooked in this comparison between protein–protein and protein–water hydrogen-bonding energy: An “unsatisfied” hydrogen bond donor or acceptor in the interior of a protein will destabilize a protein far more than 1–2 kcal/mol. Both experimental and theoretical studies concur that the enthalpic cost of breaking a hydrogen bond is at least 5–6 kcal/mol (Kresheck and Klotz 1969; Mitchell and Price 1990, 1991; Makhatadze and Privalov 1993; BenTal et al. 1997; Sheu et al. 2003).
Accordingly, unsatisfied buried polar groups are unlikely. The expected Boltzmann-weighted frequency of occurrence of an unsatisfied hydrogen bond can be estimated as
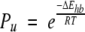 |
(1) |
where Pu is the probability of an unsatisfied hydrogen bond conformation relative to the probability of a comparable conformation with hydrogen bond satisfaction, ΔEhb is the energy of a hydrogen bond (~−5 kcal/mol), R is the gas constant, and T is the temperature. From this very approximate estimate, a conformation with an unsatisfied hydrogen bond donor or acceptor would have a relative probability (Pu) of ~0.02% at room temperature. Although in principle it is conceivable that the local energy penalty of an unsatisfied hydrogen bond could be compensated by the global system energy, this trade-off seems unlikely because proteins are energy mimimized locally (Butterfoss and Hermans 2003). In fact, proteins that do experience energetic deviations of this magnitude would be expected to be partially or even wholly unfolded, as shown in those cases that were studied (Rumbley et al. 2001).
Hydrogen-bonding hypothesis
We hypothesize that all potential hydrogen bond donors and acceptors in proteins are satisfied a significant fraction of the time, either by intramolecular hydrogen bonds or by hydrogen bonds to solvent water. In particular, the energetic cost of an unsatisfied, buried hydrogen bond is so steep that the population of nonhydrogen-bonded polar groups in the protein interior will be negligible. Here we define hydrogen bond satisfaction to mean that each donor and acceptor will have at least one hydrogen bond. A carbonyl oxygen may accept two hydrogen bonds, each directed to a lone pair of electrons, and both bonds are assumed to form with water in a fully solvated peptide group. One of these two respective hydrogen bonds is lost upon protein folding for a significant number of protein oxygen acceptors, a result previously referred to as lost hydrogen bonds (Savage et al. 1993). In small molecule crystals both single and double hydrogen bonded sp2 oxygens are frequently observed; completely unsatisfied oxygens are only very rarely observed (Taylor and Kennard 1984). In the work reported here, oxygen atoms with only one hydrogen bond are considered to be satisfied.
The hydrogen-bonding hypothesis implies that polypeptide conformations which are incompatible with complete hydrogen bond satisfaction would not contribute significantly to the population. This hypothesis can provide a powerful criterion for filtering calculated protein and peptide structures.
Reconciliation of hydrogen bond satisfaction with database surveys
If the Protein Data Bank (PDB) represents an equilibrium distribution of protein conformations (Shortle 2003), one would expect to find approximately one unsatisfied hydrogen bond per every 5000 polar groups (0.02%), not one in 10 as found in previous surveys (McDonald and Thornton 1994). To test our hypothesis, we analyzed the subset of “highly unsatisfied” protein main-chain donors and acceptors from McDonald and Thornton (1994) for which structure factors are available and electron density maps can be calculated (Kleywegt et al. 2004). In every instance, the apparent lack of satisfaction could be rationalized (Table 1). Reasons vary: Often, the group in question is occluded by a side chain with low electron density, where an alternate side-chain rotamer allows solvent access. In some cases, another experimentally indistinguishable side-chain rotamer of GLN, ASP, or THR would have provided a hydrogen bond partner for the atom in question. In several cases, a crystallographic water is situated nearby, although accessibility calculations score the donor/acceptor atom as solvent-inaccessible (see Savage et al. 1993 and Sadasivan et al. 1998).
Table 1.
Reconciling apparent main-chain donor/acceptor lack of satisfaction from the database
| PDB codea | Donor/acceptor | Rationalizationb |
| 1ake | PRO9A O | Occluding side chain (ILE116) has poor densityc |
| GLN28A N | Potential MET21 SD bondd | |
| LYS157A N | Occluding side chain (LYS157) has poor densityc | |
| 1cob | ASP81A O | Potential ARG77 NH1/NH2 bondd |
| ASN137A N | Potential THR135 OG1 bondd | |
| 1snc | LEU108 O | Potential THR41 OG1 bonde |
| 1ubq | ILE36 N | Potential ILE30 O bond (4.26 Å)e |
| 2aza | CYS3A N | Occluding side chain (GLN2) has poor densityc |
| GLU4A N | Occluding side chain (GLN2) has poor densityc | |
| 2cyp | TYR71 O | Occluding side chain (PHE77) has poor densityc |
| GLN86 N | Occluding side chain (GLN86) has poor densityc | |
| PHE91 O | Occluding side chain (LYS29) has poor densityc | |
| PRO122 O | Potential GLN117 OE1/NE2 bondd | |
| LEU161 O | Potential ASN272 OD1/ND2 bondd | |
| GLU188 N | Potential GLN222 OE1/NE2 bondd | |
| LEU238 O | Potential LEU245 N bonde | |
| 2hmz | VAL21A N | Occluding side chain (ILE20) has poor densityc |
| HIS43A N | Potential ASN40 OD1/ND2 bondd | |
| 2scp | ASN85A N | Potential MET82 O bond (4.06 Å)e |
| 4bp2 | ASN71 O | Potential GLN4 OE1/NE2 bondd |
| 5cyt | VAL20R O | Potential HOH22 bond |
| 6xia | TRP15 N | Potential ASN56 OD1/OD2 bondd |
| GLY21 N | Potential TRP19 ring bond, potential HOH6 bond | |
| PHE93 N | Potential ASN91 OD1/ND2 bondd | |
| GLY138 O | Potential ARG187 N bond (4.05 Å)e | |
| LYS148 N | Potential GLY145 O bond (4.01 Å)e | |
| ARG187 N | Potential GLY138 O bond (see above) | |
| ASN214 O | Potential GLU180 OE1/OE2 bondd |
aPDB identification code for structural models identified as having unsatisfied main-chain hydrogen bond donors and acceptors (McDonald and Thornton 1994) and for which satisfactory electron density maps could be calculated by the Uppsala electron density server (Kleywegt et al. 2004).
b The apparently unsatisfied donor or acceptor was classified as satisfied upon inspection of the PDB structure together with solvent molecules and the electron density map. In each case an explanation is noted and further described by one of the respective footnotes below.
c The side chain of a residue occluding the hydrogen bond donor or acceptor is in a region of poor electron density, and an alternate rotamer would allow solvent access to the unsatisfied group.
d A different rotamer of the occluding side chain would enable a potential intrapeptide hydrogen bond.
e Distance or orientation between N and O is slightly outside threshold criteria, but these atoms would interact weakly nevertheless.
Inspection of individual static X-ray crystallographic structures, even when combined with electron density maps, may be inadequate to evaluate hydrogen bond satisfaction. For example, the ribonuclease structure 7rsa from the PDB (Berman et al. 2000) has an unsatisfied main-chain N–H donor in residue GLU49. However, comparison of 17 available, independent X-ray crystal structures of this same molecule offers a more complete picture of how this ostensibly unsatisfied group may, in fact, interact with water. In four structures, the group is buried and unsatisfied, but it is solvent-accessible in the remaining 13 structures. The unsatisfied examples are 1fs3, 4rat, 7rsa, and 8rat; satisfied are 1bel, 1rat, 1rbx, 1rha, 1rhb, 2rat, 3rat, 3rn3, 5rat, 5rsa, 6rat, 7rat, and 9rat. This variation in solvent accessibilities for the same molecule, as represented by multiple, independent crystal structures, can be interpreted to mean that in solution, conformational variation would permit water access to the group in question a significant fraction of time. Indeed, the actual conformational fluctuations experienced by the protein in solution are likely to be even larger than those represented by a population of crystal structures, and therefore, this group should probably not be classified as unsatisfied.
It follows from these observations that most unsatisfied hydrogen bond donors and acceptors seen in database surveys are artifacts that arise from limitations in identifying hydrogen bonds by applying geometric criteria to static structures. In general, instances in which main-chain polar groups ostensibly lack hydrogen bond partners are an unavoidable consequence of basing the analysis on a time-averaged crystal structure.
Hydrogen bond satisfaction and simulations
Given the high energetic cost of an unsatisfied main-chain polar group, almost all such groups would be expected to participate in hydrogen bonds in the conformational microstates modeled by simulations. Molecular dynamics studies, which simulate time-dependent trajectories for individual molecules, should be useful for quantifying this prediction but, in fact, have provided contradictory results.
We find that molecular dynamics trajectories of small globular proteins in explicit solvent using the CHARMM force field result in conformations with more unsatisfied main-chain donors and acceptors than the respective crystal structures in the majority of the conformations sampled during simulation. Two simulations were performed at 300 K: one using the CHARMM22 force field, isobaric periodic boundary conditions (Berendsen et al. 1984), with a primitive cell of 62×62×62 Å , particle-mesh Ewald electrostatics (Darden et al. 1993), and the program NAMD (Kale et al. 1999). Bonds to hydrogen atoms were constrained with the SHAKE algorithm (Rychaert et al. 1977). The second simulation was with the CHARMM27 (MacKerell et al. 1998) force field, spherical water solvation with at least five layers of water surrounding the protein and Coulombic electrostatics with a dielectric constant of 1 and the program CHARMM (Brooks et al. 1983). Both simulations included the TIP3P water model (Jorgensen et al. 1983). After minimization, heating, and equilibration, configurations were sampled every 0.1 psec for 100 psec. Hydrogen bond satisfaction was calculated with HBPLUS using the relaxed criteria described by McDonald and Thornton to obtain “highly unsatisfied” donors and acceptors (McDonald and Thornton 1994), except that alternative ASN, GLN, and HIS orientations were explored. Explicit water was included in the analysis; both buried and accessible unsatisfied groups are found and included in the analysis.
The distributions of unsatisfied hydrogen bonds during equilibrated simulations of an 82-residue globular protein are shown in Figure 1 ▶; similar results are obtained with other proteins. Two sets of simulation conditions were investigated, and they differed in solvation systems, versions of the force field, and methods of electrostatic calculations. Both give rise to two or more unsatisfied main chain donors or acceptors in 90% of the conformations during the trajectory.
In fact, molecular mechanics force fields are known to be deficient at reproducing a satisfactory hydrogen-bonding potential with regard to orientation parameters (Lii and Allinger 1998; Grzybowski et al. 2000; Fabiola et al. 2002; Morozov et al. 2004), although the hydrogen bond electrostatic interaction energy for partial charges calculated using the CHARMM force field is consistent with a value of −5 kcal/mol (Grzybowski et al. 2000; Buck and Karplus 2001; Morozov et al. 2004). The nature of hydrogen bonds formed during molecular dynamics simulations has been previously characterized (Buck and Karplus 2001), but the preliminary results in Figure 1 ▶ suggest the need for a more comprehensive investigation into this topic.
Hydrogen bond satisfaction and the unfolded state of proteins
The hydrogen-bonding hypothesis can be used as a powerful criterion to filter unlikely conformations in the unfolded state, as demonstrated by a simple computational experiment. Adopting penta-alanine as a model system (Hummer et al. 2001; Margulis et al. 2002; Mu et al. 2005), we generated a population of sterically allowed structures of Ala5 using Monte Carlo backbone torsion angle sampling; these conformations were scored for hydrogen bond satisfaction. In this exercise, the entire backbone torsional space (−180≤φ,ψ≤+180) was sampled at random for each residue, resulting in 51,727 sterically allowed peptide conformations in 107 attempts. Intrapeptide hydrogen bonds were identified using criteria similar to those described by Kortemme et al. (2003) (ψ≥90°, θ110°, O–N distance ≤4.5 Å ), while peptide-solvent hydrogen bonds were identified by probing five different positions within the cone of approach around either the N–H or C=O vectors with a pseudo-water oxygen atom, as described previously (Fleming et al. 2005). The peptide was classified as hydrogen-bonded to water when a water oxygen was sterically allowed in an orientation compatible with hydrogen bonding. This latter method can discriminate between conformations that can form strong hydrogen bonds with water and those that cannot, regardless of the possible presence of accessible N–H or C=O surface. Thus, our method is more stringent than the criteria of McDonald and Thornton (1994), where any accessible surface was scored as a successful solvent–peptide hydrogen bond. More stringent criteria are appropriate when culling unlikely structures from simulations, where atom positions do not represent averages, as they do in crystal structures. Of the 51,727 sterically allowed penta-alanine conformers, 28,558 were found to be hydrogen-bond satisfied, i.e., 45% of the sterically allowed population could be rejected as energetically infeasible. The absolute rejection rate will depend on polypeptide chain length, but hydrogenbond satisfaction is a useful metric of energetic feasibility at any chain length.
We have focused on satisfaction of main chain polar groups, ignoring side chain donors and acceptors. However, the argument can be extended to side chain groups as well. Hydrogen bond satisfaction can also be an effective criterion for the evaluation of protein structures determined by NMR (Lipsitz et al. 2002) and X-ray crystallography (Savage et al. 1993; Hooft et al. 1996; Fabiola et al. 2002) in addition to its use in assessing the unfolded population (Lindorff-Larsen et al. 2004).
Summary
Protein hydrogen bonds are ubiquitous, directional, and largely local, partitioning the polypeptide chain into α- and 310-helices, β-sheet, and β-turns. Together, these hydrogen-bonded backbone structures account for at least 75% of the conformation, on average, with remaining residues participating in both additional intramolecular hydrogen bonding and hydrogen bonding to water.
Unsatisfied backbone polar groups are energetically expensive, to the degree that they almost never occur. Previous database surveys found that ~10% of these groups fail to form hydrogen bonds, either internally or with water. However, prompted by the hydrogen-bonding hypothesis, we argue that these exceptions can be rationalized convincingly. In retrospect, Pauling’s instincts about the importance of hydrogen bonds in protein conformation seem well justified.
The difference between ~90% and ~100% hydrogen bond satisfaction is tantamount to the difference between a statistical trend and a rule. We suggest that this rule can serve as a powerful filter for assessing the merit of experimental structures and the validity of simulated conformations.
Acknowledgments
We thank Nicholas Panasik, Timothy Street, and Haipeng Gong for critical discussions; Nicholas Fitzkee for both discussion and coding help; and Buzz Baldwin, Ross Shiman, and Karen Fleming for critical reading of the manuscript and helpful suggestions. Robert Schleif performed the water droplet molecular dynamics simulations used in our analysis. Simulations with periodic boundary conditions were performed using NAMD, developed by the Theoretical and Computational Biophysics Group in the Beckman Institute for Advanced Science and Technology at the University of Illinois at Urbana–Champaign. Support from the Mathers Foundation is gratefully acknowledged.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051454805.
References
- Avbelj, F., Luo, P., and Baldwin, R.L. 2000. Energetics of the interaction between water and the helical peptide group and its role in determining helix propensities. Proc. Natl. Acad. Sci. 97 10786–10791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, E.N. and Hubbard, R.E. 1984. Hydrogen bonding in globular proteins. Prog. Biophys. Mol. Biol. 44 97–179. [DOI] [PubMed] [Google Scholar]
- Baldwin, R.L. 2003. In search of the energetic role of peptide hydrogen bonds. J. Biol. Chem. 278 17581–17588. [DOI] [PubMed] [Google Scholar]
- Baldwin, R.L. and Rose, G.D. 1999. Is protein folding hierarchic? I. Local structure and peptide folding. Trends Biochem. Sci. 24 26–33. [DOI] [PubMed] [Google Scholar]
- BenTal, N., Sitkoff, D., Topol, I.A., Yang, A.S., Burt, S.K., and Honig, B. 1997. Free energy of amide hydrogen bond formation in vacuum, in water, and in liquid alkane solution. J. Phys. Chem. B 101 450–457. [Google Scholar]
- Berendsen, H.J.C., Postma, J.P.M., van Gunsteren, W.F., Di Nola, A., and Haak, J.R. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81 3684–3690. [Google Scholar]
- Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., and Bourne, P.E. 2000. The Protein Data Bank. Nucleic Acids Res. 28 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks, B.R., Bruccoleri, R.E., Olafson, B.D., States, D.J., Swaminathan, S., and Karplus, M. 1983. CHARMM: A program for macromolecular energy, minimization and dynamics calculations. J. Comp. Chem. 4 187–217. [Google Scholar]
- Buck, M. and Karplus, M. 2001. Hydrogen bond energetics: A simulation and statistical analysis of N-methyl acetamide (NMA), water and human lysozyme. J. Phys. Chem. B 105 11000–11015. [Google Scholar]
- Butterfoss, G.L. and Hermans, J. 2003. Boltzmann-type distribution of side-chain conformation in proteins. Protein Sci. 12 2719–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden, T., Yourk, D.M., and Pedersen, L. 1993. Particle Mesh Ewald— An Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 98 10089–10092. [Google Scholar]
- Fabiola, F., Bertram, R., Korostelev, A., and Chapman, M.S. 2002. An improved hydrogen bond potential: Impact on medium resolution protein structures. Protein Sci. 11 1415–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez, A., Colubri, A., and Berry, R.S. 2002. Three-body correlations in protein folding: The origin of cooperativity. Physica A 307 235–259. [Google Scholar]
- Fersht, A.R. and Serrano, L. 1993. Principles of protein stability derived from protein engineering experiments. Curr. Opin. Struct. Biol. 3 75–83. [Google Scholar]
- Fleming, P.J., Fitzkee, N.C., Mezei, M., Srinivasan, R., and Rose, G.D. 2005. A novel method reveals that solvent water favors polyproline II over β-strand conformation in peptides and unfolded proteins: Conditional hydrophobic accessible surface area (CHASA). Protein Sci. 14 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzybowski, B.A., Ishchenko, A.V., DeWitte, R.S., Whitesides, G.M., and Shakhnovich, E.I. 2000. Development of a knowledge-based potential for crystals of small organic molecules: Calculation of energy surfaces for C=O–H–N hydrogen bonds. J. Phys. Chem. B 104 7293–7298. [Google Scholar]
- Honig, B. and Yang, A.S. 1995. Free energy balance in protein folding. Adv. Protein Chem. 46 27–58. [DOI] [PubMed] [Google Scholar]
- Hooft, R.W., Sander, C., and Vriend, G. 1996. Positioning hydrogen atoms by optimizing hydrogen-bond networks in protein structures. Proteins 26 363–376. [DOI] [PubMed] [Google Scholar]
- Hummer, G., Garcia, A.E., and Garde, S. 2001. Helix nucleation kinetics from molecular simulations in explicit solvent. Proteins 42 77–84. [PubMed] [Google Scholar]
- Jorgensen, W.L., Chandrasekhar, J., Madura, J.D., Impey, R.W., and Klein, M.L. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79 926–935. [Google Scholar]
- Kale, L., Skeel, R., Bhandarkar, M., Brunner, R., Gursoy, A., Krawetz, N., Phillips, J., Shinozake, A., Varadarajan, K., and Shchulten, K. 1999. NAMD2: Greater scalability for parallel molecular dynamics. J. Comput. Phys. 151 283–312. [Google Scholar]
- Kang, B.S., Devedjiev, Y., Derewenda, U., and Derewenda, Z.S. 2004. The PDZ2 domain of syntenin at ultra-high resolution: Bridging the gap between macromolecular and small molecule crystallography. J. Mol. Biol. 338 483–493. [DOI] [PubMed] [Google Scholar]
- Kauzmann, W. 1959. Factors in interpretation of protein denaturation. Adv. Protein Chem. 14 1–62. [DOI] [PubMed] [Google Scholar]
- Kleywegt, G.J., Harris, M.R., Zou, J.Y., Taylor, T.C., Wahlby, A., and Jones, T.A. 2004. The Uppsala electron-density server. Acta Crystallogr. D Biol. Crystallogr. 60 2240–2249. [DOI] [PubMed] [Google Scholar]
- Klotz, I.M. and Franzen, J.S. 1962. Hydrogen bonds between model peptide groups in solution. J. Am. Chem. Soc. 84 3461–3466. [Google Scholar]
- Kortemme, T., Morozov, A.V., and Baker, D. 2003. An orientation-dependent hydrogen bonding potential improves prediction of specificity and structure for proteins and protein–protein complexes. J. Mol. Biol. 326 1239–1259. [DOI] [PubMed] [Google Scholar]
- Kresheck, G.C. and Klotz, I.M. 1969. The thermodynamics of transfer of amides from an apolar to an aqueous solution. Biochemistry 8 8–12. [DOI] [PubMed] [Google Scholar]
- Lazaridis, T., Archontis, G., and Karplus, M. 1995. The enthalpic contribution to protein stability: Insights from atom-based calculations and statistical mechanics. Adv. Protein Chem. 47 231–306. [DOI] [PubMed] [Google Scholar]
- Lii, J.H. and Allinger, N.L. 1998. Directional hydrogen bonding in the MM3 force field: II. J. Comp. Chem. 19 1001–1016. [Google Scholar]
- Lindorff-Larsen, K., Kristjansdottir, S., Teilum, K., Fieber, W., Dobson, C.M., Poulsen, F.M., and Vendruscolo, M. 2004. Determination of an ensemble of structures representing the denatured state of the bovine acyl-coenzyme a binding protein. J. Am. Chem. Soc. 126 3291–3299. [DOI] [PubMed] [Google Scholar]
- Lipsitz, R.S., Sharma, Y., Brooks, B.R., and Tjandra, N. 2002. Hydrogen bonding in high-resolution protein structures: A new method to assess NMR protein geometry. J. Am. Chem. Soc. 124 10621–10626. [DOI] [PubMed] [Google Scholar]
- MacKerell, A., Bashford, D., Bellott, M., Dunbrack, R., Evanseck, J., Field, M., Fischer, S., Gao, J., Guo, H., Ha, S., et al. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. 102 3586–3616. [DOI] [PubMed] [Google Scholar]
- Makhatadze, G.I. and Privalov, P.L. 1993. Contribution of hydration to protein folding thermodynamics. I. The enthalpy of hydration. J. Mol. Biol. 232 639–659. [DOI] [PubMed] [Google Scholar]
- Margulis, C.J., Stern, H.A., and Berne, B.J. 2002. Helix unfolding and intramolecular hydrogen bond dynamics in small α-helices in explicit solvent. J. Phys. Chem. B 106 10748–10752. [Google Scholar]
- McDonald, I.K. and Thornton, J.M. 1994. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 238 777–793. [DOI] [PubMed] [Google Scholar]
- Mirsky, A.E. and Pauling, L. 1936. On the structure of native, denatured, and coagulated proteins. Proc. Natl. Acad. Sci. 22 439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, J.B.O. and Price, S.L. 1990. The nature of the N–H O=C hydrogen-bond—An intermolecular perturbation-therory study of the formamide formaldehyde complex. J. Comp. Chem. 11 1217–1233. [Google Scholar]
- ———. 1991. On the relative strengths of amide...amide and amide...water hydrogen-bonds. Chem. Phys. Lett. 180 517–523. [Google Scholar]
- Morozov, A.V., Kortemme, T., Tsemekhman, K., and Baker, D. 2004. Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations. Proc. Natl. Acad. Sci. 101 6946–6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu, Y., Nguyen, P.H., and Stock, G. 2005. Energy landscape of a small peptide revealed by dihedral angle principal component analysis. Proteins 58 45–52. [DOI] [PubMed] [Google Scholar]
- Myers, J.K. and Pace, C.N. 1996. Hydrogen bonding stabilizes globular proteins. Biophys. J. 71 2033–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauling, L. and Corey, R.B. 1951. The pleated sheet, a new layer configuration of polypeptide chains. Proc. Natl. Acad. Sci. 37 251–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauling, L., Corey, R.B., and Branson, H.R. 1951. The structure of proteins; two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. 37 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson, J.M., Lopez, M.M., and Makhatadze, G.I. 2005. Enthalpy of helix-coil transition: Missing link in rationalizing the thermodynamics of helix-forming propensities of the amino acid residues. Proc. Natl. Acad. Sci. 102 1413–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose, G.D. and Wolfenden, R. 1993. Hydrogen bonding, hydrophobicity, packing, and protein folding. Annu. Rev. Biophys. Biomol. Struct. 22 381–415. [DOI] [PubMed] [Google Scholar]
- Roseman, M.A. 1988. Hydrophilicity of polar amino acid side-chains is markedly reduced by flanking peptide bonds. J. Mol. Biol. 200 513–522. [DOI] [PubMed] [Google Scholar]
- Rumbley, J., Hoang, L., Mayne, L., and Englander, S.W. 2001. An amino acid code for protein folding. Proc. Natl. Acad. Sci. 98 105–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychaert, J.P., Ciccotti, G., and Berendsen, H.J.C. 1977. Numerical integration of the Cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 23 327–341. [Google Scholar]
- Sadasivan, C., Nagendra, H.G., and Vijayan, M. 1998. Plasticity, hydration and accessibility in ribonuclease A. The structure of a new crystal form and its low-humidity variant. Acta Crystallogr. D Biol. Crystallogr. 54 1343–1352. [DOI] [PubMed] [Google Scholar]
- Savage, H.J., Elliott, C.J., Freeman, C.M., and Finney, J.L. 1993. Lost hydrogen bonds and buried surface area: Rationalising stability in globular proteins. J. Chem. Soc. Faraday Trans. 89 2609–2617. [Google Scholar]
- Schellman, J.A. 1955. The stability of hydrogen-bonded peptide structures in aqueous solution. C. R. Trav. Lab. Carlsberg [Chim] 29 230–259. [PubMed] [Google Scholar]
- Scholtz, J.M., Marqusee, S., Baldwin, R.L., York, E.J., Stewart, J.M., Santoro, M., and Bolen, D.W. 1991. Calorimetric determination of the enthalpy change for the α-helix to coil transition of an alanine peptide in water. Proc. Natl. Acad. Sci. 88 2854–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu, S.Y., Yang, D.Y., Selzle, H.L., and Schlag, E.W. 2003. Energetics of hydrogen bonds in peptides. Proc. Natl. Acad. Sci. 100 12683–12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirley, B.A., Stanssens, P., Hahn, U., and Pace, C.N. 1992. Contribution of hydrogen bonding to the conformational stability of ribonuclease T1. Biochemistry 31 725–732. [DOI] [PubMed] [Google Scholar]
- Shortle, D. 2003. Propensities, probabilities, and the Boltzmann hypothesis. Protein Sci. 12 1298–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stickle, D.F., Presta, L.G., Dill, K.A., and Rose, G.D. 1992. Hydrogen bonding in globular proteins. J. Mol. Biol. 226 1143–1159. [DOI] [PubMed] [Google Scholar]
- Susi, H. and Ard, J.S. 1969. Hydrophobic interactions and hydrogen bonding of epsilon–caprolactam in aqueous solution. J. Phys. Chem. 73 2440–2441. [Google Scholar]
- Takano, K., Yamagata, Y., Funahashi, J., Hioki, Y., Kuramitsu, S., and Yutani, K. 1999. Contribution of intra- and intermolecular hydrogen bonds to the conformational stability of human lysozyme. Biochemistry 38 12698–12708. [DOI] [PubMed] [Google Scholar]
- Taylor, R. and Kennard, O. 1984. Hydrogen-bond geometry in organic crystals. Acc. Chem. Res. 17 320–326. [Google Scholar]