

Abstract
Eukaryotic cells have evolved DNA damage checkpoints in response to genome damage. They delay the cell cycle and activate repair mechanisms. The kinases at the heart of these pathways and the accessory proteins, which localize to DNA lesions and regulate kinase activation, are conserved from yeast to mammals. For Saccharomyces cerevisiae Rad9, a key adaptor protein in DNA damage checkpoint pathways, no clear human ortholog has yet been described in mammals. Rad9, however, shares localized homology with both human BRCA1 and 53BP1 since they all contain tandem C-terminal BRCT (BRCA1 C-terminal) motifs. 53BP1 is also a key mediator in DNA damage signaling required for cell cycle arrest, which has just been reported to possess a tandem Tudor repeat upstream of the BRCT motifs. Here we show that the major globular domain upstream of yeast Rad9 BRCT domains is structurally extremely similar to the Tudor domains recently resolved for 53BP1 and SMN. By expressing several fragments encompassing the Tudor-related motif and characterizing them using various physical methods, we isolated the independently folded unit for yeast Rad9. As in 53BP1, the domain corresponds to the SMN Tudor motif plus the contiguous HCA predicted structure region at the C terminus. These domains may help to further elucidate the structural and functional features of these two proteins and improve knowledge of the proteins involved in DNA damage.
Keywords: protein domain, Tudor domain, yeast Rad9, mouse 53BP1, DNA damage, cell cycle checkpoint
Whatever the stress causing it, DNA damage threatens cellular homeostasis and life. Cellular response must then be fast, efficient, and coordinated. DNA repair and checkpoint signaling pathways, in particular, crosstalk and are interdependent (Weinert and Hartwell 1989; Zhou and Elledge 2000; Ward et al. 2003; Motoyama and Naka 2004). Kinases lie at the heart of DNA damage checkpoint signaling pathways. Adapter/mediator proteins, which facilitate interactions between upstream and downstream checkpoint kinases and regulate kinase activation, are also essential and conserved from yeast to mammals (Melo and Toczyski 2002; Nyberg et al. 2002; Rouse and Jackson 2002).
Many specific nuclear functions are performed by relatively small three-dimensional structures that have evolved independently and might be shared by related proteins. Furthermore, from a biochemical point of view, domains are considered to be the basic units of protein folding, evolution, and function (Wetlaufer 1973; Janin and Chothia 1985). They can be determined using computer methods (Doerks et al. 2002). As a large protein, 53BP1 (1972 amino acids) is composed of several domains, some of which have yet to be mapped and resolved to improve our understanding of its functions. The central region of 53BP1 encompassing fragment 1487–1532, which was predicted by PSIBLAST (Iwabuchi et al. 2003) to possess a Tudor motif, was indeed recently shown to consist of Tudor tandem repeats (Charier et al. 2004b; Huyen et al. 2004). The Tudor domain was named from the Drosophila Tudor protein in which it was first identified as a 10-fold repeat (Callebaut and Mornon 1997b; Ponting 1997a,b). It is a domain of ~50–70 amino acids found in many eukaryotic proteins that colocalize with ribonucleoproteins or single-stranded DNA-associated complexes in the nucleus, mitochondrial membrane, or kinetochores (Kawahire et al. 1997) (Prosite entry PDOC50304). Its topology is characterized by a five β-strand motif and four connecting loops forming a barrellike five-stranded structure, as established by NMR and X-ray crystallography resolution of the human SMN Tudor domain (Selenko et al. 2001; Sprangers et al. 2003a,b). Other domains that are evolutionarily related to the Tudor domain have also been recently described (Maurer-Stroh et al. 2003). These include the PWWP domain (Stec et al. 2000), which has been structurally characterized (Qiu et al. 2002) and can be defined as a five-stranded, anti-parallel β-barrel, very similar to the β-barrel in the Tudor domain, with a C-terminal α-helix packing onto it (Slater et al. 2003).
In this paper we focus our attention on Rad9, the first eukaryote DNA damage protein discovered in the yeast Saccharomyces cerevisiae (Sc) some 15 yr ago (Schiestl et al. 1989; Weinert and Hartwell 1989). ScRad9 was reported to protect cells from genomic instability by delaying progress in the cell cycle (Siede et al. 1994; Weinert et al. 1994). It is also involved in another signal linked to DNA repair, participating in the transcriptional response to DNA damage by controlling induction of a large “regulon” of repair, replication, and recombination genes (Aboussekhra et al. 1996). A wealth of functional data is available for ScRad9 but no direct mammalian ortholog has yet been reported.
On the basis of their common BRCT domains, both 53BP1 and BRCA1 were positioned as potential human orthologs of ScRad9 protein (Zhou and Elledge 2000). BRCT domains, initially described in mammalian tumor suppressor protein BRCA1 (Koonin et al. 1996), have now been identified in >50 different proteins taken from the various species involved in the cell’s response to DNA damage repair and cell cycle checkpoints (Bork et al. 1997; Callebaut and Mornon 1997a; Huyton et al. 2000). Moreover, both proteins have characteristics allowing the comparison with ScRad9 in terms of cell cycle delay and transcriptional response. In response to DNA damage, both BRCA1 and 53BP1 are indeed hyperphosphorylated by ATM and colocalize in DNA repair foci (DiTullio et al. 2002; Fernandez-Capetillo et al. 2002; Wang et al. 2002). Both proteins have also been shown to enhance transcriptional activation of p53 (Iwabuchi et al. 1998; Zhang et al. 1998).
Based on a bioinformatics study, we predicted that ScRad9 would include a Tudor motif, highly homologous with mouse (human) 53BP1 and SMN Tudor domains. To prove the existence of a Tudor-related domain for ScRad9 with a high degree of homology with the corresponding 53BP1 domain, we designed, purified to homogeneity, and characterized several fragments encompassing the Tudor-related motif. We thus succeeded in isolating a new structural domain for ScRad9, located, as is 53BP1, upstream of the BRCT domains.
Results
A Tudor-related domain in ScRad9 and m53BP1
The proteins involved in DNA damage response pathways are conserved from yeast to mammals. In the conserved family, including budding yeast Rad9 and fission yeast Crb2, the tandem BRCT repeat at the ScRad9 C terminus, is the only region showing appreciable sequence identity with other proteins. For this reason, hBRCA1 and h53BP1, which also share a number of regulatory similarities with yeast Rad9 (Lowndes and Murguia 2000), are possible human ortholog candidates. The schematic representations in Figure 1A ▶ show that, despite their large size, only a few domains have been identified and isolated in ScRad9, h53BP1, and hBRCA1. For further research into sequence similarities between these proteins, a secondary structure prediction was obtained through the PSIpred v2.4 Web-interface facilities (McGuffin et al. 2000). This predicted the presence of a large structural region upstream of the BRCT motifs for both ScRad9 (residues 724–977 and m53BP1 (residues 1433–1637, 94% identical to the corresponding region of human 53BP1) but no such structural region for hBRCA1 (residues 1438–1643). Hydrophobic cluster analysis (HCA) was used to compare the secondary structures. The HCA plots for these structured regions (Fig. 1B ▶) show similarities in hydrophobic cluster distribution. Many of the hydrophobic cluster motifs are vertically shaped, corresponding to β-sheet structures. This segment [1433–1637] of m53BP1 includes the region that allows 53BP1 to form foci in response to DNA damage and contains a predicted Tudor domain (five β-strand barrel-like fold), conserved motif found in SMN protein. Multiple alignment of the SMN Tudor domain, the m53BP1 PSI-BLAST predicted Tudor domain, and the putative ScRad9 Tudor domain (five β-strands) was generated using ClustalW (Thompson et al. 1994) (Fig. 2A ▶). The three fragments display ~25% sequence similarity (over 91 amino acids) in addition to a striking resemblance regarding their secondary structure prediction. Of the residues distributed along the length of the motif, seven are identical, nine show conserved substitution, and seven show semiconserved substitution: Ala100, Trp102, Tyr109, Asp117, Gly129, Asp140, and Glu147; Gln91, Val94, Ile108, Ile113, Val125, Val127, Glu135, Leu138, and Leu142; and Gly95, Ser103, Glu104, Pro110, Ala111, Ser143, and Ala149, respectively, with the numbers referring to the human SMN sequence. These amino acids mostly indicate Tudor domain markers. The conserved Ala100, Ala111, Ile113, and Val125 are structurally important residues stabilizing the Tudor domain structure through the formation of a hydrophobic core. The conserved aromatic residues Trp102 and Tyr109 are also involved in Tudor domain structural organization. Furthermore, the predicted β-sheet structures perfectly match the experimental structural elements of SMN Tudor. HCA plots of the corresponding SMN Tudor sequences (Fig. 2B ▶) were split into four parts (a, b, c, and d, respectively) according to the multiple alignment limits based on those of the experimental SMN Tudor secondary structure elements (black vertical line). This segmentation clearly showed a similar distribution of hydrophobic clusters with similar characteristics between fragments m53BP1[1466–1535] and ScRad9[764–947] and the SMN Tudor domain. All these elements supported the existence of a similar three dimensional fold.
Figure 1.

(A). Schematic representations of S. cerevisiae Rad9, human 53BP1, and human BRCA1 primary sequence showing the location of conserved domains: (i) for ScRad9, two C-terminal BRCT domains—an [S/T]Q cluster domain (SCD) and a Chk1 activation domain (CAD). The region reported to interact with Rad53 FHA2 is indicated by a black horizontal line. (ii) for h53BP1, the PSI-Blast-predicted Tudor domain and two C-terminal BRCT domains. The regions reported to allow 53BP1 to form foci in response to DNA damage, and to bind to phosphorylated H2AX (-H2AX) in vitro, are indicated by a black horizontal line. (iii) for hBRCA1, two C-terminal BRCT domains, an SQ cluster domain, and an N-terminal RING finger domain. (B) Hydrophobic cluster analysis plots of the regions ScRad9[724–977] and m53BP1[1433–1637], with the positions of the experimental constructs. m53BP1[1433–1637] is 94% identical to the corresponding h53BP1 [1448–1652] segment. The standard one-letter code for amino acids is used except for proline (regular secondary structure breaker), glycine (the less constrained amino acid), serine, and threonine, which are represented by a star, a diamond, an empty rectangle, and a rectangle containing a black rectangle, respectively.
Figure 2.
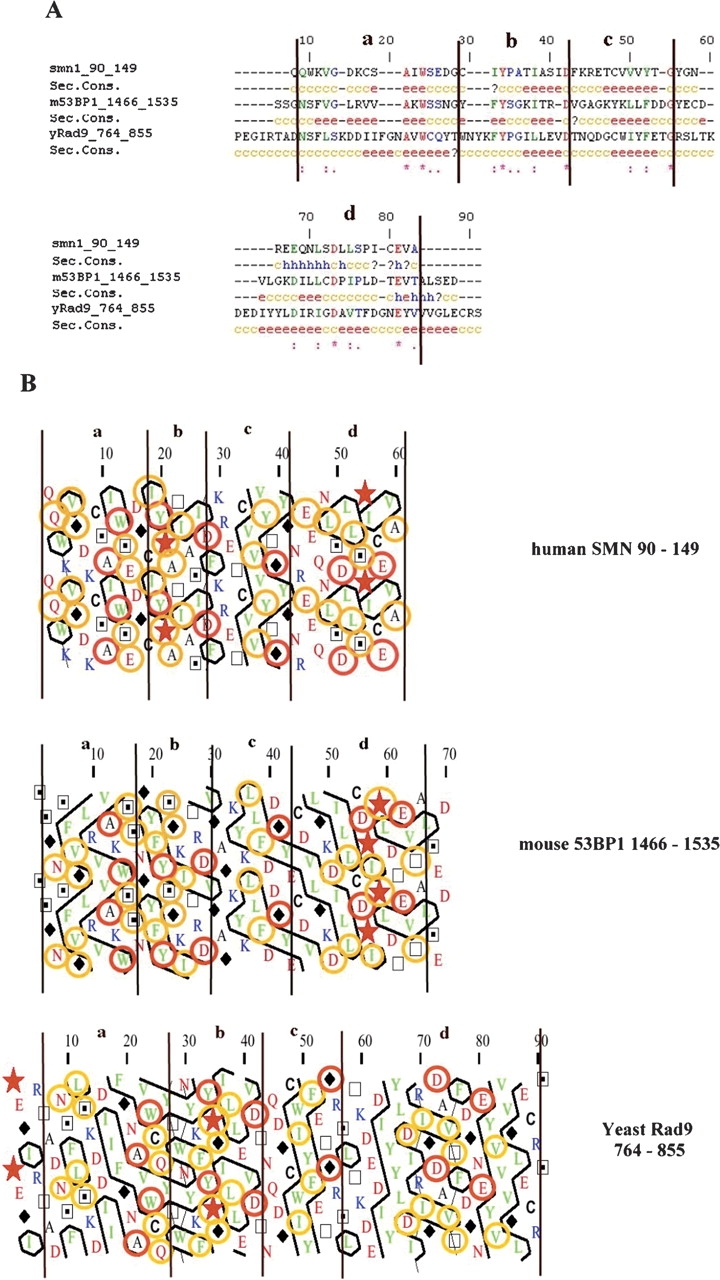
Multiple alignment and hydrophobic cluster analysis plots of the Tudor and Tudor putative motif of hSMN, m53BP1, and ScRad9. (A) ClustlalW multialignment of hSMN[90–149] Tudor domain, m53BP1[1466–1535] PSI-Blast-predicted Tudor motif, and ScRad9[764–855] putative Tudor motif. Secondary structure predictions are indicated below each sequence. e, extended or β-strand structure; c, coiled structure; h, helix. (B) HCA plots of hSMN[90–149], m53BP1[1466–1535], and ScRad9[764–855] . The vertical lines are based on the multialignment and correspond to four conserved parts: a, b, c, and d. The orange circles represent amino acids displaying similarity, and the red circles show conserved amino acids.
The presence of a highly structured region contiguous to the C-terminal Tudor motif was noted in both m53BP1 and ScRad9 (Fig. 1B ▶). In the case of the PWWP domain, the five-stranded anti-parallel β-barrel, which is very similar to the β-barrel in the Tudor domain, was incapable of independent folding when the C-terminal α-helix was missing (Slater et al. 2003). This analogy with the PWWP domain then also led to the consideration of larger segments as a possible structural module.
Molecular constructs: Design and cloning
To isolate the minimal region able to independently fold, we delimited two fragments for m53BP1: [1466– 1535] and [1463–1617]. Three fragments were studied for yeast Rad9: [764–855], [764–947], and [754–947]. All these fragments displayed a high probability for the formation of inclusion bodies as suggested by their high scores in the Wilkinson and Harrison (1991) equation. The insolubility percentages (IB score) were 59%, 56%, and 95%, respectively, for ScRad9 fragments [754–947], [764–947], and [764–855], and 52% and 80%, respectively, for m53BP1 fragments [1466–1535] and [1463–1617]. A fusion protein strategy was therefore used to increase the fraction of soluble gene products in the cytoplasm (Swartz 2001) and produce a suitable quantity for structural determination. There is no universal fusion tag for this purpose, so the most common tags were used: 13 kDa thioredoxin (TRX) (LaVallie et al. 1993) from Escherichia coli, known to dramatically increase the solubility of heterologous protein synthesis in E. coli translation efficiency, 26 kDa glutathione-S-transferase (GST) (Smith and Johnson 1988) from Schistosoma japonicum, and the very commonly used 6His tag (Hochuli 1988).
Recombinant plasmids encoding the selected fragments were obtained using either the Gateway cloning system (Walhout et al. 2000) to insert 6His and GST at the N terminus or ligation-independent cloning (LIC technology) (Aslanidis and de Jong 1990; Haun and Moss 1992; Haun et al. 1992) to obtain the TRX fusion partner. To remove the large fusion protein, the cleavage site for tobacco etch virus (TEV) was inserted into the Gateway vectors (Invitrogen) during insertion of the fragments obtained by PCR amplification, using forward primers modified at their 5′ end with the nucleotides that encode the “ENLYFQG” TEV canonical recognition site (Parks et al. 1994). Furthermore, the LIC vector used (Novagen) has internally different protease sites for TRX fusion tag cleavage.
Soluble fragment expression
Optimal growth conditions were first determined by testing induction at 37°C and 30°C, respectively, for 4 h and at 18°C overnight on small-scale cultures. For each condition, soluble/insoluble fraction distribution was qualitatively analyzed by SDS-PAGE and the yield of soluble fraction quantitatively assessed by small-scale purification using an affinity spin column (data not shown). If required, further optimization of induction conditions was investigated before scaling up the cultures. The results of the 3- to 8-L experiments are given in Table 1. Concerning short Tudor motif expressing fragments, a higher proportion of soluble expression was obtained from the TRX expression vectors by induction at low temperature for 15–22 h. The long fragments, both m53BP1[1463–1617] and ScRad9[754–947], were expressed as soluble with a high yield at 30°C in the form of GST fusion protein. The latter fragment was also expressed with a high yield at 30°C with the TRX fusion partner. Paradoxically, expression must be run at 18°C for 48 h to obtain soluble ScRad9[764–947] fused to GST with a medium yield.
Table 1.
Expression of m53BP1 and ScRad9 fragments in E. coli BL21star (DE3)
| Fragment boundaries | N-term fusiona | Expressionb | Yieldc |
| m53BP1[1466–1535] | 6-His | S (16°C, 22 h) | 2–5 mg/L |
| TRX-Stag | S (16°C, 15 h) | 65 mg/L | |
| GST | I | - | |
| m53BP1[1463–1617] | 6-His | I | - |
| GST | S (30°C, 15 h) | 70 mg/L | |
| scRad9[764–855] | 6-His | I | - |
| TRX-Stag | S (16°C, 22 h) | 2–5 mg/L | |
| GST | I | - | |
| scRad9[764–947] | 6-His | I | - |
| GST | S (16°C, 48 h) | 7 mg/L | |
| scRad9[754–947] | 6-His | I | - |
| TRX-Stag | S (30°C, 4 h) | 65 mg/L | |
| GST | S (30°C, 4 h) | 40 mg/L |
a(TRX) Thioredoxin; (GST) glutathione-S-transferase.
b (S) Soluble protein expression; (I) inclusion bodies or mostly aggregated after cell lysis. Optimal temperature and time conditions for IPTG induction are given in parentheses.
c Expression level of soluble protein after affinity chromatography.
Purification and proteolytic cleavage
For GST fusion protein fragments, clarified lysate binding to glutathione sepharose resin was efficient and specific. For 6His-tagged protein fragments, the efficiency of Ni-NTA resin purification appeared to depend strongly on the expression level and was straightforward for high-yield expressions only (50–70 mg/L protein level).
Biotinylated thrombin and 6His-TEV proteases were then used for site-specific cleavage of GST and TRX fusion protein fragments, respectively. With the knowledge that cleavage efficiency depends on the protein fused to the C terminus at the protease cleavage site, we optimized enzymatic conditions for each reaction (Table 2). We observed that, whereas a low enzyme-to-substrate ratio of 1% was adequate for complete GST-Sc Rad9[754–947] and GST-m53BP1[1463–1617] cleavage, a much higher proportion of TEV enzyme, i.e., 30% instead of 1%, was needed to cleave GST from GSTScRad9[764–947]. Moreover, the latter fragment appeared to be very difficult to isolate because it was prone to precipitation and aggregation from solution. To remove the TRX affinity tag efficiently, the corresponding fusion fragments were processed with 1 unit and 1.6 units of thrombin per milligram of target protein, which was generally sufficient, with 3–16 h incubation at 20°C, as recommended by the supplier. Under these low enzyme concentration conditions, TRX-ScRad9[754–947] was efficiently cleaved but not in the expected fragments. A higher enzyme-to-substrate ratio (25- to 30-fold) and a long incubation time (at least 20 h at 20°C) was required to get the right size fragments (data not shown).
Table 2.
Cleavage conditions for tag removal
| Protease | Fragmenta boundaries | E/S ratiob w/w (U/mg) | Reaction conditions | Efficiencyc |
| Thrombin | TRX-Stag yRad9[764–855] | 0.08% (1.6 U/mg) | 16 h, 20°C | ~Complete |
| TRX-Stag yRad9[754–947] | 1.85% (37 U/mg) | 16 h, 20°C | ~Complete | |
| TRX-Stag m53BP1[1466–1535] | 0.05% (1 U/mg) | 3 h, 20°C | ~Complete | |
| rTEV | GST scRad9[764–947] | 30% (3000 U/mg) | 16 h, 30°C | ~70%–80% |
| GST scRad9[754–947] | 1%–2% (150 U/mg) | 3 h, 30°C | ~Complete | |
| GST m53BP1[1463–1617] | 1%–2% (150 U/mg) | 3 h, 30°C | ~Complete |
a (TRX) Thioredoxin; (GST) glutathione-S-transferase.
b (E/S) Enzyme-to-substrate ratio.
cSDS-PAGE estimation of cleavage efficiency.
The protease was eliminated and the fragments recovered by affinity chromatography. Purification was quite straightforward for the TEV proteolyzed fragments. The residual GST was eliminated during subsequent gel filtration, when required. On the other hand, following thrombin proteolysis, difficulties were encountered for quantitative separation of the resulting cleaved Stag fragments from the TRX-6His N terminal, which was found to elute at a lower imidazole concentration than expected. Fragment identity was finally confirmed by Edman N-terminal amino-acid sequencing (data not shown).
Physical studies
The 6His-m53BP1[1466–1535], Stag-m53BP1[1466–1535], and ScRad9[764–947] fragments were difficult to characterize in detail because they are prone to aggregation and precipitation. Consequently, for these fragments, it was not always possible to obtain solutions at the concentration required for some of the physical measurements.
All the produced fragments were probed using size-exclusion chromatography, since this analytical study can be performed with solutions of micromolar range concentration. An analysis of the size-exclusion chromatography data is given in Figure 3A ▶ except for 6His-m53BP1[1466–1535], which did not emerge from the column. m53BP1[1463–1617] (17.4 kDa), ScRad9[754–947] (22.4 kDa), and Stag-ScRad9[754–947] (25.6 kDa) eluted as a single peak at elution volumes close to those expected for proteins of their molecular mass. A shift was observed for ScRad9[764–947], which eluted at a volume corresponding to a much higher molecular mass than expected (~60 kDa instead of 21 kDa). The short fragments, Stag-ScRad9[764–855] (13.9 kDa) and Stag-m53BP1[1466–1535] (11 kDa), eluted at the column void volume, which indicates the formation of high-order aggregates.
Figure 3.
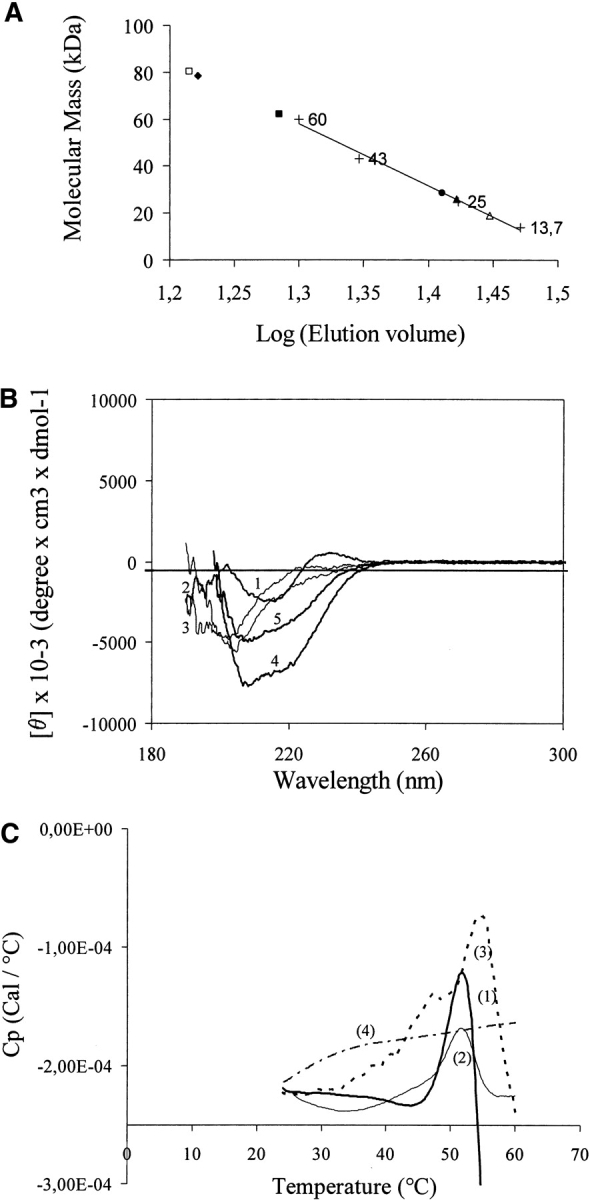
Physicochemical characteristics of Tudor-related fragments. (A) Size-exclusion chromatography of Tudor-related fragments from ScRad9 and m53BP1. The elution volume vs. molecular mass is on a logarithmic scale. The column (Superdex 75) was calibrated with globular proteins (+): ribonuclease A (13.7 kDa), chymotrypsin (25 kDa), ovalbumin (43 kDa), and albumin (60 kDa). The standard curve was drawn from molecular weight marker data, whereas the experimental elution volume of each fragment is spotted on the abscissa scale.Δ, m53BP1[1463–1617]; □, Stag-m53BP1[1466–1535]; ▴, ScRad9 [754–947]; •, Stag-ScRad9[754–947]; ▪, ScRad9[764–947]; ♦, Stag-ScRad9[764–855]. (B) Far-UV CD spectra of (1) m53BP1[1463–1617], (2) ScRad9[754–947], (3) Stag ScRad9[754–947], (4) Stag ScRad9[764–855], and (5) Stag-m53BP1[1466–1535]. (C) Microcalorimetric heat transitions are depicted as dependence of heat capacity on temperature for (1) m53BP1[1463–1617], (2) ScRad9[754– 947], (3) Stag-ScRad9[754–947], and (4) Stag-ScRad9[764–855]. Concentration of the heated protein solutions was 0.05mg•mL−1 (i.e., 2.9 μM, 2.2 μM, 1.95 μM, and 3.6 μM, respectively). Heat transitions exist for 1, 2, and 3.
To test for the presence of secondary structures, far-UV circular dichroism (CD) spectroscopy was used. The far-UV CD spectra measured for all the fragments are given in Figure 3B ▶ except for ScRad9[764–947]. It is noteworthy that all the spectra reflected the existence of secondary structure elements. Moreover, the CD spectra differed for the short and long fragments of ScRad9 and m53BP1, respectively. The results of estimated secondary structure content are given in Table 3. m53BP1[1463–1617] and ScRad9[754–947] spectra (with or without Stag at its N terminus) appeared to consist mainly of β structural components (~70%–75%), whereas the short fragments, Stag-m53BP1[1466–1535] and Stag-ScRad9[764–855], displayed β-sheet, α-helix, and β-turn distribution. For these latter, their spectra recorded a somewhat stronger minimum at 220 nm, indicating a higher average helical content.
Table 3.
Estimation of secondary structure content in the fragments
| Fragment | Helix (% content) | β (% content) | Turn (% content) | Random (% content) |
| m53BP1[1463–1617] | 6 ± 3 | 74 ± 0 | 0 | 19 ± 3 |
| Stag m53BP1 [1466–1535] | 12.4 ± 2 | 45 ± 5 | 14 ± 2 | 28.6 ± 3 |
| yRad9[754–947] | 0 ± 0 | 73 ± 3 | 0 ± 0 | 27 ± 3 |
| Stag yRad9[754–947] | 0 ± 0 | 69 ± 1 | 6 ± 0 | 25 ± 1 |
| yRad9[764–947] | N.D. | N.D. | N.D. | N.D. |
| Stag yRad9[764–855] | 22 ± 0 | 22 ± 7 | 24 ± 7 | 33 ± 0 |
The values were obtained by analyzing the CD spectra shown in Figure 3B ▶ according to Dr. Yang’s program (reference method of JASCO 810 apparatus). Values are given as the mean (± SD) of two experiments. (N.D.) Nondetermined.
The dependence of heat capacity on temperature reflects conformational transitions. The thermodynamic stability of the fragments produced was therefore measured by differential scanning calorimetry (DSC). A DSC scan for m53BP1[1463–1617], ScRad9[754–947], Stag-ScRad9[754–947], and Stag-ScRad9[764–855] fragments, respectively, is given in Figure 3C ▶ (curves 1–4). m53BP1[1463–1617] (curve 1), ScRad9 [754–947](curve 2), and Stag-ScRad9 [754–947] (curve 3) display cooperative transition upon melting. Their heat transitions can be approached by an irreversible two-state process, with midpoints of 52°, 52°, and 54°C, respectively. On the other hand, the DSC melting curve for ScRad9[764–855] did not display any cooperative heat absorption.
One-dimensional 1H NMR spectra in TBS or PBS were also recorded for fragments m53BP1[1463–1617], ScRad9[754–947], Stag-ScRad9[754–947], and Stag-ScRad9[764–855]. For the first three fragments, good dispersion of the amide and methyl proton signals revealed that these protein fragments are folded. On the contrary, no such signal was found in the short ScRad9 fragment (data not shown). 1H-15N HSQC spectra of uniformly 15N-labeled ScRad9[754–947] and m53BP1[1463–1617] were then recorded to confirm that the fragments were folded and to assess whether a three-dimensional structural determination could be made by NMR. For heteronuclear NMR structural determination, solutions of a fairly high concentration are necessary. Although a concentrated solution of the m53BP1fragment was easily obtained with phosphate and Tris buffer containing 150 mM NaCl, this was much more difficult for ScRad9[754–947]. To solve the problem, different buffers were tested. Finally a 50-mM MES buffer at pH 6.0, 50 mM NaCl, in which a stable concentration as high as 12–15 mg/mL could be obtained, was chosen. The CD spectrum of Rad9[754–947] recorded in this buffer showed insignificant alteration compared with the spectrum recorded in PBS at pH 7.3 (used for 1H NMR), indicating no particular structural modification (data not shown). NMR samples of 0.5–0.8 mM were therefore prepared in 50 mM Tris-HCl at pH 7.2 containing 150 mM NaCl form53BP1[1463–1617] and inMES atpH6.0, 50mM NaCl, for ScRad9[754–947]. Both 1H-15N HSQC spectra displayed good dispersion of the amide signals, as shown in Figure 4 ▶.
Figure 4.

1H-15N Heteronuclear Single-Quantum Coherence (HSQC) spectra. (A) Spectrum of m53BP1[1463–1617]. (B) Spectrum of ScRad9[754– 947].
Discussion
From the sequence, secondary structure prediction revealed that the ScRad9 segment [765–935] displayed globular domain characteristics and was predicted to contain a putative Tudor domain in the same way as the m53BP1 segment [1480–1616]. HCA analysis, which is known to work efficiently in detecting structural homology even with low sequence homology (Callebaut and Mornon 1997b), further confirmed the possible structural similarity. The Tudor domain (or SH3-like domain), like any other domain, was characterized by conserved residues identified from resolved Tudor structures (SMN, Tudor protein). Five residues (Ala100, Trp102, Tyr109, Gly129, and Asp 140, numbers referring to the SMN sequence), conserved in all SMN homologs and many other Tudor domain-containing proteins, were indeed found in m53BP1[1466–1535] and ScRad9[764–855] fragments.
To isolate the module capable of independent folding, two types of fragments were selected by hydrophobic cluster distribution. The short-type fragment was strongly homologous to the SMN Tudor domain. The long-type fragment included the contiguous region immediately following the Tudor fold as a supplementary C-terminal half. This latter fragment, predicted to contain a helical region, is reminiscent of PWWP domains. Short-fragment limits were determined considering the strict Tudor domain defined by hydrophobic residue characteristics. To avoid missing any structurally important residues, they were extended with additional residues at both ends to ScRad9[764–855] and m53BP1[1466–1535]. The long fragments were selected from the HCA plots: m53BP1[1463–1617] and ScRad9[764–947]. For ScRad9, a second long fragment [754–947] was also selected to take into account a hydrophobic cluster upstream of the Tudor domain.
Expression level
As previously reported (Schein 1990; Baneyx 1999), a lower growth temperature promoted the formation of soluble recombinant fragments. TRX-ScRad9[765–855], GST-ScRad9[764–947], 6His-m53BP1[1466–1535], and TRX-m53BP1[1466–1535] expression as soluble proteins was thus only observed at 16°C with a rather low yield except for the last one. Whatever the fragment, both TRX and/or GST clearly helped fragment expression in the form of soluble polypeptides, if not domains. On the other hand, the 6His tag, which does not modify the hydrophobic nature of the fragments, gave, at best, far lower expression yields than the GST or the TRX tag. The two ScRad9[754–947] and m53P1[1463–1617] fragments were expressed as soluble, no matter what the temperature and tag at very low levels (observable by Western blot with colorimetric detection) with the 6His tag, whereas the corresponding GST and/or TRX fusion proteins reached high levels in E. coli cytoplasm.
Cleavage
For structural studies, the fragment should be obtained free of its fusion partner. GST fusion protein was easily cleaved from GST-ScRad9[754–947] and GSTm53BP1[ 1463–1617] fusion proteins. On the other hand, a correct but more difficult cleavage was observed for GST-ScRad9[764–947]. This fragment was processed to 70%–80% at best, using a high enzyme-to-substrate ratio. This difficult cleavage may be linked to the existence of a proline in the P′2 position for this fragment. This position has been said to make a great contribution to rhinovirus 3C protease specificity (Cordingley et al. 1990), and this was found to be the case for TEV, a 3C-like protease. The TRX fusion partner was cleaved by thrombin, giving 33 extra amino acids at the N-terminal end of the cleaved fragments, whereas just one extra Gly amino acid residue was contributed to the N terminus by TEV digestion. The LVPR↓GS primary recognition sequence found downstream of the six histidines on the TRX fragments was cleaved very quickly. However, upstream of the six histidines, the IR↓G bond constituted a secondary site (Chang 1985) that could also be cleaved. A structural difference between TRX-ScRad9[754–947] on the one hand and TRX-m53BP1[1466–1535] or TRX-ScRad9[765–855] on the other could explain the need for more enzyme to access the primary cleavage site. The presence of the second cleavage site helps explain the difficulty in purifying the thrombin-cleaved fragments. The N-terminal TRX fragment generated probably also lost its 6His tag due to cleavage at the alternative site as it eluted at low imidazole concentration and polluted part of the Stag-fragment fractions.
Physical characterization
The hydrodynamic dimensions of a protein molecule are easily tested by gel filtration using a column calibrated with globular proteins of known molecular mass. Gel filtration clearly showed that ScRad9[754–947], Stag-ScRad9[754–947], and m53BP1[1463–1617] are monomeric and globular in solution. For the other fragments, the large apparent molecular masses mainly indicate a strong tendency toward self-association, leading to the formation of high-order aggregates, soluble aggregates in the case of ScRad9[764–855]. The CD results indicated that all the tested fragments had a secondary structure, even the short fragments present as aggregates, implying that they are not fully unfolded. The long fragments, 53BP1[1463–1617], Rad9[754–947], and Stag-Rad9[754–947], appear to have a thermodynamically stable ordered structure, since they displayed cooperative transition upon melting, a characteristic of a rigidly packed protein structure (Privalov and Potekhin 1986). In contrast, the DSC scan for the short fragment, ScRad9[765–855], confirmed that its structure is very flexible without rigid packing.
The one-dimensional 1H NMR spectra led to the conclusion that only ScRad9[754–947], with or without Stag, and m53BP1[1463–1617] were folded fragments, confirming the conclusions of size-exclusion and DSC analyses. Both 1H-15N HSQC spectra indicated a compact globular protein, compatible with resonance assignment for structural determination.
Conclusion
HCA was used effectively to investigate the threedimensional similarities of the globular region present in both m53BP1 and ScRad9 proteins, upstream of their BRCT motifs. The existence of a Tudor fold within this region of ScRad9 was strongly supported by its high degree of homology with both the human SMN Tudor domain and the predicted Tudor motif of m53BP1, the N-terminal Tudor motif in the now-resolved structure. An N-terminal fusion strategy was used to produce, at best, several fragments encompassing the putative Tudor fold. Size-exclusion chromatography, DSC, and CD analyses were performed to characterize the different fragments. The results demonstrated that for both proteins, the single Tudor motif cannot be expressed in soluble form: Indeed, either an insoluble protein or an oligomeric species was generated. The stable unit that can fold autonomously for m53BP1 and ScRad9 was identified as the extended Tudor fragment. Their folded nature was proved by NMR spectroscopy to correlate with the size-exclusion chromatography and DSC results.
The m53BP1 Tudor domain consisted of fragment [1463–1617]. The resonance assignments, determined with the aid of both triple resonance experiments and three-dimensional 15N or 13C-edited NOESY spectra, have been published recently (Charier et al. 2004a). The three-dimensional structure of the solution has just been resolved by both heteronuclear multidimensional NMR spectroscopy and X-ray crystallography (Charier et al. 2004b). The hypothesis of a Tudor β-barrel fold was confirmed, but unexpectedly this domain displayed a new structural motif composed of two tightly packed Tudor domains and a C-terminal α-helix. This domain, shown to be adequate for targeting 53BP1 to sites with doublestranded DNA breaks, is included in the kinetochore-binding domain (Jullien et al. 2002) and corresponds perfectly to the minimal domain recently found to focus formation on DNA damage (Iwabuchi et al. 2003). Previously described as displaying chromatin-binding activity in vivo, the m53BP1 tandem-Tudor domain was found to bind methylated histone H3 (Huyen et al. 2004).
For ScRad9, the putative Tudor domain consisted of residues 754–947 in the Rad53 FHA2 interacting region (Durocher et al. 1999). Its 1H-15N HSQC spectrum displayed signal dispersion, promoting structural resolution by NMR analysis. The presence of some superposition was, however, indicatory of difficulties for assignment. The ScRad9 module [754–947] 1H, 15N, 13C assignment is in progress. Concerning this hypothesis, three-dimensional structural conservation reported between m53BP1 tandem Tudor domain and ScRad9[750–917] favors Tudor folding. Moreover, the ScRad9[750–917] fragment was also observed to bind methylated histone H3 in vitro (Huyen et al. 2004). ScRad9 belongs to the group of adaptor proteins that bind effector serine/threonine kinases and are involved in their activation. In this group, the claspins subgroup (Kumagai and Dunphy 2000; Tanaka and Russell 2001) is well conserved in yeast and vertebrates. The ScRad9 subgroup has so far been found to be poorly conserved outside the BRCT motif. The discovery of this new domain, by combining biochemical, structural, and genetic experimental approaches, will help improve understanding of adaptor protein function.
Materials and methods
Chemical, biological, and microbiological reagents
The chemicals used in this study were of analytical grade and obtained from Sigma unless stated otherwise. Oligonucleotide primers were from Genaxys and Genset (Proligo). Ampli-Taq DNA polymerase was from Applied Biosystems. Chromatography reagents were purchased from Amersham Bioscience unless stated otherwise. Nickel-nitrolotriacetic acid (Ni-NTA) Superflow and Ni-NTA spin columns were available from Qiagen. Proteinase inhibitors were purchased from Roche. Bugbuster reagent, benzonase nuclease biotinylated thrombin, streptavidin-agarose, and Xa/LIC pET32 vector encoding an N-terminal TRX-tag were obtained from Novagen (VWR). TEV recombinant protease; E. coli strains; and pDONR201, pDEST15, and pDEST17 vectors were from Invitrogen.
Most of the cultures were grown in Luria broth (LB) medium, in flasks or in a pH-regulated fermentor. Media were supplemented with the following antibiotics when necessary: ampicillin (100 μg/mL), tetracycline (10 μg/mL), chloramphenicol (20 μg/mL), and kanamycin (50 μg/mL).
FPLC purification was performed on an Äkta Purifier system (Pharmacia). SDS/PAGE gel electrophoresis was performed on precast polyacrylamide mini-gel systems (Biorad and NuPAGE Bis-Tris from Invitrogen) with Coomassie blue or silver staining. Centriplus 20 or microcon YM3 filtration units (Amicon, Millipore) were used for concentration and buffer exchange that was also performed either by dialysis or gel filtration. Protein concentrations were determined with a Bradford assay or BCA protein assay.
ScRad9 and m53BP1 were subcloned from the Ycp50-Rad9 and pEGFP-m53BP1 plasmids kindly provided by Dr. Carl Mann (CEA-Saclay). The usual DNA manipulations were performed according to standard procedures. The reference sequences used for ScRad9, BRCA1, and m53BP1 were SwissProt entries P14737 and P38398 and TrEMBL entry Q91YC9, respectively.
Plasmid construction
For LIC technology, the primers were designed with the appropriate 5′ extensions: 5′-GGTATTGAGGGTCGC at the 5′-end of the sense primers and 5′-AGAGGAGAGTTA GAGCC and a stop codon for the anti-sense primer. PCR-amplified products ScRad9[764–855], ScRad9[764–947], and ScRad9[754–947], treated according to the supplier protocol, were cloned into an Xa/LIC pET32 vector. The constructs express a protein with the sequence/structure of 1Ser- - - - -112Met(His)6Ser-Ser-Gly-Leu-Val-Pro-Arg-Gly-Ser-Gly-Met-Stag at the N terminal. The underlined dipeptide is a thrombin cleavage site; 1Ser- - - - -112Met represents the 109-residue portion of TRX-tag thioredoxin protein. A Factor Xa cleavage site is also present.
For Gateway technology, the 5′-sense primers were designed to incorporate a 5′-terminal attB1 sequence and encode the seven-amino-acid ENLYFQG of the tobacco etch virus (TEV) protease recognition site, with 3′-anti-sense primers to incorporate a 5′-terminal attB2 sequence and add a stop codon for translation termination. PCR-amplified products, ScRad9[764–855], ScRad9[764–947], ScRad9[754–947], m53BP[1466–1535], and m53BP[1463–1617], were cloned by recombination into a pDONR201 vector according to the supplier’s protocol. Several colonies were screened by PCR analysis. Plasmid DNA obtained from positive clones and confirmed by sequencing for sequence accuracy (see concerned paragraph) was also used to transfer the fragments into pDEST15 and pDEST17 vectors, respectively, via a second recombination reaction (LR reaction, according to the supplier’s instructions). The expression plasmids were used without further sequencing.
DNA sequencing
Nucleotide sequencing of both DNA strands was carried out using a Quick Start Beckman Coulter DTCS sequencing kit with double-stranded templates. Samples containing fluorescence-labeled dideoxynucleotide terminators were cleaned by ethanol precipitation and processed on a CEQ2000 XL Beckman Coulter capillary-based automated sequencer. Sequences were compiled and analyzed with the Vector NTI software package (Informax).
Protein expression
E. coli strains BL21-star(DE3), BL21-(DE3), and BL21(DE3) pLysS, previously transformed with the different expression plasmids, were stored in GSB medium at −80°C. Cells were grown at 37°, 30°, and 16°C in LB medium, supplemented with 100 μg/mL ampicillin in baffled flasks or in a 4-L Labfors fermentor (Infors). Protein expression was induced by adding 0.5 mM IPTG when optical density (A600 nm) reached 0.6–0.8 (flasks or fermentor). After induction time, cells were harvested at 4000g for 20 min at 4°C and washed once with 50 mM Tris-HCl buffered at pH 8 and 0.5 mM PMSF. The cell pellet was finally frozen at −80°C.
For small-scale expression (<500-mL volume), cell pellets were treated with bugbuster HT or sonicated, whereas on a larger scale, lysis was by disruption on a Cell D disruptor (BioCell apparatus) at 0.9 kbar breaking pressure. In all cases, the lysed pellets were clarified by centrifugation at 20,000g for 20 min at 4°C to remove cell debris and aggregated proteins. For GST fusion proteins, phosphate buffer saline 10 mM at pH 7.3, was used as a lysis buffer, whereas 20 mM Tris-HCl at pH 8, 20 mM NaCl, or 20 mM KH2PO4/Na2HPO4 buffered at pH 7.5 was used for purification on nickel-charged resin.
Protein purification
Small-scale purification of 6His and TRX-tagged proteins
Small-scale purification was performed using Ni-NTA spin columns, whereas the microspin GST purification module was used for GST-tagged proteins, according to the manufacturer’s instructions in both cases.
Batch purification of TRX-m53BP1[1466–1535] and TRX-ScRad9[754–947] proteins
The whole procedure was performed at 4°C. The supernatant was added to Ni-NTA resin, equilibrated beforehand in TBS (25 mM Tris-HCl at pH 8, 300 mM NaCl), and mixed gently by shaking overnight at 4°C. The lysate-resin mixture was loaded into an empty column and the flow-through collected. After column washing by 9 mM, 30 mM, and 80 mM imidazole in TBS successively, the TRX fusion protein was eluted with the same buffer, including 300 mM imidazole.
FPLC purification of 6His-m53BP1[1466–1535] protein
The lysate solution was applied to a 5-mL NiSO4-loaded Hi-Trap chelating Sepharose column, equilibrated with buffer A (20 mM phosphate at pH 7.5, 500 mM NaCl) plus 20 mM imidazole. After washing (10-column volumes), a 12-column volume linear gradient from 20 to 500 mM imidazole was applied. Recombinant 6His-m53BP1[1466–1535] was eluted between 220 mM and 500 mM imidazole and desalted by overnight dialysis against 10 mM phosphate at pH 8.
FPLC purification of TRX-ScRad9[764–855]
The lysate was prepurified by precipitation at 4°C with 33% saturated ammonium sulfate. The precipitate was then dissolved and extensively dialyzed at 4°C against 20 mM KH2PO4/Na2HPO4 buffer at pH 7.5. After centrifugation at 4000g for 30 min at 4°C, the supernatant was applied to a NiSO4-loaded chelating Sepharose Fast Flow column for purification with an eight-column volume step gradient from 50 to 300 mM imidazole in buffer A. TRX-ScRad9[764–855] protein was eluted at both 200 mM and 300 mM imidazole. After concentration and dialysis against 20 mMTris-HCl at pH 8.2, 150 mM NaCl (TBS 20 mM buffer at pH 8.2), the purified protein pools were stored at −20°C at concentrations of 0.5–1 mg/mL until use.
Batch purification of GST-ScRad9[754–947] protein
The lysate was added to glutathione resin pre-equilibrated in PBS at pH 7.2 and the mixture incubated for 2 h at room temperature with gentle shaking. The lysate-resin mixture was loaded into an empty column and the flow-through collected. After column washing with 10 volumes of PBS at pH 7.2, the GST fusion protein was eluted with four volumes of elution buffer (10 mM Tris-HCl at pH 8, 10 mM reduced glutathione). After pooling, the elution fraction was dialyzed in a spectrapor MW 6–8 kDa against 50 mM Tris-HCl at pH 8, 150 mM NaCl, and stored at −20°C as aliquots.
FPLC purification of GST-ScRad9[764–947] and GST-m53BP1[1463–1617]
The PBS lysates were applied to a glutathione Sepharose column pre-equilibrated with PBS buffer at 4°C. After extensive washing with PBS, GST-ScRad9[764–947] was eluted with a two-column volume of elution buffer, dialyzed against TBS 50 mM at pH 8, and stored at −20°C.
Proteolytic cleavage
For GST fusion proteins (0.2–6 mg/mL in concentration), the reactions were performed at 30°C in TEV protease buffer. The concentration of TEV protease varied depending on the efficiency with which each GST fusion protein was processed. For TRX fusion proteins (0.2 mg/mL), the reactions were performed in thrombin protease buffer at 20°C (Table 3). The cleaved fragments were purified by affinity (as previously described) followed by gel filtration if necessary.
N-terminal amino acid sequencing
N-terminal amino acid sequencing was by Edman degradation using a Procise protein sequencing system (Applied Biosystems) coupled with a 140C microgradient pump system (Applied Biosystems) and a Series 200 UV-visible detector (Perkin Elmer Life Sciences).
Gel filtration analysis
The globular and monomeric state of the fragment was evaluated by size exclusion chromatography using an analytical Superdex-75 column (HR 10/30) and 50 mM PBS buffer (pH 7.3). Protein standards used to calibrate the column were ribonuclease A (15.8 kDa), chymotrypsinogen A (21.2 kDa), ovalbumin (49.4 kDa), and albumin (69.8 kDa).
CD spectral measurements
Circular dichroism spectra were recorded at 25°C between 190 and 350 nm with a J810 Jasco spectro-dichrograph, using a quartz cuvette of 1-mm pathlength, with an integration time of 1 sec for each 0.1-nm step and a bandwidth of 1 nm. Spectra were scanned at 50 nm•min−1. Spectral analysis of secondary structure was performed using Dr. Yang J.T.’s program, described in Methods in Enzymology(1986) and suupplied with JASCO 810 apparatus.
DSC measurements
Temperature-induced melting of soluble protein fragments was monitored with a differential scanning microcalorimeter VP-DSC from MicroCal. Prior to calorimetric analysis, the samples were diluted in the reference buffer used in the experiment (50 mM or 10 mM KH2PO4/Na2HPO4 buffer at pH 7.3 containing 150 mM NaCl) and then degassed. Calorimetric scans were performed between 23°C and 60°C–80°C with a heating scan rate of 40°C•h−1 in a 0.51-mL volume microcalorimeter cell, against the reference buffer. The heat capacity curves were plotted after subtracting the baseline.
Nuclear magnetic resonance spectroscopy
1H-NMR spectra
NMR samples of unlabeled protein fragments were prepared at a concentration of 0.1 mM in a 10-mM KH2PO4/Na2HPO4 buffer, 150 mM NaCl.
1H-15N NMR spectra
The uniformly 15N-labeled GST-ScRad9[754–947] and GST-m53BP1[1463–1617] were produced in a minimum medium containing 1 g/L 15NH4Cl. After purification, the cleaved fragments were concentrated to 0.3–0.5 mM in 50 mM Tris, 150 mM NaCl at pH 7.2 for m53BP1[1463–1617] and in 50 mM MES, 50 mM NaCl at pH 6.0 for ScRad9[754–947]. All NMR experiments were performed at 300K for m53BP1 and 303K for ScRad9, on 500- and 600-MHz Bruker spectrophotometers equipped with triple-resonance (1H, 13C, 15N) probes including shielded z-gradients. The spectra were processed using XwinNMR(Bruker) and treated with the Felix97 software (Accelrys).
Acknowledgments
We are grateful to A. Dedieu and I. Dany for mass spectroscopy analyses and to J.-C. Gaillard for protein N-terminal sequencing. We are grateful to Philippe Savarin and Flavio Toma who kindly lent their 600-MHz spectrometer. We thank D. Jullien for providing m53BP1 cDNA, C. Mann for yeast Ycp50-Rad9 plasmid, and G. Stier for TEV protease cDNA.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041305205.
References
- Aboussekhra, A., Vialard, J.E., Morrison, D.E., de la Torre-Ruiz, M.A., Cernakova, L., Fabre, F., and Lowndes, N.F. 1996. A novel role for the budding yeast RAD9 checkpoint gene in DNA damage-dependent transcription. EMBO J. 15 3912–3922. [PMC free article] [PubMed] [Google Scholar]
- Aslanidis, C. and de Jong, P.J. 1990. Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res. 18 6069–6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baneyx, F. 1999. Recombinant protein expression in Escherichia coli. Curr. Opin. Biotechnol. 10 411–421. [DOI] [PubMed] [Google Scholar]
- Bork, P., Hofmann, K., Bucher, P., Neuwald, A.F., Altschul, S.F., and Koonin, E.V. 1997. A superfamily of conserved domains in DNA damage-responsive cell cycle checkpoint proteins. FASEB J. 11 68–76. [PubMed] [Google Scholar]
- Callebaut, I. and Mornon, J.P. 1997a. From BRCA1 to RAP1: A widespread BRCT module closely associated with DNA repair. FEBS Lett. 400 25–30. [DOI] [PubMed] [Google Scholar]
- ———. 1997b. The human EBNA-2 coactivator p100: Multidomain organization and relationship to the staphylococcal nuclease fold and to the tudor protein involved in Drosophila melanogaster development. Biochem. J. 321 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, J.Y. 1985. Thrombin specificity. Requirement for apolar amino acids adjacent to the thrombin cleavage site of polypeptide substrate. Eur. J. Biochem. 151 217–224. [DOI] [PubMed] [Google Scholar]
- Charier, G., Alpha-Bazin, B., Couprie, J., Callebaut, I., Berenguer, F., Quemeneur, E., Gilquin, B., and Zinn-Justin, S. 2004a. Letter to the Editor: 1H, 13C and 15N resonance assignments of the region 1463–1617 of the mouse p53 binding protein 1 (53BP1). J. Biomol. NMR 28 303–304. [DOI] [PubMed] [Google Scholar]
- Charier, G., Couprie, J., Alpha-Bazin, B., Meyer, V., Quemeneur, E., Guerois, R., Callebaut, I., Gilquin, B., and Zinn-Justin, S. 2004b. The Tudor tandem of 53BP1: A new structural motif involved in DNA and RG-rich peptide binding. Structure (Camb.) 12 1551–1562. [DOI] [PubMed] [Google Scholar]
- Cordingley, M.G., Callahan, P.L., Sardana, V.V., Garsky, V.M., and Colonno, R.J. 1990. Substrate requirements of human rhinovirus 3C protease for peptide cleavage in vitro. J. Biol. Chem. 265 9062–9065. [PubMed] [Google Scholar]
- DiTullio Jr., R.A., Mochan, T.A., Venere, M., Bartkova, J., Sehested, M., Bartek, J., and Halazonetis, T.D. 2002. 53BP1 functions in an ATM-dependent checkpoint pathway that is constitutively activated in human cancer. Nat. Cell Biol. 4 998–1002. [DOI] [PubMed] [Google Scholar]
- Doerks, T., Copley, R.R., Schultz, J., Ponting, C.P., and Bork, P. 2002. Systematic identification of novel protein domain families associated with nuclear functions. Genome Res. 12 47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durocher, D., Henckel, J., Fersht, A.R., and Jackson, S.P. 1999. The FHA domain is a modular phosphopeptide recognition motif. Mol. Cell 4 387–394. [DOI] [PubMed] [Google Scholar]
- Fernandez-Capetillo, O., Chen, H.T., Celeste, A., Ward, I., Romanienko, P.J., Morales, J.C., Naka, K., Xia, Z., Camerini-Otero, R.D., Motoyama, N., et al. 2002. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat. Cell Biol. 4 993–997. [DOI] [PubMed] [Google Scholar]
- Haun, R.S. and Moss, J. 1992. Ligation-independent cloning of glutathione S-transferase fusion genes for expression in Escherichia coli. Gene 112 37–43. [DOI] [PubMed] [Google Scholar]
- Haun, R.S., Serventi, I.M., and Moss, J. 1992. Rapid, reliable ligation-independent cloning of PCR products using modified plasmid vectors. Biotechniques 13 515–518. [PubMed] [Google Scholar]
- Hochuli, E. 1988. Large-scale chromatography of recombinant proteins. J. Chromatogr. 444 293–302. [DOI] [PubMed] [Google Scholar]
- Huyen, Y., Zgheib, O., Ditullio Jr., R.A., Gorgoulis, V.G., Zacharatos, P., Petty, T.J., Sheston, E.A., Mellert, H.S., Stavridi, E.S., and Halazonetis, T.D. 2004. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature 432 406–411. [DOI] [PubMed] [Google Scholar]
- Huyton, T., Bates, P.A., Zhang, X., Sternberg, M.J., and Freemont, P.S. 2000. The BRCA1 C-terminal domain: Structure and function. Mutat. Res. 460 319–332. [DOI] [PubMed] [Google Scholar]
- Iwabuchi, K., Li, B., Massa, H.F., Trask, B.J., Date, T., Fields, S., Bartel, P.L., and Marraccino, R. 1998. Stimulation of p53-mediated transcriptional activation by the p53-binding proteins, 53BP1 and 53BP2 two cellular proteins that bind to wild-type but not mutant p53. J. Biol. Chem. 273 26061–26068. [DOI] [PubMed] [Google Scholar]
- Iwabuchi, K., Basu, B.P., Kysela, B., Kurihara, T., Shibata, M., Guan, D., Cao, Y., Hamada, T., Imamura, K., Jeggo, P.A., et al. 2003. Potential role for 53BP1 in DNA end-joining repair through direct interaction with DNA. J. Biol. Chem. 278 36487–36495. http://www.jbc.org. [DOI] [PubMed] [Google Scholar]
- Janin, J. and Chothia, C. 1985. Domains in proteins: Definitions, location, and structural principles. Methods Enzymol. 115 420–430. [DOI] [PubMed] [Google Scholar]
- Jullien, D., Vagnarelli, P., Earnshaw, W.C., and Adachi, Y. 2002. Kinetochore localisation of the DNA damage response component 53BP1 during mitosis. J. Cell Sci. 115 71–79. [DOI] [PubMed] [Google Scholar]
- Kawahire, S., Takeuchi, M., Gohshi, T., Sasagawa, S., Shimada, M., Takahashi, M., Abe, T.K., Ueda, T., Kuwano, R., Hikawa, A., et al. 1997. cDNA cloning of nuclear localization signal binding protein NBP60, a rat homologue of lamin B receptor, and identification of binding sites of human lamin B receptor for nuclear localization signals and chromatin. J. Biochem. (Tokyo) 121 881–889. [DOI] [PubMed] [Google Scholar]
- Koonin, E.V., Altschul, S.F., and Bork, P. 1996. BRCA1 protein products... Functional motifs. Nat. Genet. 13 266–268. [DOI] [PubMed] [Google Scholar]
- Kumagai, A. and Dunphy, W.G. 2000. Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts. Mol. Cell 6 839–849. [DOI] [PubMed] [Google Scholar]
- LaVallie, E.R., DiBlasio, E.A., Kovacic, S., Grant, K.L., Schendel, P.F., and McCoy, J.M. 1993. A thioredoxin gene fusion expression system that circumvents inclusion body formation in the E. coli cytoplasm. Biotechnology (N Y) 11 187–193. [DOI] [PubMed] [Google Scholar]
- Lowndes, N.F. and Murguia, J.R. 2000. Sensing and responding to DNA damage. Curr. Opin. Genet. Dev. 10 17–25. [DOI] [PubMed] [Google Scholar]
- Maurer-Stroh, S., Dickens, N.J., Hughes-Davies, L., Kouzarides, T., Eisenhaber, F., and Ponting, C.P. 2003. The Tudor domain ‘Royal Family’: Tudor, plant Agenet, Chromo, PWWP and MBT domains. Trends Biochem. Sci. 28 69–74. [DOI] [PubMed] [Google Scholar]
- McGuffin, L.J., Bryson, K., and Jones, D.T. 2000. The PSIPRED protein structure prediction server. Bioinformatics 16 404–405. [DOI] [PubMed] [Google Scholar]
- Melo, J. and Toczyski, D. 2002. A unified view of the DNA-damage checkpoint. Curr. Opin. Cell Biol. 14 237–245. [DOI] [PubMed] [Google Scholar]
- Motoyama, N. and Naka, K. 2004. DNA damage tumor suppressor genes and genomic instability. Curr. Opin. Genet. Dev. 14 11–16. [DOI] [PubMed] [Google Scholar]
- Nyberg, K.A., Michelson, R.J., Putnam, C.W., and Weinert, T.A. 2002. Toward maintaining the genome: DNA damage and replication checkpoints. Annu. Rev. Genet. 36 617–656. [DOI] [PubMed] [Google Scholar]
- Parks, T.D., Leuther, K.K., Howard, E.D., Johnston, S.A., and Dougherty, W.G. 1994. Release of proteins and peptides from fusion proteins using a recombinant plant virus proteinase. Anal. Biochem. 216 413–417. [DOI] [PubMed] [Google Scholar]
- Ponting, C.P. 1997a. P100, a transcriptional coactivator, is a human homologue of staphylococcal nuclease. Protein Sci. 6 459–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1997b. Tudor domains in proteins that interact with RNA. Trends Biochem. Sci. 22 51–52. [DOI] [PubMed] [Google Scholar]
- Privalov, P.L. and Potekhin, S.A. 1986. Scanning microcalorimetry in studying temperature-induced changes in proteins. Methods Enzymol. 131 4–51. [DOI] [PubMed] [Google Scholar]
- Qiu, C., Sawada, K., Zhang, X., and Cheng, X. 2002. The PWWP domain of mammalian DNA methyltransferase Dnmt3b defines a new family of DNA-binding folds. Nat. Struct. Biol. 9 217–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouse, J. and Jackson, S.P. 2002. Interfaces between the detection, signaling, and repair of DNA damage. Science 297 547–551. [DOI] [PubMed] [Google Scholar]
- Schein, C.H. 1990. Solubility as a function of protein structure and solvent components. Biotechnology (N Y) 8 308–317. [DOI] [PubMed] [Google Scholar]
- Schiestl, R.H., Reynolds, P., Prakash, S., and Prakash, L. 1989. Cloning and sequence analysis of the Saccharomyces cerevisiae RAD9 gene and further evidence that its product is required for cell cycle arrest induced by DNA damage. Mol. Cell. Biol. 9 1882–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selenko, P., Sprangers, R., Stier, G., Buhler, D., Fischer, U., and Sattler, M. 2001. SMN tudor domain structure and its interaction with the Sm proteins. Nat. Struct. Biol. 8 27–31. [DOI] [PubMed] [Google Scholar]
- Siede, W., Friedberg, A.S., Dianova, I., and Friedberg, E.C. 1994. Characterization of G1 checkpoint control in the yeast Saccharomyces cerevisiae following exposure to DNA-damaging agents. Genetics 138 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater, L.M., Allen, M.D., and Bycroft, M. 2003. Structural variation in PWWP domains. J. Mol. Biol. 330 571–576. [DOI] [PubMed] [Google Scholar]
- Smith, D.B. and Johnson, K.S. 1988. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene 67 31–40. [DOI] [PubMed] [Google Scholar]
- Sprangers, R., Groves, M.R., Sinning, I., and Sattler, M. 2003a. High-resolution X-ray and NMR structures of the SMN Tudor domain: Conformational variation in the binding site for symmetrically dimethylated arginine residues. J. Mol. Biol. 327 507–520. [DOI] [PubMed] [Google Scholar]
- Sprangers, R., Selenko, P., Sattler, M., Sinning, I., and Groves, M.R. 2003b. Definition of domain boundaries and crystallization of the SMN Tudor domain. Acta Crystallogr. D Biol. Crystallogr. 59 366–368. [DOI] [PubMed] [Google Scholar]
- Stec, I., Nagl, S.B., van Ommen, G.J., and den Dunnen, J.T. 2000. The PWWP domain: A potential protein-protein interaction domain in nuclear proteins influencing differentiation? FEBS Lett. 473 1–5. [DOI] [PubMed] [Google Scholar]
- Swartz, J.R. 2001. Advances in Escherichia coli production of therapeutic proteins. Curr. Opin. Biotechnol. 12 195–201. [DOI] [PubMed] [Google Scholar]
- Tanaka, K. and Russell, P. 2001. Mrc1 channels the DNA replication arrest signal to checkpoint kinase Cds1. Nat. Cell Biol. 3 966–972. [DOI] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walhout, A.J., Temple, G.F., Brasch, M.A., Hartley, J.L., Lorson, M.A., van den Heuvel, S., and Vidal, M. 2000. GATEWAY recombinational cloning: Application to the cloning of large numbers of open reading frames or ORFeomes. Methods Enzymol. 328 575–592. [DOI] [PubMed] [Google Scholar]
- Wang, B., Matsuoka, S., Carpenter, P.B., and Elledge, S.J. 2002. 53BP1, a mediator of the DNA damage checkpoint. Science 298 1435–1438. http://www.sciencemag.org. [DOI] [PubMed] [Google Scholar]
- Ward, I.M., Minn, K., van Deursen, J., and Chen, J. 2003. p53 Binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol. Cell. Biol. 23 2556–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert, T. and Hartwell, L. 1989. Control of G2 delay by the rad9 gene of Saccharomyces cerevisiae. J. Cell Sci. Suppl. 12 145–148. [DOI] [PubMed] [Google Scholar]
- Weinert, T.A., Kiser, G.L., and Hartwell, L.H. 1994. Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes & Dev. 8 652–665. [DOI] [PubMed] [Google Scholar]
- Wetlaufer, D.B. 1973. Nucleation, rapid folding, and globular intrachain regions in proteins. Proc. Natl. Acad. Sci. 70 697–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson, D.L. and Harrison, R.G. 1991. Predicting the solubility of recombinant proteins in Escherichia coli. Biotechnology (N Y) 9 443–448. [DOI] [PubMed] [Google Scholar]
- Zhang, H., Somasundaram, K., Peng, Y., Tian, H., Bi, D., Weber, B.L., and El-Deiry, W.S. 1998. BRCA1 physically associates with p53 and stimulates its transcriptional activity. Oncogene 16 1713–1721. [DOI] [PubMed] [Google Scholar]
- Zhou, B.B. and Elledge, S.J. 2000. The DNA damage response: Putting checkpoints in perspective. Nature 408 433–439. [DOI] [PubMed] [Google Scholar]