

Abstract
Methylenetetratetrahydromethanopterin reductase (Mer) is involved in CO2 reduction to methane in methanogenic archaea and catalyses the reversible reduction of methylenetetrahydromethanopterin (methylene-H4MPT) to methyl-H4MPT with coenzyme F420H2, which is a reduced 5′-deazaflavin. Mer was recently established as a TIM barrel structure containing a nonprolyl cis-peptide bond but the binding site of the substrates remained elusive. We report here on the crystal structure of Mer in complex with F420 at 2.6 Å resolution. The isoalloxazine ring is present in a pronounced butterfly conformation, being induced from the Re-face of F420 by a bulge that contains the non-prolyl cis-peptide bond. The bindingmode of F420 is very similar to that in F420-dependent alcohol dehydrogenase Adf despite the low sequence identity of 21%. Moreover, binding of F420 to the apoenzyme was only associated with minor conformational changes of the polypeptide chain. These findings allowed us to build an improved model of FMN into its binding site in bacterial luciferase, which belongs to the same structural family as Mer and Adf and also contains a nonprolyl cis-peptide bond in an equivalent position.
Keywords: Methylenetetrahydromethanopterin reductase, bacterial luciferase, crystal structure, nonprolyl cis-peptide bond coenzyme F420, FMN
Methylenetetrahydromethanopterin reductase (Mer) catalyzes the reversible reduction of N5, N10-methylenetetrahydromethanopterin (methylene-H4MPT) to N5-methyltetrahydromethanopterin (methyl-H4MPT) with the concomitant oxidation of F420H2 to oxidized F420 (Fig. 1 ▶). F420 is a 5′ deazaflavin derivative and H4MPT, a tetrahydrofolate analog. Mer is found in methanogenic and sulfate reducing archaea (Ma and Thauer 1990a, b; te Brommelstroet et al. 1990; Ma et al. 1991; Vaupel and Thauer 1995; Shima et al. 2002). In the methanogens Mer is involved in CO2 reduction to CH4, inmethyl group oxidation to CO2, and in autotrophic CO2 fixation (Shima et al. 2000). In the sulphate-reducers the enzyme participates in acetate oxidation to CO2 with sulphate and in autotrophic CO2 fixation (Schmitz et al. 1991; Vorholt et al. 1995, 1997). The cytoplasmic enzyme is composed of only one type of subunit with an apparent molecular mass between 35kDaand 40 kDa. It lacks a prosthetic group and exhibits a ternary complex mechanism. Mer is not phylogenetically related to methylenetetrahydrofolate reductase (MetF), although both enzymes catalyze analogous reactions (Guenther et al. 1999).
Figure 1.

(A) F420-dependent methylenetetrahydromethanopterin reductase (Mer) catalyzes the reversible reduction of N5, N10-methylenetetrahydromethanopterin (Methylene-H4MPT) to N5-methyltetrahydromethanopterin (Methyl-H4MPT) with the concomitant oxidation ofF420H2 toF420. The enzyme is Si-face specificwith respect to the C5 atom of F420. (B) F420 is a deazaflavin derivative and tetrahydromethanopterin (H4MPT) a tetrahydrofolate analog. (R′) in H4MPT consists of an aminophenyl, a 1-deoxyribose, a ribose, a phosphate and a 2-hydroxyglutarate group. This figure was produced with ChemWindow (Bio-Rad Laboratories).
The crystal structures of Mer from two thermophilic methanogens, Methanopyrus kandleri (kMer) and Methanothermobacter marburgensis (tMer), in the absence of their substrates, have been reported (Shima et al. 2000). The subunits have a TIM barrel structure and exhibit an unusual nonprolyl cis-peptide bond where the active site is presumed. Attempts to obtain the crystal structures of tMer and kMer in complex with substrates have failed so far. Neither the cocrystallization of the enzyme with F420 under various screening conditions nor soaking of the crystals with the substrates yielded crystals of the complex.
Mer belongs to the bacterial luciferase family that involves FMN- and F420-dependent oxidoreductases using diverse substrates (Aufhammer et al. 2004). Mer, FMN-dependent bacterial luciferase (LuxAB) (Baldwin et al. 1979), FMN-dependent alkanesulfonate monoxygenase (SsuD) (van Der Ploeg et al. 1999), F420-dependent alcohol dehydrogenase (Adf) (Aufhammer et al. 2004), F420- dependent glucose-6-phosphate dehydrogenase from Mycobacteria (Purwantini and Daniels 1998), F420- dependent oxidoreductase from Streptomyces (Peschke et al. 1995), and F420-dependent hydride transferase 1 from Rhodococcus (Heiss et al. 2002) are known so far as members of this family. Crystal structures of LuxAB(Fisher et al. 1995, 1996), Mer (Shima et al. 2000), SsuD (Eichhorn et al. 2002), and Adf (Aufhammer et al. 2004) have already been solved. The four enzymes with low sequence identity (<30%) show a similar (αβ)8 barrel fold (TIMbarrel fold) (Aufhammer et al. 2004). The unusual cis-peptide bond found in Mer (Shima et al. 2000) is also present in LuxAB (Fisher et al. 1996) and Adf (Aufhammer et al. 2004) but is absent in SsuD (Eichhorn et al. 2002).
Of the bacterial luciferase enzyme family only the crystal structure of Adf in complex with its coenzyme has been determined (Aufhammer et al. 2004). The nonprolyl cis-peptide bond was identified as an essential part of a bulge that serves as backstop to the Re-face of the F420isoalloxazine ring. Here, we present the structure of Mer from a mesophilic methanogen, Methanosarcina barkeri (bMer), in complex with F420. Unfortunately all attempts to obtain a crystal structure with methylene-H4MPT have failed. The substrate was therefore modeled into the Mer structure assuming the conformation of methylene-H4MPT to be the same as in the crystal structure of the formaldehyde activating enzyme (Fae) (Acharya et al. 2005).
Results
Crystal structure
The crystal structure of bMer was solved at 2.6 Å resolution by the molecular replacement method. The model was refined to Rcryst- and Rfree-factors of 18.5% and 22.1%, respectively, in the resolution range 2.6–50 Å. bMer is present as a homotetramer in agreement with gel filtration results (Ma and Thauer 1990a). The arrangement of the subunits corresponds to that of kMer (Shima et al. 2000). The subunit of bMer consists of an (αβ)8barrel core and three insertion regions named IR1 (Pro94-Pro111), IR2 (Gly127-Pro152), and IR3 (Lys216- Pro285) (Figs. 2 ▶, 3 ▶). IR1 and IR3 consist mainly of helix α4-1 and the helical subdomains α7-1–α7-5, respectively, and constitute the walls of the F420 and methylene-H4MPT binding cleft. Again, the electron density clearly indicates a well-ordered nonprolyl cis-peptide bond between Gly61 and Val62 that is located in a bulge at the end of strand β3. A superposition of the Mer subunits resulted in a root-mean- square deviation (RMSD) of 1.1 Å (bMer to tMer) and of 1.3 Å (bMer to kMer) for all Cαpositions indicating their strong structural relationship. The difference between the (αβ)8barrel cores is significantly smaller (RMSD 0.6 Å) than between the insertion regions. Structural differences of IR1 can be partly accounted for by the fact that bMer is present in complex with F420 and tMer and kMer without the coenzyme (see below).
Figure 2.

Stereo plot of the barrel fold of F420-dependent methylenetetrahydromethanopterin reductase from Methanosarcina barkeri in the monomeric form. The (αβ)8 barrel core is presented in gray. Insertion regions (IR) are indicated in green (IR1 and IR3) and in blue (IR2). IR1 and IR3 consist mainly of helix α4-1 and the helical subdomains α7-1–α7-5, respectively. IR2 is composed of a β-hairpin like segment between α4 and β5. These insertion regions have important functions in either binding of F420 (illustrated in red) and methylene-H4MPT (not shown) or in forming the interface between the subunits. This stereoplot was prepared with PYMOL (DeLano Scientific LLC).
Figure 3.

Sequence alignment of F420-dependent methylenetetrahydromethanopterin reductase from Methanosarcina barkeri (bMer), F420- dependent alcohol dehydrogenase from Methanoculleus thermophilicus (Adf) and FMN-dependent bacterial luciferase from Vibrio harveyi (LuxA). Conserved residues are colored in red. Arrows and bars above indicate the secondary structure assignments of each sequence. Residues involved in F420 or FMN binding are highlighted in yellow. The amino acid residues at non-prolyl cis-peptide bonds are indicated with asterisks. His36, which is bound to the F420 isoalloxazine ring, is boxed in green. His44, Cys106, and Arg107 of LuxA are underlined. One segment of the structure was not visible in the electron density of LuxA.
The structural analysis of bMer revealed two features in the electron density that are not interpretable as protein residues, F420, or water. The first electron density is located close to residue His36 in the substrate binding cleft of one of the subunits in the asymmetric unit and could be assigned to a polyethylene glycol (PEG) molecule (n=5). The PEG molecule is primarily linked to the protein by a hydrogen bond between its oxygen atom and the His36-Nɛ atom and by hydrophobic interaction to Phe231, which is reoriented upon PEG binding. Bound PEG molecules have also been reported in other protein structures (Becker et al. 1998; Shima et al. 2000; Pylypenko et al. 2003). The second electron density patch is 6 Å long and is attached to the C5atom of F420 and protrudes perpendicularly to its Si-face (Fig. 4 ▶). The presence of a covalent F420-adduct at the Si-face of the isoalloxazine ring indicates that C5of F420 is in an sp3 hybridization state. A similar covalent adduct was observed in the structure of the Adf–F420 complex (Aufhammer et al. 2004) and in the short-chain alcohol dehydrogenase from Drosophila spec. (Benach et al. 1999) in which the additional molecule was characterized as an acetone. The mother liquor of the bMer crystals contained isopropanol, which is easily autoxidized to acetone, but an acetone molecule would be too small to fill out the electron density. The adduct must therefore be derived from the polyethylene glycol, which is known to be contaminated by formaldehyde and to contain various polyethylene glycol degradation products.
Figure 4.

The 2Fobs − Fcalc electron density (contour level 1.0σ) of the part of F420 isoalloxazine ring in the 2.6 Å crystal structure of bMer. Additional electron density was found at the C5of the ring. Only a part of the unknown molecule is shown. The drawing was rendered with Bobscript (Esnouf 1997) and Raster 3D (Bacon and Anderson 1988).
F420 binding mode
F420 binds rigidly with an high occupancy (see electron density in Figs. 4 ▶ and 5 ▶) into a cleft built up of strands β2, β5, β6, β7, and IR1. The 5′ deazaisoalloxazine ring is in a pronounced butterfly conformation, the bending angle being 27° (Fig. 4 ▶). The central pyridine ring is kept in its position outside the plane by the nonprolyl cis-peptide oxygen of Gly61 and by the side chain of Val62 pointing toward the Re-site and acting as a backstop (Fig. 5 ▶). Conformation of the 5′ deazaisoalloxazine ring in bMer is maintained at the Si-face by fixing the pyrimidine and hydroxybenzyl rings via hydrogen bonds to Asp35 and His36 and via hydrogen bonds to Leu174, respectively. The rest consisting of a ribitole, phosphate, lactate and two glutamates is buried inside the protein up to the first glutamate and contributes significantly to coenzyme binding. The second partly solvent-exposed glutamate is anchored to the protein matrix by interaction with the peptide nitrogens of Lys161 and Ala112 located at the N-terminal end of helix α4 (Fig. 5 ▶).
Figure 5.

Stereo plot showing the interactions between the coenzyme F420 molecule and bMer was drawn using Bobscript (Esnouf 1997) and Raster 3D (Bacon and Anderson 1988). The isoalloxazine ring is hydrogen-bonded to Asp35, His36, and Leu174. The nonprolyl cis-peptide bond formed by Gly61 and Val62 holds the isoalloxazine ring at its Re-face. The ribitol group is hydrogen-bonded to Gly95 and the phosphate is in hydrogen-bond distance to Gln158 and Gly159. Trp107 provides hydrophobic interaction to the lactate part of F420. The first glutamate of the tail is bound to Met162 and Lys161 and the second glutamate to Asp112 by hydrogen bonds.
In comparison, Adf that deviates by anRMSD of 2.3 Å from bMer using 302 of the 330 Cαatoms for superposition (DALI) (Holm and Sander 1993) showed a highly related F420 binding mode (Fig. 6 ▶) (Aufhammer et al. 2004). In particular, binding of the 5′ deazaisoalloxazine ring from the Re-face is very similar. Differences in amino acid residues are found at the Si-face of F420; as in bMer, sufficient space is required to allow the approach of methylene-H4MPT to the Si-site of C5, whereas in Adf only space for the small secondary alcohol is needed. Additionally, differences are observed around the hydroxybenzyl group, which in bMer is hydrogen-bonded to the peptide carbonyl-oxygen of Leu174, whereas in Adf it only weakly interacts with Ser173. The reason might be that in bMer the bent conformation of the 5′ deazaflavin ring is predominately maintained by electrostatic interactions with the hydroxybenzyl ring and in Adf by steric restraints.
Figure 6.

Superposition of the Cα chains of bMer (red) and Adf (blue) within the F420 binding regions. The nonprolyl cis-peptide bonds (indicated by an arrow), formed by Gly61 and Val62 in bMer and by Cys72 and Ile73 in Adf are shown as ball-and-stick representations. This figure was created using Molscript (Kraulis 1991) and Raster 3D (Bacon and Anderson 1988).
Comparison of the structures of Mer with and without F420 bound
Unfortunately only the structure ofbMer with F420 bound can be compared with the structures of tMer and kMer without the coenzyme bound. A structure of bMer without F420 is not available. However, because of the close structural relationship of the three enzymes (see above) it is nevertheless possible to draw some conclusions. The structural comparison clearly reveals that the F420 binding site is preconstructed in the apoenzyme. The only significant main chain alteration involves IR1, which moves at most 2 Å toward the coenzyme upon F420 binding. IR1 is conformationally variable in tMer and kMer, while the four crystallographically unrelated bMer molecules (RMSD ~0.8 Å) revealed an identical conformation of IR1 reflecting the rigidity of IR1 upon F420 binding. The observed movement places residues like Trp107 and Pro110 of the loop following helix α4-1 in van der Waals distance to the substituent of F420. Interestingly, the side chain of Asp96 in IR1, which probably is involved in methylene-H4MPT binding, changes its conformation upon F420 binding. Asp96 is hydrogen-bonded to Ala98 and Thr99 before F420-binding, whereas after coenzyme binding it points toward the bulk solvent in front of F420. Remarkable is also that Asp96, Gln158, and Asn176 showed higher B factors upon F420 binding, which might correlate with a higher flexibility of these residues.
The methylene-H4MPt binding site of Mer
Methylene-H4MPT is not found in the crystal structure of bMer, although the crystals are grown in the presence of methylene-H4MPT and F420. We assume that the unknown bulky molecule at the C5position of F420 partly occupies the binding site of the pterin and imidazolidine rings and thus prevents the binding of methylene- H4MPT. However, analyses of the Mer structures provide us with a variety of information, which can be used to postulate how methylene-H4MPT binds (Figs. 2 ▶, 7 ▶).
Figure 7.

The proposed binding mode of methylene-H4MPT in Mer. Both substrates, coenzyme F420 and methylene-H4MPT are shown. F420 is colored in red and C/O/N/P atoms of methylene-H4MPT are colored in white, red, blue, and magenta, respectively. The 2Fobs − Fcalc electron density (green) is that of an unspecifically bound polyethylene glycol molecule (only electron density map is shown), which outlines the course of the pterin-substituent. Selected protein residues lining in the binding site and discussed in the text are shown as ball-and-stick representation and are labeled. Possible intermolecular hydrogen bonds between the pterin-ring and the protein are shown by black dashed lines. Helical subdomains α7-1 and α4-1 are shown in green.
The structure of bMer revealed a cleft generated by the (αβ)8 barrel core (β1+β8), IR1 (helix α4-1) and IR3 (helix α7-1) (Fig. 2 ▶). This cleft offers itself as binding site for methylene-H4MPT. The participation of IR3 in methylene-H4MPT binding was previously proposed (Shima et al. 2000). The side of the cleft formed by IR3 is characterized by several solvent-exposed hydrophobic side chains (e.g., Val227 and Phe231 shown in Fig. 7 ▶). These rather hydrophobic residues are involved in binding a PEG molecule, approximately outlining the binding site for the substituent of methylene-H4MPT.
The position of F420 serves as fixed point to position the head group of methylene-H4MPT as its C14aatom has to be within a distance of 3.0–3.5 Å of the C5atom of F420 for optimal hydride transfer. For sterical reasons the bulky head group has to be placed at the Si-face of F420. A hydride transfer at the Si-face is also in agreement with previous studies (Kunow et al. 1993).
As mentioned above, Asp96, Gln158, and Asn176 showed an increased B-factor upon F420 binding, and might be fixed via a hydrogen bond to a bound methylene-H4MPT (Fig. 7 ▶). This is substantiated by the finding that Glu6, Asp96, and Gln158 of bMer are similarly arranged as the corresponding residues Glu25, Asp120, and Gln183 of methylenetetrahydrofolate reductase (MetF). The latter residues in MetF were shown to be involved in methylenetetrahydrofolate binding by site-specific mutagenesis experiments (Trimmer et al. 2001). A similar function of these residues in both enzymes is predicted.
Based on all these factors, a model of methylene- H4MPT was built (Fig. 7 ▶) by taking the conformation of methylene-H4MPT bound in the formaldehydeactivating enzyme (Fae). Fae is the only H4MPT-specific enzyme whose structure with methylene-H4MPT bound has been solved (Acharya et al. 2005). Interestingly, in this conformation the benzyl, ribitol, and ribose moieties of methylene-H4MPT approximately superimposes with the PEG found in the structure (Fig. 7 ▶). This coincidence supports the reliability of the model.
Discussion
The structure of bMer in complex with F420 reported here is the second enzyme in the bacterial luciferase family where the 5′ deazaflavin derivative could be seen in the electron density. Despite the low sequence identity between Mer and Adf (21%) and a few modifications of the F420-binding site, the position and conformation of F420 is remarkably conserved. Moreover, F420 binding induced only minor conformational changes of the polypeptide such that the F420 binding site can be considered to be preconstructed prior to F420 binding. Both findings prompted us to model the FMN molecule into the structure of bacterial luciferase subunit LuxA (Fig. 7 ▶).
Modeling of FMN into LuxA
Despite an insignificant sequence identity between LuxA from Vibrio harveyi (Fisher et al. 1995) and bMer (16%), both enzymes are structurally related as indicated by an RMSD of 2.7 Å when using 292 of the 340 Cαatoms for superposition by DALI (Holm and Sander 1993) (Fig. 8A ▶). Two crystal structures of LuxA are available: 1BRL at 2.5 Å resolution and 1LUC at 1.5 Å resolution. Although the general accuracy of the 1.5 Å structure is improved, allowing the detection of the nonprolyl cis-peptide bond, we used the 2.5 Å structure for constructing a model since the presence of phosphate bound to the enzyme better reflects the protein conformation after FMN binding. Accordingly, modeling of FMN into LuxA is straightforward by transferring F420 from bMer into the superimposed LuxA and by changing it to FMN (Fig. 8B ▶). Except for Leu109, the distances between FMN and the protein atoms are longer than 2 Å, indicating only minor interference. After modeling a cis-peptide bond between Ala74 and Ala75 and the side chain of Leu109 according to the 1.5 Å structure of luciferase His44 was rotated into the position found in bMer. Finally, for adjusting the optimal van der Waals distances between the polypeptide chain and FMN the model was subjected to a short energy minimization procedure using CNS (Brunger et al. 1998; Hagemeier et al. 2003) that did not, however, alter the global results of the experimentally based “flavin transfer modelling” from bMer to luciferase.
Figure 8.
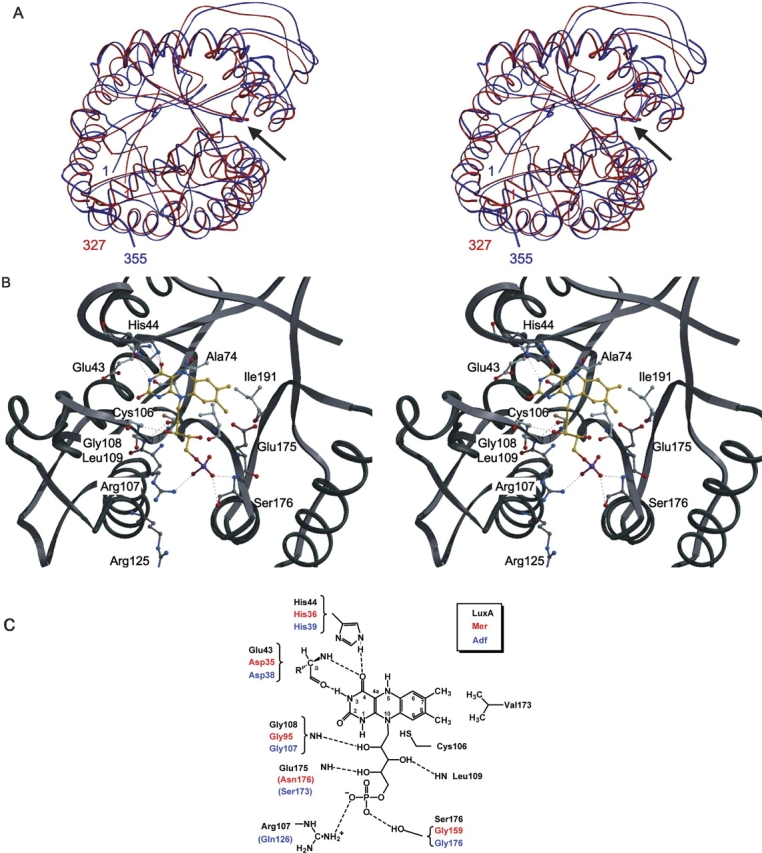
Modeling of FMN into the bacterial luciferase α subunit (LuxA) from Vibrio harveyi. (A) Superposition of the Cαchains of bMer (red) and LuxA (blue). The arrows show the protein segments for the F420/FMN binding pockets. Figure created with Molscript (Kraulis 1991). (B) Stereo views of the modeled complex between LuxA and FMN. Figure created with Bobscript and Raster 3D. (C) Scheme of the proposed FMN binding mode in LuxA. Residues contacting F420 in Mer and Adf are indicated in red and blue, respectively. Conserved residues contacting the coenzyme (F420 or FMN) are shown by brackets. Intermolecular hydrogen bonds between the coenzyme molecule and the protein are shown by dashed lines. Some additional specific contacts are found in the three enzymes. The cis-peptide bond-forming residues are not shown. This figure was produced with ChemWindow (Bio-Rad Laboratories).
As visualized in Figure 8C ▶, the essential flavin–protein interactions conserved between bMer and Adf are maintained in luciferase. However, the surrounding of the dimethylbenzyl group is significantly changed compared to that of the hydroxybenzyl group of Mer. Val173 and Ile191 provide hydrophobic contacts with the methyl groups at C7 and C8 of FMN and might also play a role in maintaining the butterfly conformation (Fig. 8B ▶). The crucial role of Val173 is confirmed by mutations at Val173 that affected the color of bioluminescence emission and/or the reaction kinetics of luciferase catalysis (Lin et al. 2004). Other mutational studies (Baldwin et al. 1995) comprising the residues Asp113, Ser227, Trp194, and Trp250 contacting the isoalloxazine ring are completely compatible with our model (these residues are not shown in Fig. 8 ▶ for clarity). Interestingly, the phosphate molecule found in LuxA is only 1.2 Å and 0.8 Å apart from the independently modeled phosphate groups of F420 in bMer and Adf, respectively. Mutation of Arg107, which is hydrogen bonded to the phosphate group in our FMN model (Fig. 8B, C ▶), to neutral or negatively charged amino acids led to an increased Km value for reduced FMN (Moore et al. 1999; Lin et al. 2001) thereby supporting the correct positioning of the phosphate group.
The new FMN model and its implication for the LuxA reaction
LuxA catalyzes the reaction of reduced FMN, molecular oxygen, and a long-chain aliphatic aldehyde to yield oxidized FMN, aliphatic carboxylic acid, and blue-green light (Baldwin et al. 1979). The binding mode for FMN and aliphatic aldehyde substrates are still not established experimentally, but a model for FMN was proposed based on computer-assisted conformational search methods (Lin et al. 2001) with incorporation of the mutational data and the inorganic phosphate binding site (Xin et al. 1991; Abu-Soud et al. 1993; Moore et al. 1999; Lin et al. 2001). The FMN binding position derived by modelling based on the structure of the bMer-F420 complex is similar to that obtained via computations. However, the orientation of the isoalloxazine ring differs substantially. For example, the nonprolyl cis-peptide oxygen approximately points toward atom N5 of FMN, whereas this oxygen serves as a backstop for the ring in our model. Furthermore, the conformation of the reduced isoalloxazine ring in LuxA was considered to be planar based on nuclear magnetic resonance (NMR) spectra (Vervoort et al. 1986). We favor, in contrast, a bent conformation of the flavin ring in LuxA, as observed in bMer and Adf (Figs. 5 ▶, 6 ▶). A bent conformation of FMN would be compatible with the conformation of FMN and FAD found in other enzymes (Ahn et al. 2004; Edmondson et al. 2004) and with energy calculations of flavins (Hall et al. 1987).
The new model of FMN also provides us with new information about the function of crucial residues that might contribute to a better understanding of the luciferase reaction. The most striking observation is the pronounced butterfly conformation of the isoalloxazine ring that increases the space in front of atom C4a. The bending of flavin ring raises C4aout of the plane and forces it to rehybridize. With binding the hydroperoxy or hydroxy group in its intermediate states, a sp3 hybridization is reached. Whether the bent conformation and its potential alteration can be correlated with the bioluminescence property of luciferase is, of course, an open question. The importance of the nonprolyl cis-peptide bond for isoalloxazine binding was recognized previously (Lin et al. 2001), but its specific function to maintain a bent conformation is only now evident. Accordingly, the dramatic effect of the Cys106Val mutant might be due to a collision between the side chains of Val106 and cis-peptide forming Ala75, thereby destroying the butterfly conformation of FMN. In the bMer derived model, the thiol group of Cys106 has a distance of 3.2 Å to the N1atom of F420 and is not directly involved in the bioluminescence reaction in agreement with previous results (Abu-Soud et al. 1993). Nevertheless, Cys106 of Vibrio harveyi luciferase could stabilize the deprotonated N1atom, increasing the nucleophilicity of atoms N5 and C4a and stabilizing the C4a-hydroperoxy and hydroxy-intermediate states of the flavin (Xi et al. 1990; Abu-Soud et al. 1993). From the structural point of view the most interesting residue is His44, which was rotated toward the Si-site of the isoalloxazine ring in accordance with the conformation found in bMer and Adf. In this position the imidazole ring would be directly in contact with the postulated covalent flavin adducts and might act as a catalytic acid or a catalytic base. These results are in agreement with mutagenesis experiments that already showed the important role of His44 for bioluminescence activity and on aldehyde consumption (Xin et al. 1991). The function of His44 as a catalytic base was also previously proposed (Huang and Tu 1997).
Materials and methods
Materials
Methanosarcina barkeri strain Fusaro (DSMZ 804) was obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen. Blue Sepharose CL-6B and Resource Q columns were from Amersham Biosciences. Coenzyme F420 and tetrahydromethanopterin (H4MPT) were isolated from Methanothermobacter marburgensis (DSMZ 2133) (Shima and Thauer 2001). Initial screening experiments were performed with the Hampton Research crystallization kit (Jancarik and Kim 1991).
Preparation of bMer
M. barkeri was grown in a medium containing 1% methanol at 37°C and cells were harvested in the exponential phase (Karrasch et al. 1989). Cell extracts were prepared in 50 mM Tris/HCl (pH 7.6). All purification steps were performed in an anaerobic chamber (Coy) due to the inactivation of the enzyme under aerobic conditions. bMer was purified using Blue Sepharose CL-6B and Resource Q chromatography columns as previously described (Ma and Thauer 1990a). Additionally, a Ceramic Hydroxyapatit column (BioRad) (14 mL) was applied, which was equilibrated with 30 mL 0.03 M sodium phosphate buffer (pH 6.0). bMer was eluted using a linear gradient of a sodium phosphate buffer (0.03–0.5 M, 200 mL). The enzyme was recovered in the fractions at 0.5 M sodium phosphate. About 4 mg pure enzyme, with a specific activity of 250 U/mg at 55°C under standard assay conditions (Ma and Thauer 1990a) could be isolated from 10 l culture. The purified enzyme was desalted and concentrated in a Centricon 10 microconcentrator (Amicon) and adjusted to a concentration of 13 mg/mL in MOPS/KOH (pH 7.0) supplemented with 0.1 M NaCl for subsequent crystallization. Precipitations that occurred during the enzyme concentration were removed by filtration (pore size 0.45 μm).
Crystallization and data collection
Lyophilised F420 and H4MPT were dissolved in 120 mM sodium phosphate buffer (pH 6.0) under anaerobic conditions. Methylene-H4MPT was synthesized from the spontaneous reaction of formaldehyde with H4MPT (Ma and Thauer 1990a). Shortly before crystallization, the enzyme was mixed with these substrates. The protein concentration was 9 mg/mL; the concentrations of F420 and of methylene-H4MPT were 1.5 mM. The search for crystallization conditions was carried out in an anaerobic chamber at a temperature of 8°C using the hanging drop vapor diffusion method. Crystals grew best in a drop consisting of 1 μL enzyme solution containing the substrates and 1 μL of reservoir solution consisting of 0.1 M Tris/ HCl (pH 8.5), 20% polyethylene glycol (PEG) 4000, and 10% isopropanol. As freezing buffer, the reservoir solution containing 20% glycerol was applied. Crystals were mounted on a fiber loop and flash frozen in a gaseous nitrogen stream (100 K). The space group was P21 and the lattice parameters were a=81.8 Å, b=83.4 Å, c=99.2 Å, and β=91.2° best compatible with one tetramer in the asymmetric unit (Matthews 1968). X-ray data sets were collected at the European Synchroton Radiation Facility (ESRF), Grenoble, France, at the ID29 beamline using an ADSC Quantum 4 CCD detector. The data collection statistics for the data set (Kabsch 1988) are summarized in Table 1.
Table 1.
Data collection and refinement statistics
| Data set | Mer in complex with F420 |
| Wavelength (Å) | 0.9792 |
| Space group | P21 |
| cell (Å,°) | 81.8, 83.4, 99.2, 91.2 |
| Resolution (Å) | 50-2.6 |
| Measured reflectionsa | 160,780 (20,509) |
| Unique reflectionsa | 40,607 (5,785) |
| Completeness (%)a | 97.1 (86.4) |
| Rsym(%)a | 8.5 (21.4) |
| I/σ (I)a | 14.2 (6.8) |
| Rfactor/Rfreeb | 0.185/0.221 |
| No. of protein residues, F420 | 4 × 327, 4 |
| PEG, water molecules, and ions | 1, 239, 2 |
| r.m.s.d. bonds (Å) | 0.012 |
| r.m.s.d. bonds angles (°) | 1.61 |
| Average B factor (Å2) | 28.8 |
aRmerge=∑hkl ∑i|Ii(hkl) − <I(hkl)>|/ ∑hkl ∑iIi(hkl); values in parentheses correspond to the highest resolution shell.
bRfactor=∑|Fobs − Fcalc|/∑Fobs; Rfree calculated with 5% of randomly selected data.
Phase determination, model building, and refinement
Phases of bMer crystals were determined by the method of molecular replacement with the programs AmoRe (Navaza 1994) and EPMR (Kissinger et al. 1999), using the coordinates of a tMer dimer (1F07) as model. For refinement the sequence of bMer was built into the structure of tMer using the SwissProt server (Schwede et al. 2003). Coordinates were refined with the crystallography and NMR system program (CNS) (Brunger et al. 1998) using standard protocols. Manual corrections of the model were performed by examination of the (2Fo − Fc ) and (Fo − Fc ) maps with the program O (Jones et al. 1991). A total of 239 solvent molecules were added using the water pick protocol implemented in CNS. The quality of the model was checked within PROCHECK (Laskowski et al. 1993). The Ramachandran plot showed all of the nonglycine or nonproline residues in the most favored (91.1%) or additionally allowed regions (8.5%) except for one.
Accession number
The structure factors and atomic coordinates of bMer from M. barkerihave been deposited in the RCSB Data Bank with the accession code 1Z69.
Acknowledgments
This work was supported by the Max Planck Society and by the Fonds der Chemischen Industrie. We thank Hartmut Michel for generous support; Peter Haebel for introducing us to molecular modeling; Erica J. Lyon for reading of the manuscript; and Emanuela Screpanti, Carola Hunte, and the members of the ID29 beam line at ESRF for help during data collection.
Abbreviations
FMN, flavin mononucleotide
FAD, flavin adenine dinucleotide
F420, coenzyme F420
NADP, nicotinamide adenine dinucleotide phosphate
H4 MPT, tetrahydromethanopterin
Mer, N5, N10- methylenetetrahydromethanopterin reductase
bMer, Mer from Methanosarcina barkeri, tMer, Mer from Methanothermobacter marburgensis
kMer, Mer from Methanopyrus kandleri
Adf, F420-dependent alcohol dehydrogenase
Lux, bacterial luciferase
SsuD, FMN-dependent alkane-sulfonate monooxygenase
MetF, NADP-dependent FAD harboring N5, N10-methylenetetrahydrofolate reductase
IR, insertion regions in the Mer structures
PEG, polyethylene glycol.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041289805.
References
- Abu-Soud, H.M., Clark, A.C., Francisco, W.A., Baldwin, T.O., and Raushel, F.M. 1993. Kinetic destabilization of the hydroperoxy flavin intermediate by site-directed modification of the reactive thiol in bacterial luciferase. J. Biol. Chem. 268 7699–7706. [PubMed] [Google Scholar]
- Acharya, P., Goenrich, M., Hagemeier, C.H., Demmer, U., Vorholt, J., Thauer, R.K., and Ermler, U. 2005. How an enzyme binds the C1-carrier tetrahydromethanopterin: Structure of the tetrahydromethanopterin dependent formaldehyde-activating enzyme Fae from Methylobacterium extorquens AM1. J. Biol. Chem. (in press). [DOI] [PubMed]
- Ahn, H.J., Yoon, H.J., Lee 2d, B., and Suh, S.W. 2004. Crystal structure of chorismate synthase: A novel FMN-binding protein fold and functional insights. J. Mol. Biol. 336 903–915. [DOI] [PubMed] [Google Scholar]
- Aufhammer, S.W., Warkentin, E., Berk, H., Shima, S., Thauer, R.K., and Ermler, U. 2004. Coenzyme binding in F420-dependent secondary alcohol dehydrogenase, a member of the bacterial luciferase family. Structure 12 361–370. [DOI] [PubMed] [Google Scholar]
- Bacon, D.J. and Anderson, W.F. 1988. A fast algorithm for rendering space-filling molecule pictures. J. Mol. Graph. 6 219–220. [Google Scholar]
- Baldwin, T.O., Ziegler, M.M., and Powers, D.A. 1979. Covalent structure of subunits of bacterial luciferase: NH2-terminal sequence demonstrates subunit homology. Proc. Natl. Acad. Sci. 76 4887–4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin, T.O., Christopher, J.A., Raushel, F.M., Sinclair, J.F., Ziegler, M.M., Fisher, A.J., and Rayment, I. 1995. Structure of bacterial luciferase. Curr. Opin. Struct. Biol. 5 798–809. [DOI] [PubMed] [Google Scholar]
- Becker, A., Schlichting, I., Kabsch, W., Schultz, S., and Wagner, A.F. 1998. Structure of peptide deformylase and identification of the substrate binding site. J. Biol. Chem. 273 11413–11416. [DOI] [PubMed] [Google Scholar]
- Benach, J., Atrian, S., Gonzalez-Duarte, R., and Ladenstein, R. 1999. The catalytic reaction and inhibition mechanism of Drosophila alcohol dehydrogenase: Observation of an enzyme-bound NAD-ketone adduct at 1.4 Å resolution by X-ray crystallography. J. Mol. Biol. 289 335–355. [DOI] [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54 905–921. [DOI] [PubMed] [Google Scholar]
- Edmondson, D.E., Binda, C., and Mattevi, A. 2004. The FAD binding sites of human monoamine oxidases A and B. Neurotoxicology 25 63–72. [DOI] [PubMed] [Google Scholar]
- Eichhorn, E., Davey, C.A., Sargent, D.F., Leisinger, T., and Richmond, T.J. 2002. Crystal structure of Escherichia coli alkanesulfonate mono-oxygenase SsuD. J. Mol. Biol. 324 457–468. [DOI] [PubMed] [Google Scholar]
- Esnouf, R.M. 1997. An extensively modified version of MOLSCRIPT that includes greatly enhanced coloring capabilities. J. Mol. Graph. 15 133– 138. [DOI] [PubMed] [Google Scholar]
- Fisher, A.J., Raushel, F.M., Baldwin, T.O., and Rayment, I. 1995. Three dimensional structure of bacterial luciferase from Vibrio harveyi at 2.4 Å resolution. Biochemistry 34 6581–6586. [DOI] [PubMed] [Google Scholar]
- Fisher, A.J., Thompson, T.B., Thoden, J.B., Baldwin, T.O., and Rayment, I. 1996. The 1.5- Å resolution crystal structure of bacterial luciferase in low salt conditions. J. Biol. Chem. 271 21956–21968. [DOI] [PubMed] [Google Scholar]
- Guenther, B.D., Sheppard, C.A., Tran, P., Rozen, R., Matthews, R.G., and Ludwig, M.L. 1999. The structure and properties of methylenetetrahydrofolate reductase from Escherichia coli suggest how folate ameliorates human hyperhomocysteinemia. Nat. Struct. Biol. 6 359–365. [DOI] [PubMed] [Google Scholar]
- Hagemeier, C.H., Shima, S., Thauer, R.K., Bourenkov, G., Bartunik, H.D., and Ermler, U. 2003. Coenzyme F420-dependent methylenetetrahydromethanopterin dehydrogenase (Mtd) from Methanopyrus kandleri: A methanogenic enzyme with an unusual quarternary structure. J. Mol. Biol. 332 1047–1057. [DOI] [PubMed] [Google Scholar]
- Hall, L.H., Bowers, M.L., and Durfor, C.N. 1987. Further consideration of flavin coenzyme biochemistry afforded by geometry-optimized molecular orbital calculations. Biochemistry 26 7401–7409. [DOI] [PubMed] [Google Scholar]
- Heiss, G., Hofmann, K.W., Trachtmann, N., Walters, D.M., Rouviere, P., and Knackmuss, H.J. 2002. npd gene functions of Rhodococcus (opacus) erythropolis HL PM-1 in the initial steps of 2,4,6-trinitrophenol degradation. Microbiology 148 799–806. [DOI] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1993. Secondary structure comparison by alignment of distance matrices. J. Mol. Biol. 233 123–138. [DOI] [PubMed] [Google Scholar]
- Huang, S. and Tu, S.C. 1997. Identification and characterization of a catalytic base in bacterial luciferase by chemical rescue of a dark mutant. Biochemistry 36 14609–14615. [DOI] [PubMed] [Google Scholar]
- Jancarik, J. and Kim, S.-H. 1991. Sparse matrix sampling: A screening method for crystallization of proteins. J. Appl. Crystallogr. 24 409–411. [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Kabsch, W.J. 1988. Automatic indexing of rotation diffraction patterns. J. Appl. Crystallogr. 21 67–71. [Google Scholar]
- Karrasch, M., Börner, G., Enßle, M., and Thauer, R.K. 1989. Formylmethanofuran dehydrogenase from methanogenic bacteria, a molybdoenzyme. FEBS Lett. 253 226–230. [DOI] [PubMed] [Google Scholar]
- Kissinger, C.R., Gehlhaar, D.K., and Fogel, D.B. 1999. Rapid automated molecular replacement by evolutionary search. Acta Crystallogr. D Biol. Crystallogr. 55 484–491. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Kunow, J., Schworer, B., Setzke, E., and Thauer, R.K. 1993. Si-face stereospecificity at C5 of coenzyme F420 for F420-dependentN5, N10-methylenetetrahydromethanopterin dehydrogenase, F420-dependent N5, N10- methylenetetrahydromethanopterin reductase and F420 H2:dimethylnaphthoquinone oxidoreductase. Eur. J. Biochem. 214 641–646. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., Macarthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK—A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26 283–291. [Google Scholar]
- Lin, L.Y., Sulea, T., Szittner, R., Vassilyev, V., Purisima, E.O., and Meighen, E.A. 2001. Modeling of the bacterial luciferase–flavin mononucleotide complex combining flexible docking with structure–activity data. Protein Sci. 10 1563–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, L.Y., Szittner, R., Friedman, R., and Meighen, E.A. 2004. Changes in the kinetics and emission spectrum on mutation of the chromophore-binding platform in Vibrio harveyi luciferase. Biochemistry 43 3183–3194. [DOI] [PubMed] [Google Scholar]
- Ma, K. and Thauer, R.K. 1990a. N5, N10-Methylenetetrahydromethanopterin reductase from Methanosarcina barkeri. FEMS Microbiol. Lett. 70 119–124. [Google Scholar]
- ———. 1990b. Purification and properties of N5, N10-methylenetetrahydromethanopterin reductase from Methanobacterium thermoautotrophicum (strain Marburg). Eur. J. Biochem. 191 187–193. [DOI] [PubMed] [Google Scholar]
- Ma, K., Linder, D., Stetter, K.O., and Thauer, R.K. 1991. Purification and properties of N5, N10-methylenetetrahydromethanopterin reductase (coenzyme F420-dependent) from the extreme thermophile Methanopyrus kandleri. Arch. Microbiol. 155 593–600. [DOI] [PubMed] [Google Scholar]
- Matthews, B.W. 1968. Solvent content of protein crystals. J. Mol. Biol. 33 491–497. [DOI] [PubMed] [Google Scholar]
- Moore, C., Lei, B., and Tu, S.C. 1999. Relationship between the conserved α subunit arginine 107 and effects of phosphate on the activity and stability of Vibrio harveyiluciferase. Arch. Biochem. Biophys. 370 45–50. [DOI] [PubMed] [Google Scholar]
- Peschke, U., Schmidt, H., Zhang, H.Z., and Piepersberg, W. 1995. Molecular characterization of the lincomycin-production gene cluster of Streptomyces lincolnensis78-11. Mol. Microbiol. 16 1137–1156. [DOI] [PubMed] [Google Scholar]
- Purwantini, E. and Daniels, L. 1998. Molecular analysis of the gene encoding F420-dependent glucose-6-phosphate dehydrogenase from Mycobacterium smegmatis. J. Bacteriol. 180 2212–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pylypenko, O., Vitali, F., Zerbe, K., Robinson, J.A., and Schlichting, I. 2003. Crystal structure of OxyC, a cytochrome P450 implicated in an oxidative C-C coupling reaction during vancomycin biosynthesis. J. Biol. Chem. 278 46727–46733. [DOI] [PubMed] [Google Scholar]
- Schmitz, R.A., Linder, D., Stetter, K.O., and Thauer, R.K. 1991. N-5 N-10 Methylenetetrahydromethanopterin reductase coenzyme F-420-dependent and formylmethanofuran dehydrogenase from the hyperthermophile Archaeoglobus fulgidus. Arch. Microbiol. 156 427–434. [Google Scholar]
- Schwede, T., Kopp, J., Guex, N., and Peitsch, M.C. 2003. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 31 3381–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima, S. and Thauer, R.K. 2001. Tetrahydromethanopterin-specific enzymes from Methanopyrus kandleri. Methods Enzymol. 331 317–353. [DOI] [PubMed] [Google Scholar]
- Shima, S., Warkentin, E., Grabarse, W., Sordel, M., Wicke, M., Thauer, R.K., and Ermler, U. 2000. Structure of coenzyme F(420) dependent methylenetetrahydromethanopterin reductase from two methanogenic archaea. J. Mol. Biol. 300 935–950. [DOI] [PubMed] [Google Scholar]
- Shima, S., Warkentin, E., Thauer, R.K., and Ermler, U. 2002. Structure and function of enzymes involved in the methanogenic pathway utilizing carbon dioxide and molecular hydrogen. J. Biosci. Bioeng. 93 519–530. [DOI] [PubMed] [Google Scholar]
- te Brommelstroet, B.W., Hensgens, C.M., Keltjens, J.T., van der Drift, C., and Vogels, G.D. 1990. Purification and properties of 5,10-methylenetetrahydromethanopterin reductase, a coenzyme F420-dependent enzyme, from Methanobacterium thermoautotrophicum strain ΔH. J. Biol. Chem. 265 1852–1857. [PubMed] [Google Scholar]
- Trimmer, E.E., Ballou, D.P., Ludwig, M.L., and Matthews, R.G. 2001. Folate activation and catalysis in methylenetetrahydrofolate reductase from Escherichia coli: Roles for aspartate 120 and glutamate 28. Biochemistry 40 6216–6226. [DOI] [PubMed] [Google Scholar]
- van Der Ploeg, J.R., Iwanicka-Nowicka, R., Bykowski, T., Hryniewicz, M.M., and Leisinger, T. 1999. The Escherichia coli ssuEADCB gene cluster is required for the utilization of sulfur from aliphatic sulfonates and is regulated by the transcriptional activator Cbl. J. Biol. Chem. 274 29358–29365. [DOI] [PubMed] [Google Scholar]
- Vaupel, M. and Thauer, R.K. 1995. Coenzyme F420-dependent N5, N10- methylenetetrahydromethanopterin reductase (Mer) from Methanobacterium thermoautotrophicumstrain Marburg. Cloning, sequencing, transcriptional analysis, and functional expression in Escherichia coliof the mer gene. Eur. J. Biochem. 231 773–778. [DOI] [PubMed] [Google Scholar]
- Vervoort, J., Muller, F., O’Kayne, D., Lee, J., and Bacher, A. 1986. Bacterial luciferase: A carbon-13, nitrogen-15, and phosphorus-31 nuclear magnetic resonance investigation. Biochemistry 25 8067–8075. [DOI] [PubMed] [Google Scholar]
- Vorholt, J., Kunow, J., Stetter, K.O., and Thauer, R.K. 1995. Enzymes and coenzymes of the carbon monoxide dehydrogenase pathway for autotrophic CO2 fixation in Archaeoglobus lithotrophicus and the lack of carbon monoxide dehydrogenase in the heterotrophic A. profundus. Arch. Microbiol. 163 112–118. [Google Scholar]
- Vorholt, J.A., Hafenbradl, D., Stetter, K.O., and Thauer R.K. 1997. Pathways of autotrophic CO2 fixation and of dissimilatory nitrate reduction to N2 O in Ferroglobus placidus. Arch. Microbiol. 167 19–23. [DOI] [PubMed] [Google Scholar]
- Xi, L., Cho, K.W., Herndon, M.E., and Tu, S.C. 1990. Elicitation of an oxidase activity in bacterial luciferase by site-directed mutation of a noncatalytic residue. J. Biol. Chem. 265 4200–4203. [PubMed] [Google Scholar]
- Xin, X., Xi, L., and Tu, S.C. 1991. Functional consequences of site-directed mutation of conserved histidyl residues of the bacterial luciferase α subunit. Biochemistry 30 11255–11262. [DOI] [PubMed] [Google Scholar]